
VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions

 , ,
, , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling Datasets
2.2. Molecular Fingerprints
2.3. Classification Models
2.4. Model Building and Evaluation
2.5. Final Model Evaluation Metrics
2.6. Consensus Strategy and Consensus Score
3. Results and Discussion
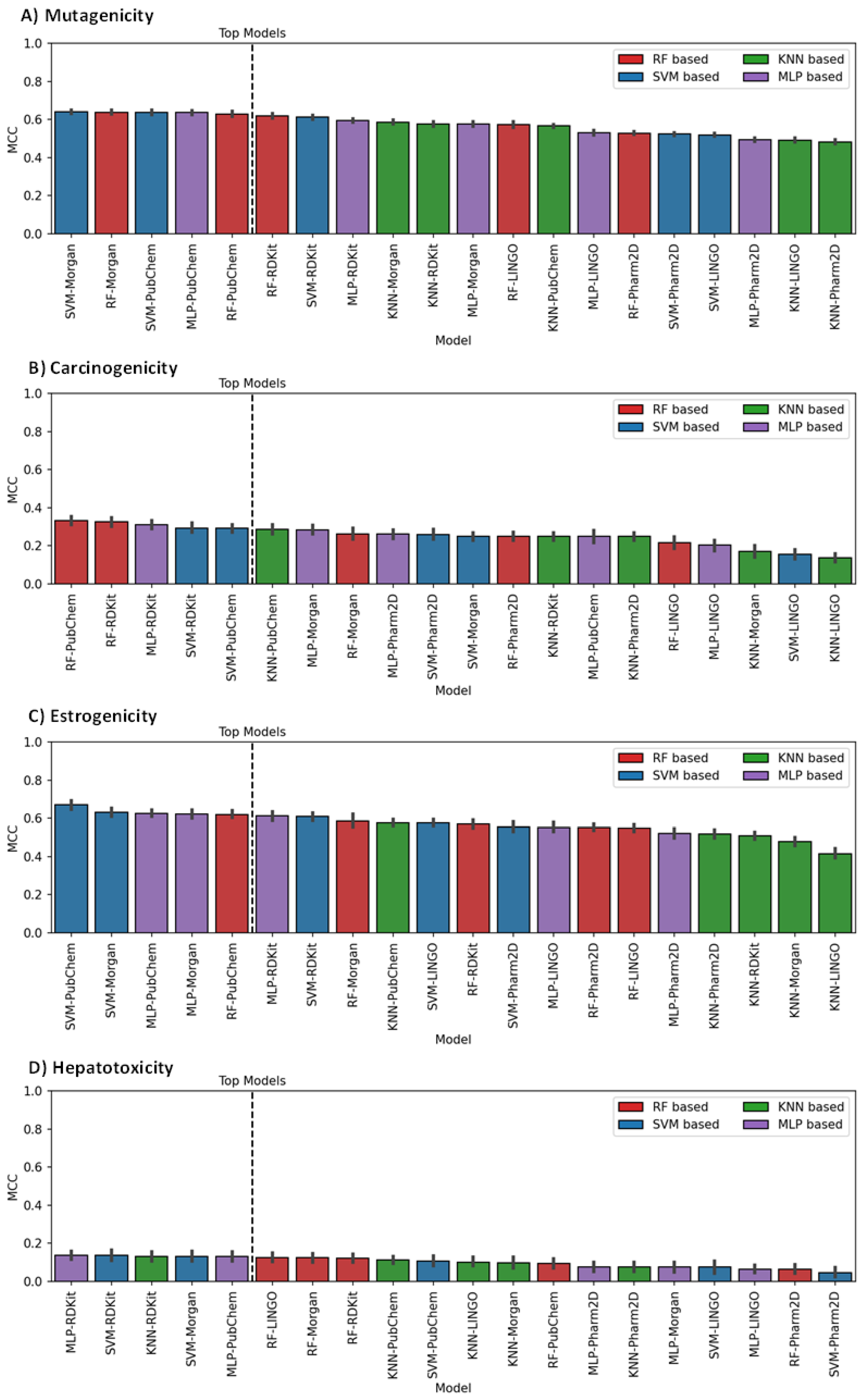
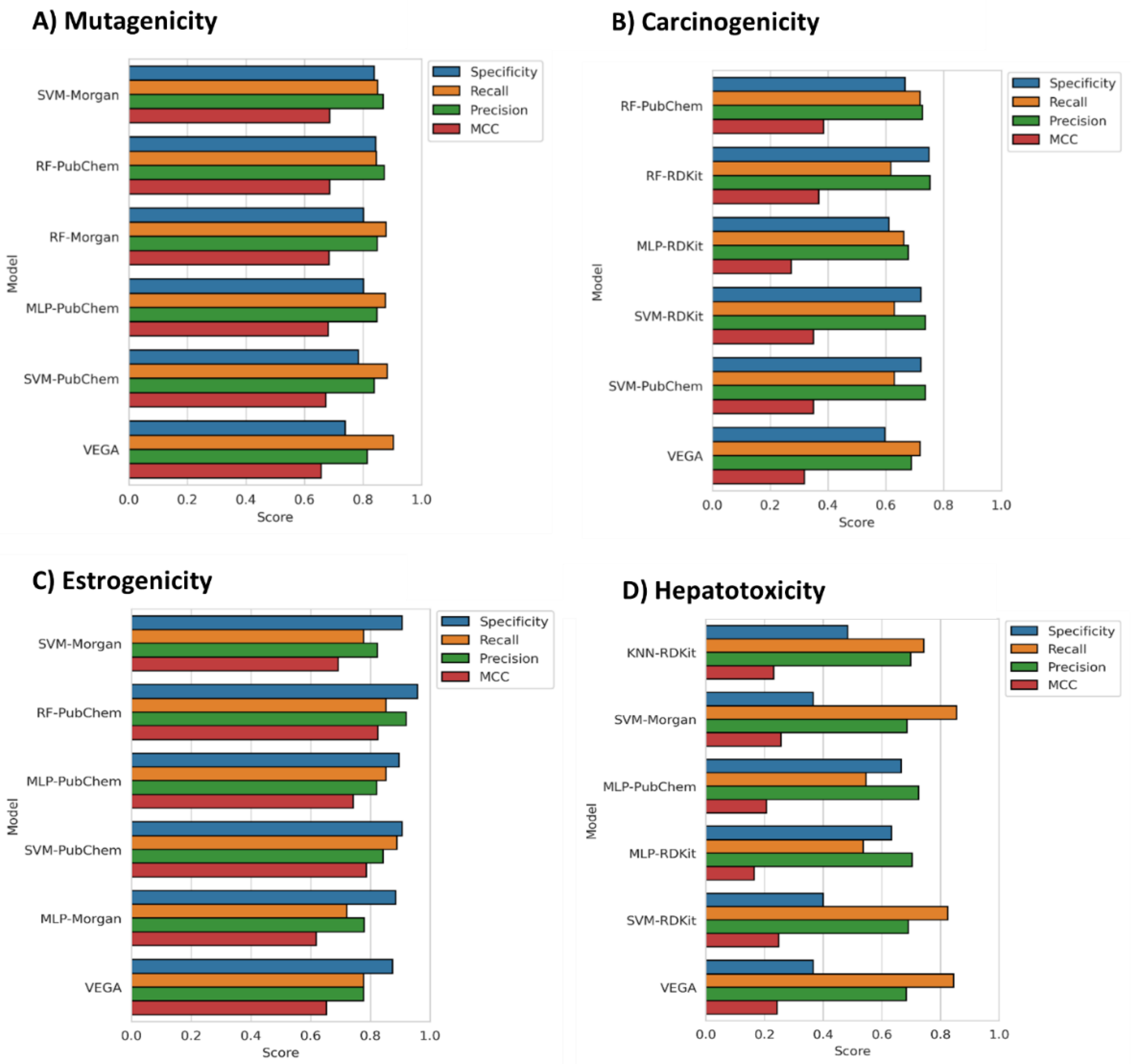
3.1. Model Generation, Optimization and Selection
3.2. Models Evaluation
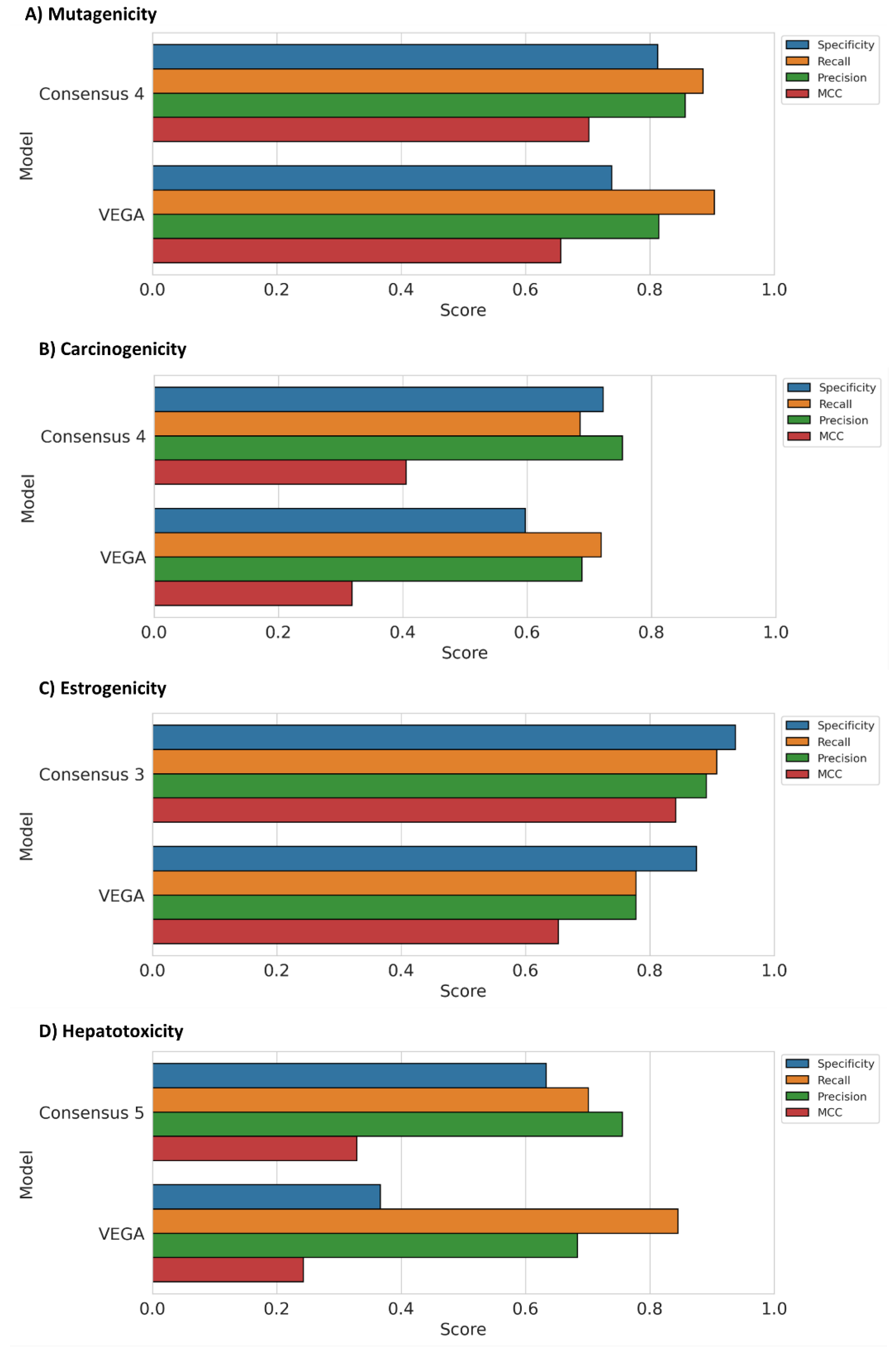
3.3. Consensus Strategy
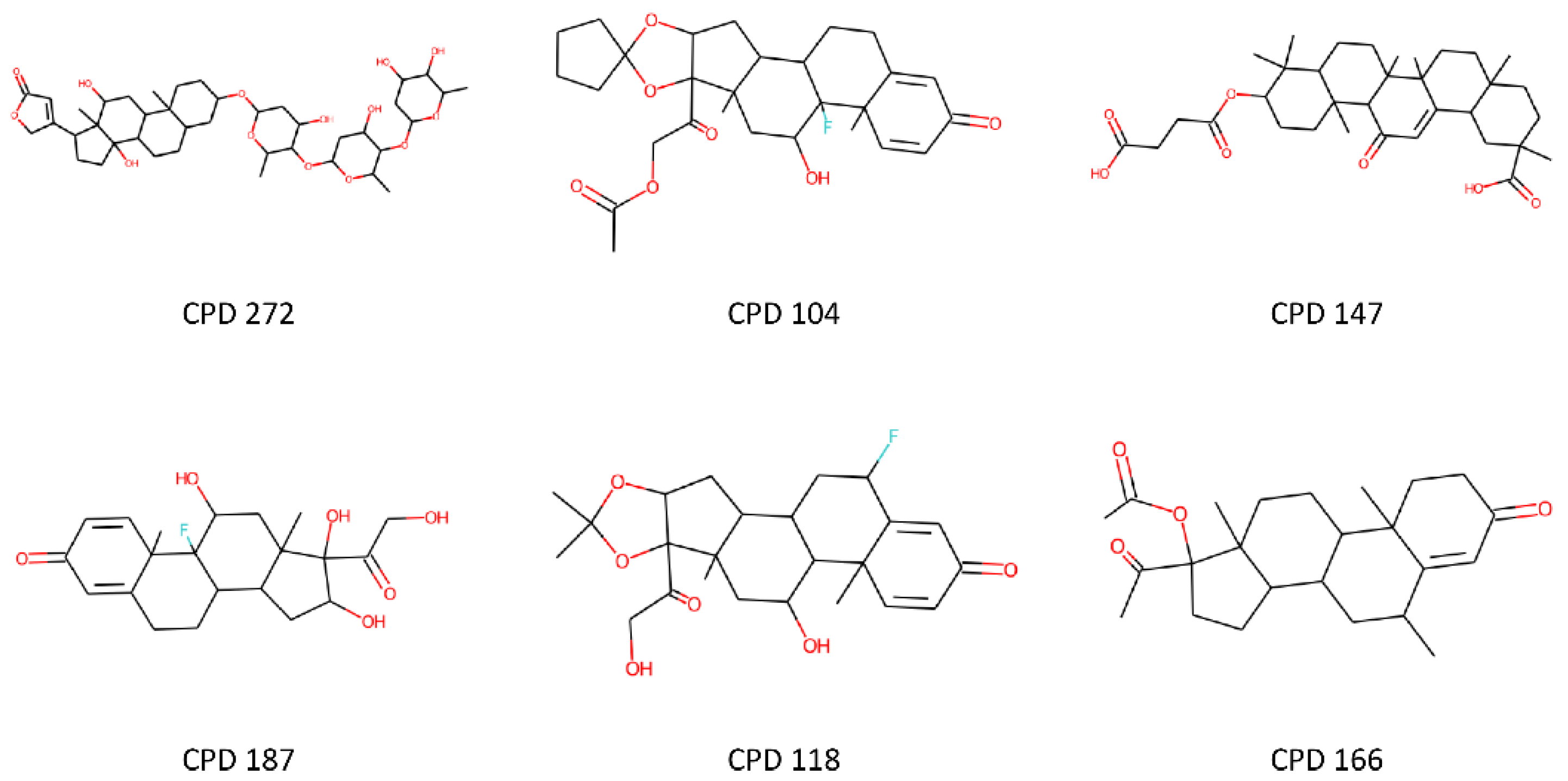
3.4. Structure-Based Analysis of Toxicity Predictions
3.5. VenomPred Web Tool
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- REACH. 2006. Available online: https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:02006R1907-20200824 (accessed on 10 February 2022).
- ECHA. 2007. Available online: https://echa.europa.eu/legislation (accessed on 10 February 2022).
- Innovative Medicines Initiative. IMI 2010. Available online: www.imi.europa.eu (accessed on 10 February 2022).
- eTOX Project. 2010–2016. Available online: http://www.e-tox.net (accessed on 10 February 2022).
- TOX21 Strategy. Available online: https://www.niehs.nih.gov/research/programs/tox21/index.cfm (accessed on 10 February 2022).
- Galati, S.; Di Stefano, M.; Martinelli, E.; Poli, G.; Tuccinardi, T. Recent Advances in In Silico Target Fishing. Molecules 2021, 26, 5124. [Google Scholar] [CrossRef] [PubMed]
- VEGAHUB. Available online: https://www.vegahub.eu (accessed on 10 February 2022).
- Ferrari, T.; Gini, G. An open source multistep model to predict mutagenicity from statistical analysis and relevant structural alerts. Chem. Cent. J. 2010, 4 (Suppl. 1), S2. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Lee, F.D.; Durston, W.E. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc. Natl. Acad. Sci. USA 1973, 70, 782–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortelmans, K.; Zeiger, E. The Ames Salmonella/microsome mutagenicity assay. Mutat. Res. 2000, 455, 29–60. [Google Scholar] [CrossRef]
- Fjodorova, N.; Vračko, M.; Novič, M.; Roncaglioni, A.; Benfenati, E. New public QSAR model for carcinogenicity. Chem. Cent. J. 2010, 4 (Suppl. 1), S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncaglioni, A.; Piclin, N.; Pintore, M.; Benfenati, E. Binary classification models for endocrine disrupter effects mediated through the estrogen receptor. SAR QSAR Environ. Res. 2008, 19, 697–733. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, F.; Lombardo, A.; Manganaro, A.; Benfenati, E. A New Structure-Activity Relationship (SAR) Model for Predicting Drug-Induced Liver Injury, Based on Statistical and Expert-Based Structural Alerts. Front. Pharmacol. 2016, 7, 442. [Google Scholar] [CrossRef] [Green Version]
- Varnek, A.; Baskin, I. Machine Learning Methods for Property Prediction in Chemoinformatics: Quo Vadis? J. Chem. Inf. Model. 2012, 52, 1413–1437. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org (accessed on 10 February 2022).
- Ji, H.; Deng, H.; Lu, H.; Zhang, Z. Predicting a Molecular Fingerprint from an Electron Ionization Mass Spectrum with Deep Neural Networks. Anal. Chem. 2020, 92, 8649–8653. [Google Scholar] [CrossRef]
- Laufkötter, O.; Sturm, N.; Bajorath, J.; Chen, H.; Engkvist, O. Combining structural and bioactivity-based fingerprints improves prediction performance and scaffold hopping capability. J. Cheminform. 2019, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Vidal, D.; Thormann, M.; Pons, M. LINGO, an Efficient Holographic Text Based Method To Calculate Biophysical Properties and Intermolecular Similarities. J. Chem. Inf. Model. 2005, 45, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Noble, W.S. What is a support vector machine? Nat. Biotechnol. 2006, 24, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; LeTiran, A.; Xiao, Y.; Golbraikh, A.; Kohn, H.; Tropsha, A. Quantitative Structure−Activity Relationship Analysis of Functionalized Amino Acid Anticonvulsant Agents Using k Nearest Neighbor and Simulated Annealing PLS Methods. J. Med. Chem. 2002, 45, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z. Introduction to machine learning: K-nearest neighbors. Ann. Transl. Med. 2016, 4, 218. [Google Scholar] [CrossRef] [Green Version]
- Cova, T.F.G.G.; Pais, A.A.C.C. Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns. Front. Chem. 2019, 7, 809. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Chen, M.; Borlak, J.; Tong, W. Predicting idiosyncratic drug-induced liver injury—Some recent advances. Expert Rev. Gastroenterol. Hepatol. 2014, 8, 721–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, W.J.; Zlokarnik, G.; Grootenhuis, P.D.J. In silico prediction of drug safety: Despite progress there is abundant room for improvement. Drug Discov. Today Technol. 2004, 1, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Forrez, I.; Boon, N.; Verstraete, W.; Carballa, M. 6.38—Biodegradation of Micropollutants and Prospects for Water and Wastewater Biotreatment; Moo-Young, M.B.T.-C.B., Second, E., Eds.; Academic Press: Burlington, NJ, USA, 2011; pp. 485–494. ISBN 978-0-08-088504-9. [Google Scholar]
- Tuccinardi, T.; Poli, G.; Romboli, V.; Giordano, A.; Martinelli, A. Extensive consensus docking evaluation for ligand pose prediction and virtual screening studies. J. Chem. Inf. Model. 2014, 54, 2980–2986. [Google Scholar] [CrossRef]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Reliability analysis and optimization of the consensus docking approach for the development of virtual screening studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Bienfait, B.; Ertl, P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashby, J.; Tennant, R.W. Chemical structure, Salmonella mutagenicity and extent of carcinogenicity as indicators of genotoxic carcinogenesis among 222 chemicals tested in rodents by the U.S. NCI/NTP. Mutat. Res. 1988, 204, 17–115. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutagenicity Model | |||
| Set | Total | Mutagen | Non-mutagen |
| Training | 3367 | 1883 | 1484 |
| Test | 798 | 446 | 352 |
| Carcinogenicity Model | |||
| Set | Total | Carcinogenic | Non-carcinogenic |
| Training | 645 | 333 | 312 |
| Test | 161 | 89 | 72 |
| Estrogenicity Model | |||
| Set | Total | Active | Inactive |
| Training | 656 | 234 | 422 |
| Test | 150 | 54 | 96 |
| Hepatotoxicity Model | |||
| Set | Total | Toxic | Non-toxic |
| Training | 760 | 408 | 352 |
| Test | 157 | 97 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galati, S.; Di Stefano, M.; Martinelli, E.; Macchia, M.; Martinelli, A.; Poli, G.; Tuccinardi, T. VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions. Int. J. Mol. Sci. 2022, 23, 2105. https://doi.org/10.3390/ijms23042105
Galati S, Di Stefano M, Martinelli E, Macchia M, Martinelli A, Poli G, Tuccinardi T. VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions. International Journal of Molecular Sciences. 2022; 23(4):2105. https://doi.org/10.3390/ijms23042105
Chicago/Turabian StyleGalati, Salvatore, Miriana Di Stefano, Elisa Martinelli, Marco Macchia, Adriano Martinelli, Giulio Poli, and Tiziano Tuccinardi. 2022. "VenomPred: A Machine Learning Based Platform for Molecular Toxicity Predictions" International Journal of Molecular Sciences 23, no. 4: 2105. https://doi.org/10.3390/ijms23042105