
An Essential Role for Alzheimer’s-Linked Amyloid Beta Oligomers in Neurodevelopment: Transient Expression of Multiple Proteoforms during Retina Histogenesis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Alzheimer’s-Linked AβOs and pTau Are Expressed in the Embryonic Retina
2.2. Verification of AβO Identification Using Mass Spectrometry
2.3. Western Blots Show the Presence of Antibody-Specific AβO Proteoforms
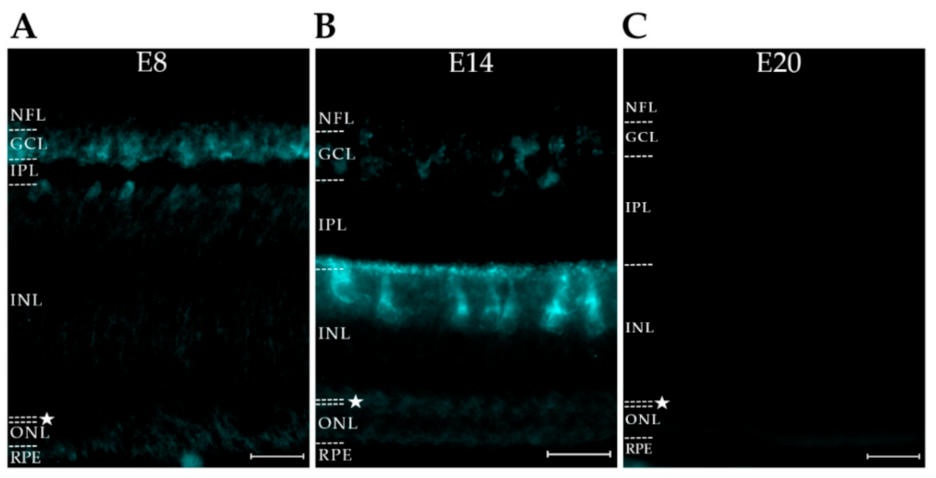
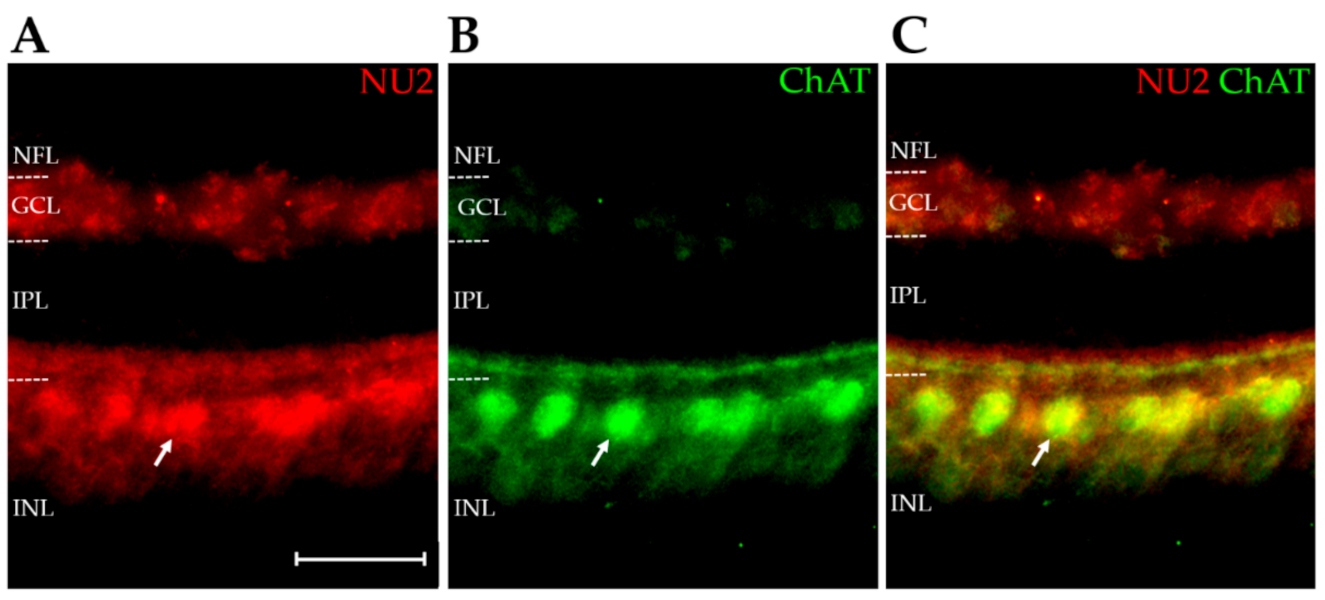
2.4. AβOs Are Expressed in a Cell-Specific Manner
2.5. AβO Proteoforms Differ in Spatiotemporal Expression
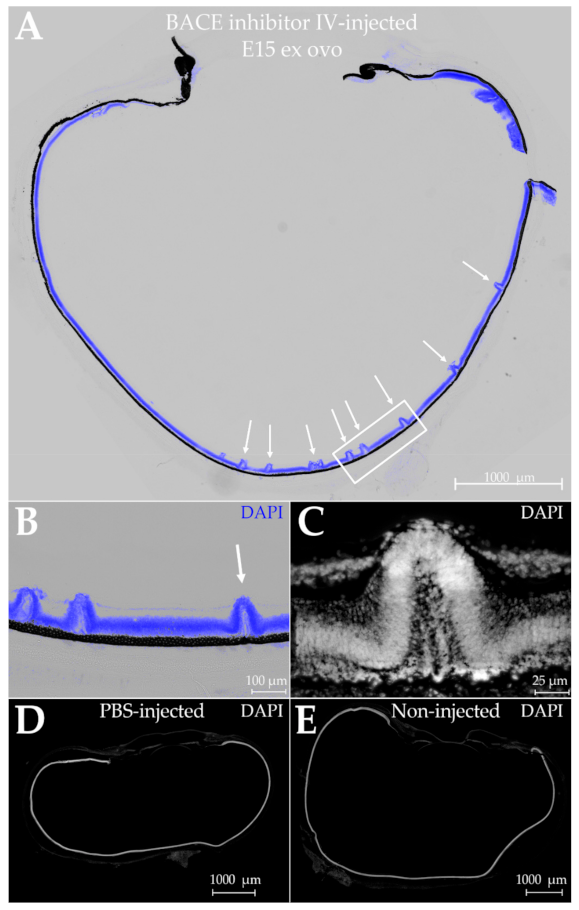
2.6. Dependence of Retina Morphogenesis on AβOs
3. Discussion
3.1. AβOs, Neurotoxins Linked to AD, May Be Essential for Neurodevelopment
3.2. Multiple Methods Substantiated the Presence of AβOs and Revealed Distinct AβO Proteoforms
3.3. The AβO Proteoforms Targeted by the Three Antibodies Show Striking Spatiotemporal Regulation, with Patterns That Were Clearly Different from Each Other
3.4. Developing Retina and AD-Related pTau
3.5. Investigations into Possible Functions of AβOs in the Embryonic Nervous System Are Just Beginning
3.6. Important Questions for the Future
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Dissection, Fixation, Sectioning, and Immunohistofluorescence
4.4. Immunoblot
4.5. Gel-Eluted Liquid-Fraction Entrapment Electrophoresis (GELFrEE)
4.6. Liquid Chromatography-Mass Spectrometry (LC-MS)
4.7. Label-Free Quantitative Top-down Analysis
4.8. Shell-Free (Ex-Ovo) Embryo Culturing
4.9. Intravitreal Injections
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.; Chang, L.; Viola, K.L.; Lacor, P.N.; Lambert, M.P.; Finch, C.E.; Krafft, G.A.; Klein, W.L. Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georganopoulou, D.G.; Chang, L.; Nam, J.M.; Thaxton, C.S.; Mufson, E.J.; Klein, W.L.; Mirkin, C.A. Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2273–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Bakhos, L.; Wang, Z.; Venton, D.L.; Klein, W.L. Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J. Mol. Neurosci. MN 2003, 20, 305–313. [Google Scholar] [CrossRef]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Tran, L.; Lambert, M.P.; Glabe, C.G.; Klein, W.L.; LaFerla, F.M. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J. Biol. Chem. 2006, 281, 1599–1604. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, M.T.; Partridge, V.; Leon, W.C.; Canneva, F.; Allard, S.; Arvanitis, D.N.; Vercauteren, F.; Houle, D.; Ducatenzeiler, A.; Klein, W.L.; et al. Transgenic mice as a model of pre-clinical Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 4–23. [Google Scholar] [CrossRef]
- Tomiyama, T.; Matsuyama, S.; Iso, H.; Umeda, T.; Takuma, H.; Ohnishi, K.; Ishibashi, K.; Teraoka, R.; Sakama, N.; Yamashita, T.; et al. A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 4845–4856. [Google Scholar] [CrossRef] [Green Version]
- Welikovitch, L.A.; Do Carmo, S.; Magloczky, Z.; Malcolm, J.C.; Loke, J.; Klein, W.L.; Freund, T.; Cuello, A.C. Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain. Proc. Natl. Acad. Sci. USA 2020, 117, 6844–6854. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Shankar, G.M.; Townsend, M.; Fadeeva, J.V.; Betts, V.; Podlisny, M.B.; Cleary, J.P.; Ashe, K.H.; Rowan, M.J.; et al. The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention. Biochem. Soc. Trans. 2005, 33, 1087–1090. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Vieira, M.N.; De Felice, F.G. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life 2007, 59, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goure, W.F.; Krafft, G.A.; Jerecic, J.; Hefti, F. Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics. Alzheimer’s Res. Ther. 2014, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Viola, K.L.; Klein, W.L. Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Wisniewski, T.; Drummond, E. Future horizons in Alzheimer’s disease research. Prog. Mol. Biol. Transl. Sci. 2019, 168, 223–241. [Google Scholar] [CrossRef]
- Ashe, K.H. The biogenesis and biology of amyloid beta oligomers in the brain. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 1561–1567. [Google Scholar] [CrossRef]
- Tolar, M.; Hey, J.; Power, A.; Abushakra, S. Neurotoxic Soluble Amyloid Oligomers Drive Alzheimer’s Pathogenesis and Represent a Clinically Validated Target for Slowing Disease Progression. Int. J. Mol. Sci. 2021, 22, 6355. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Hayden, E.Y.; Teplow, D.B. Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. JAD 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; Gyorgy, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 100, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 1982, 32, 164–168. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Lee, H.; Brott, B.K.; Kirkby, L.A.; Adelson, J.D.; Cheng, S.; Feller, M.B.; Datwani, A.; Shatz, C.J. Synapse elimination and learning rules co-regulated by MHC class I H2-Db. Nature 2014, 509, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Hughes, W.F.; McLoon, S.C. Ganglion cell death during normal retinal development in the chick: Comparisons with cell death induced by early target field destruction. Exp. Neurol. 1979, 66, 587–601. [Google Scholar] [CrossRef]
- Martin-Estebane, M.; Navascues, J.; Sierra-Martin, A.; Martin-Guerrero, S.M.; Cuadros, M.A.; Carrasco, M.C.; Marin-Teva, J.L. Onset of microglial entry into developing quail retina coincides with increased expression of active caspase-3 and is mediated by extracellular ATP and UDP. PLoS ONE 2017, 12, e0182450. [Google Scholar] [CrossRef] [Green Version]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. Embo J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forny-Germano, L.; Lyra e Silva, N.M.; Batista, A.F.; Brito-Moreira, J.; Gralle, M.; Boehnke, S.E.; Coe, B.C.; Lablans, A.; Marques, S.A.; Martinez, A.M.; et al. Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 13629–13643. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.; Enam, S.A.; Bawa, N.; Miller, B.E.; Ghanbari, H.A.; Klein, W.L. Phosphorylated tau epitope of Alzheimer’s disease is coupled to axon development in the avian central nervous system. Exp. Neurol. 1993, 120, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Rosner, H.; Rebhan, M.; Vacun, G.; Vanmechelen, E. Expression of a paired helical filament tau epitope in embryonic chicken central nervous system. Neuroreport 1994, 5, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Rosner, H.; Rebhan, M.; Vacun, G.; Vanmechelen, E. Developmental expression of tau proteins in the chicken and rat brain: Rapid down-regulation of a paired helical filament epitope in the rat cerebral cortex coincides with the transition from immature to adult tau isoforms. Int. J. Dev. Neurosci. 1995, 13, 607–617. [Google Scholar] [CrossRef]
- Burack, M.A.; Halpain, S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience 1996, 72, 167–184. [Google Scholar] [CrossRef]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [Green Version]
- Esselmann, H.; Maler, J.; Kunz, N.; Otto, M.; Paul, S.; Lewczuk, P.; Rüther, E.; Kornhuber, J.; Wiltfang, J. Lithium decreases secretion of Aβ1–42 and C-truncated species Aβ1–37/38/39/40 in chicken telencephalic cultures but specifically increases intracellular Aβ1–38. Neurodegener. Dis. 2004, 1, 236–241. [Google Scholar] [CrossRef]
- Mileusnic, R.; Rose, S. The chick as a model for the study of the cellular mechanisms and potential therapies for Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, 2010, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barnes, N.Y.; Li, L.; Yoshikawa, K.; Schwartz, L.M.; Oppenheim, R.W.; Milligan, C.E. Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 5869–5880. [Google Scholar] [CrossRef] [Green Version]
- Caswell, M.D.; Mok, S.S.; Henry, A.; Cappai, R.; Klug, G.; Beyreuther, K.; Masters, C.L.; Small, D.H. The amyloid beta-protein precursor of Alzheimer’s disease is degraded extracellularly by a Kunitz protease inhibitor domain-sensitive trypsin-like serine protease in cultures of chick sympathetic neurons. Eur J. Biochem. 1999, 266, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shepardson, N.; Yang, T.; Chen, G.; Walsh, D.; Selkoe, D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 5819–5824. [Google Scholar] [CrossRef] [Green Version]
- Seward, M.E.; Swanson, E.; Norambuena, A.; Reimann, A.; Cochran, J.N.; Li, R.; Roberson, E.D.; Bloom, G.S. Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 2013, 126, 1278–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weysse, A.W.; Burgess, W.S. Histogenesis of the retina. Am. Nat. 1906, 40, 611–637. [Google Scholar] [CrossRef] [Green Version]
- Tsui, H.C.; Pope, W.B.; Kim, C.S.; Klein, W.L. Transient expression of adheron molecules during chick retinal development. J. Neurobiol. 1992, 23, 720–738. [Google Scholar] [CrossRef]
- Grunwald, G.B.; Fredman, P.; Magnani, J.L.; Trisler, D.; Ginsburg, V.; Nirenberg, M. Monoclonal antibody 18B8 detects gangliosides associated with neuronal differentiation and synapse formation. Proc. Natl. Acad. Sci. USA 1985, 82, 4008–4012. [Google Scholar] [CrossRef] [Green Version]
- Daniloff, J.K.; Chuong, C.M.; Levi, G.; Edelman, G.M. Differential distribution of cell adhesion molecules during histogenesis of the chick nervous system. J. Neurosci. Off. J. Soc. Neurosci. 1986, 6, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.O.; Yamawaki, R.M.; Shatz, C.J. Synaptic Contacts and the Transient Dendritic Spines of Developing Retinal Ganglion Cells. Eur. J. Neurosci. 1992, 4, 1387–1397. [Google Scholar] [CrossRef]
- Harris, W.A. Cellular diversification in the vertebrate retina. Curr. Opin. Genet. Dev. 1997, 7, 651–658. [Google Scholar] [CrossRef]
- Chow, R.L.; Lang, R.A. Early eye development in vertebrates. Annu. Rev. Cell Dev. Biol. 2001, 17, 255–296. [Google Scholar] [CrossRef] [Green Version]
- Reis, R.A.; Ventura, A.L.; Kubrusly, R.C.; de Mello, M.C.; de Mello, F.G. Dopaminergic signaling in the developing retina. Brain Res. Rev. 2007, 54, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Kostadinov, D.; Sanes, J.R. Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. eLife 2015, 4, e08964. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sanes, J.R. Cellular and Molecular Analysis of Dendritic Morphogenesis in a Retinal Cell Type That Senses Color Contrast and Ventral Motion. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 12247–12262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raeisossadati, R.; Ferrari, M.F.R.; Kihara, A.H.; AlDiri, I.; Gross, J.M. Epigenetic regulation of retinal development. Epigenetics Chromatin 2021, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.A.; Jiang, D.; Mackin, R.D.; Samuel, M.A. Development and maintenance of vision’s first synapse. Dev. Biol 2021, 476, 218–239. [Google Scholar] [CrossRef]
- Yamagata, M.; Yan, W.; Sanes, J.R. A cell atlas of the chick retina based on single-cell transcriptomics. eLife 2021, 10, e63907. [Google Scholar] [CrossRef]
- Goldberg, S.; Coulombre, A.J. Topographical development of the ganglion cell fiber layer in the chick retina. A whole mount study. J. Comp. Neurol. 1972, 146, 507–517. [Google Scholar] [CrossRef]
- Drenhaus, U.; Morino, P.; Veh, R.W. On the development of the stratification of the inner plexiform layer in the chick retina. J. Comp. Neurol. 2003, 460, 1–12. [Google Scholar] [CrossRef]
- Thangaraj, G.; Greif, A.; Bachmann, G.; Layer, P.G. Intricate paths of cells and networks becoming “cholinergic” in the embryonic chicken retina. J. Comp. Neurol. 2012, 520, 3181–3193. [Google Scholar] [CrossRef]
- Robles, E.; Baier, H. Assembly of synaptic laminae by axon guidance molecules. Curr. Opin. Neurobiol. 2012, 22, 799–804. [Google Scholar] [CrossRef]
- Amini, R.; Rocha-Martins, M.; Norden, C. Neuronal Migration and Lamination in the Vertebrate Retina. Front. Neurosci. 2017, 11, 742. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Crowther, R.; Six, J.; Lübke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.; Lee, V. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Natl. Acad. Sci. USA 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussbaum, J.M.; Schilling, S.; Cynis, H.; Silva, A.; Swanson, E.; Wangsanut, T.; Tayler, K.; Wiltgen, B.; Hatami, A.; Ronicke, R.; et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 2012, 485, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Umeda, T.; Ono, K.; Sakai, A.; Yamashita, M.; Mizuguchi, M.; Klein, W.L.; Yamada, M.; Mori, H.; Tomiyama, T. Rifampicin is a candidate preventive medicine against amyloid-beta and tau oligomers. Brain A J. Neurol. 2016, 139, 1568–1586. [Google Scholar] [CrossRef] [Green Version]
- Carrodeguas, J.A.; Rodolosse, A.; Garza, M.V.; Sanz-Clemente, A.; Perez-Pe, R.; Lacosta, A.M.; Dominguez, L.; Monleon, I.; Sanchez-Diaz, R.; Sorribas, V.; et al. The chick embryo appears as a natural model for research in beta-amyloid precursor protein processing. Neuroscience 2005, 134, 1285–1300. [Google Scholar] [CrossRef]
- Chromy, B.A.; Nowak, R.J.; Lambert, M.P.; Viola, K.L.; Chang, L.; Velasco, P.T.; Jones, B.W.; Fernandez, S.J.; Lacor, P.N.; Horowitz, P.; et al. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry 2003, 42, 12749–12760. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.H.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of Abeta. J. Neurochem. 2007, 100, 23–35. [Google Scholar] [CrossRef]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Knight, E.M.; Kim, S.H.; Kottwitz, J.C.; Hatami, A.; Albay, R.; Suzuki, A.; Lublin, A.; Alberini, C.M.; Klein, W.L.; Szabo, P.; et al. Effective anti-Alzheimer Abeta therapy involves depletion of specific Abeta oligomer subtypes. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulekar, M.H. Bioinformatics in Life and Environmental Sciences. In Bioinformatics: Applications in Life and Environmental Sciences; Fulekar, M.H., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 1–11. [Google Scholar]
- Mey, J.; Thanos, S. Development of the visual system of the chick: I. Cell differentiation and histogenesis. Brain Res. Rev. 2000, 32, 343–379. [Google Scholar] [CrossRef]
- Dunn, B.E. Technique of shell-less culture of the 72-hour avian embryo. Poult. Sci. 1974, 53, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, R.; Kubai, L.; Knighton, D.; Folkman, J. A simple procedure for the long-term cultivation of chicken embryos. Dev. Biol. 1974, 41, 391–394. [Google Scholar] [CrossRef]
- Yalcin, H.C.; Shekhar, A.; Rane, A.A.; Butcher, J.T. An ex-ovo chicken embryo culture system suitable for imaging and microsurgery applications. JoVE (J. Vis. Exp.) 2010, 44, e2154. [Google Scholar] [CrossRef] [Green Version]
- Datar, S.; Bhonde, R.R. Shell-less chick embryo culture as an alternative in vitro model to investigate glucose-induced malformations in mammalian embryos. Rev. Diabet. Stud. 2005, 2, 221. [Google Scholar] [CrossRef] [Green Version]
- Dohle, D.S.; Pasa, S.D.; Gustmann, S.; Laub, M.; Wissler, J.H.; Jennissen, H.P.; Dünker, N. Chick ex ovo culture and ex ovo CAM assay: How it really works. JoVE (J. Vis. Exp.) 2009, 33, e1620. [Google Scholar]
- Schomann, T.; Qunneis, F.; Widera, D.; Kaltschmidt, C.; Kaltschmidt, B. Improved method for ex ovo-cultivation of developing chicken embryos for human stem cell xenografts. Stem Cells Int. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hamburger, V.; Hamilton, H.L. A series of normal stages in the development of the chick embryo. J. Morphol. 1951, 88, 49–92. [Google Scholar] [CrossRef] [PubMed]
- Jesberg, D.O. Ocular malformations of the chick embryo produced by photocoagulation. Investig. Ophthalmol. 1962, 1, 348–354. [Google Scholar]
- Manuel, M.; Pratt, T.; Liu, M.; Jeffery, G.; Price, D.J. Overexpression of Pax6 results in microphthalmia, retinal dysplasia and defective retinal ganglion cell axon guidance. BMC Dev. Biol. 2008, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Néron, B.; Marx, M.; Crisanti, P. Role of QN1 protein in cell proliferation arrest and differentiation during the neuroretina development. Mech. Dev. 2001, 102, 107–117. [Google Scholar] [CrossRef]
- Nambu, H.; Taomoto, M.; Ogura, E.; Tsubura, A. Time-specific action of N-methyl-N-nitrosourea in the occurrence of retinal dysplasia and retinal degeneration in neonatal mice. Pathol. Int. 1998, 48, 199–205. [Google Scholar] [CrossRef]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. Defects in the outer limiting membrane are associated with rosette development in the Nrl-/- retina. PLoS ONE 2012, 7, e32484. [Google Scholar] [CrossRef]
- Finnegan, S.; Robson, J.; Hocking, P.M.; Ali, M.; Inglehearn, C.F.; Stitt, A.; Curry, W.J. Proteomic profiling of the retinal dysplasia and degeneration chick retina. Mol. Vis. 2010, 16, 7–17. [Google Scholar]
- Bevilaqua, M.C.; Andrade-da-Costa, B.L.; Fleming, R.L.; Dias, G.P.; da Silveirada Luz, A.C.; Nardi, A.E.; de Mello, F.G.; Gardino, P.F.; Calaza, K.C. Retinal development impairment and degenerative alterations in adult rats subjected to post-natal malnutrition. Int. J. Dev. Neurosci. 2015, 47, 172–182. [Google Scholar] [CrossRef]
- Willem, M.; Lammich, S.; Haass, C. Function, regulation and therapeutic properties of beta-secretase (BACE1). Semin. Cell Dev. Biol. 2009, 20, 175–182. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; De Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of beta-amyloid: Where the Alzheimer’s disease protein meets physiology. Rev. Neurosci. 2010, 21, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Tharp, W.G.; Sarkar, I.N. Origins of amyloid-beta. BMC Genom. 2013, 14, 290. [Google Scholar] [CrossRef] [Green Version]
- Lambert, M.P.; Velasco, P.T.; Viola, K.L.; Klein, W.L. Targeting generation of antibodies specific to conformational epitopes of amyloid beta-derived neurotoxins. CNS Neurol. Disord. Drug Targets 2009, 8, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Wicklund, L.; Lacor, P.N.; Klein, W.L.; Nordberg, A.; Marutle, A. Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol. Aging 2012, 33, 825.e1–825.e13. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjo, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, W.; Melnik, M.; Huang, C.; Teter, B.; Chandra, S.; Zhu, C.; McIntire, L.B.; John, V.; Gylys, K.H.; Bilousova, T. Multi-Omics Analysis of Microglial Extracellular Vesicles From Human Alzheimer’s Disease Brain Tissue Reveals Disease-Associated Signatures. Front. Pharmacol. 2021, 12, 766082. [Google Scholar] [CrossRef]
- Wooff, Y.; Cioanca, A.V.; Chu-Tan, J.A.; Aggio-Bruce, R.; Schumann, U.; Natoli, R. Small-Medium Extracellular Vesicles and Their miRNA Cargo in Retinal Health and Degeneration: Mediators of Homeostasis, and Vehicles for Targeted Gene Therapy. Front. Cell. Neurosci. 2020, 14, 160. [Google Scholar] [CrossRef]
- Schubert, D.; LaCorbiere, M.; Klier, F.G.; Birdwell, C. A role for adherons in neural retina cell adhesion. J. Cell Biol. 1983, 96, 990–998. [Google Scholar] [CrossRef]
- Schubert, D. A Brief History of Adherons: The Discovery of Brain Exosomes. Int. J. Mol. Sci. 2020, 21, 7673. [Google Scholar] [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Silverstein, A.M.; Osburn, B.I.; Prendergast, R.A. The pathogenesis of retinal dysplasia. Am. J. Ophthalmol. 1971, 72, 13–21. [Google Scholar] [CrossRef]
- Lahav, M.; Albert, D.M.; Craft, J.L. Light and electron microscopic study of dysplastic rosette-like structures occurring in the disorganized mature retina. Albrecht Von Graefe’s Arch. Clin. Exp. Ophthalmol. 1975, 195, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.R.; Iaboni, D.S.M.; Seamone, M.E.; Sarraf, D. Inner, outer, and full-thickness retinal folds after rhegmatogenous retinal detachment repair: A review. Surv. Ophthalmol. 2019, 64, 135–161. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Coleman, P.D.; Flood, D.G. Neuron numbers and dendritic extent in normal aging and Alzheimer’s disease. Neurobiol. Aging 1987, 8, 521–545. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- Terry, R.D. Alzheimer’s disease and the aging brain. J. Geriatr. Psychiatry Neurol. 2006, 19, 125–128. [Google Scholar] [CrossRef]
- Johnson, E.M., Jr.; Deckwerth, T.L. Molecular mechanisms of developmental neuronal death. Annu. Rev. Neurosci. 1993, 16, 31–46. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Faust, T.E.; Gunner, G.; Schafer, D.P. Mechanisms governing activity-dependent synaptic pruning in the developing mammalian CNS. Nat. Rev. Neurosci. 2021, 22, 657–673. [Google Scholar] [CrossRef]
- Limoni, G. Modelling and Refining Neuronal Circuits with Guidance Cues: Involvement of Semaphorins. Int. J. Mol. Sci. 2021, 22, 6111. [Google Scholar] [CrossRef] [PubMed]
- Djurisic, M.; Vidal, G.S.; Mann, M.; Aharon, A.; Kim, T.; Ferrao Santos, A.; Zuo, Y.; Hubener, M.; Shatz, C.J. PirB regulates a structural substrate for cortical plasticity. Proc. Natl. Acad. Sci. USA 2013, 110, 20771–20776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Vidal, G.S.; Djurisic, M.; William, C.M.; Birnbaum, M.E.; Garcia, K.C.; Hyman, B.T.; Shatz, C.J. Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 2013, 341, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Breuzard, G.; Pagano, A.; Bastonero, S.; Malesinski, S.; Parat, F.; Barbier, P.; Peyrot, V.; Kovacic, H. Tau regulates the microtubule-dependent migration of glioblastoma cells via the Rho-ROCK signaling pathway. J. Cell Sci. 2019, 132, jcs.222851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito-Moreira, J.; Paula-Lima, A.C.; Bomfim, T.R.; Oliveira, F.B.; Sepulveda, F.J.; De Mello, F.G.; Aguayo, L.G.; Panizzutti, R.; Ferreira, S.T. Abeta oligomers induce glutamate release from hippocampal neurons. Curr. Alzheimer Res. 2011, 8, 552–562. [Google Scholar] [CrossRef]
- Nunes-Tavares, N.; Santos, L.E.; Stutz, B.; Brito-Moreira, J.; Klein, W.L.; Ferreira, S.T.; de Mello, F.G. Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid-beta peptide oligomers. J. Biol. Chem. 2012, 287, 19377–19385. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, R.S.; Hausman, R.E. Cloning of chicken choline acetyltransferase and its expression in early embryonic retina. Brain Res. Mol. Brain Res. 2004, 129, 54–66. [Google Scholar] [CrossRef]
- Zheng, J.Q.; Felder, M.; Connor, J.A.; Poo, M.M. Turning of nerve growth cones induced by neurotransmitters. Nature 1994, 368, 140–144. [Google Scholar] [CrossRef]
- Katz, L.C.; Shatz, C.J. Synaptic activity and the construction of cortical circuits. Science 1996, 274, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Ford, K.J.; Feller, M.B. Assembly and disassembly of a retinal cholinergic network. Vis. Neurosci. 2012, 29, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojeda, J.; Avila, A. Early Actions of Neurotransmitters During Cortex Development and Maturation of Reprogrammed Neurons. Front. Synaptic Neurosci. 2019, 11, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Mao, X. Role of Retinal Amyloid-beta in Neurodegenerative Diseases: Overlapping Mechanisms and Emerging Clinical Applications. Int. J. Mol. Sci. 2021, 22, 2360. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chen, L.; Chen, X.; Liu, X. Soluble amyloid beta oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma. Med. Hypotheses 2008, 71, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Su, S.; Jiang, S.; Sun, X.; Wang, J. Role of amyloid β-peptide in the pathogenesis of age-related macular degeneration. BMJ Open Ophthalmol. 2021, 6, e000774. [Google Scholar] [CrossRef]
- Lynn, S.A.; Johnston, D.A.; Scott, J.A.; Munday, R.; Desai, R.S.; Keeling, E.; Weaterton, R.; Simpson, A.; Davis, D.; Freeman, T.; et al. Oligomeric Abeta1-42 Induces an AMD-Like Phenotype and Accumulates in Lysosomes to Impair RPE Function. Cells 2021, 10, 413. [Google Scholar] [CrossRef]
- Bitel, C.L.; Kasinathan, C.; Kaswala, R.H.; Klein, W.L.; Frederikse, P.H. Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J. Alzheimer’s Dis. JAD 2012, 32, 291–305. [Google Scholar] [CrossRef]
- Arendt, T.; Stieler, J.; Ueberham, U. Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 2017, 143, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Khan, U.A.; Liu, L.; Provenzano, F.A.; Berman, D.E.; Profaci, C.P.; Sloan, R.; Mayeux, R.; Duff, K.E.; Small, S.A. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat. Neurosci. 2014, 17, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wessel, D.; Flugge, U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Belov, M.E.; Damoc, E.; Denisov, E.; Compton, P.D.; Horning, S.; Makarov, A.A.; Kelleher, N.L. From protein complexes to subunit backbone fragments: A multi-stage approach to native mass spectrometry. Anal. Chem. 2013, 85, 11163–11173. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartley, S.C.; Proctor, M.T.; Xia, H.; Ho, E.; Kang, D.S.; Schuster, K.; Bicca, M.A.; Seckler, H.S.; Viola, K.L.; Patrie, S.M.; et al. An Essential Role for Alzheimer’s-Linked Amyloid Beta Oligomers in Neurodevelopment: Transient Expression of Multiple Proteoforms during Retina Histogenesis. Int. J. Mol. Sci. 2022, 23, 2208. https://doi.org/10.3390/ijms23042208
Bartley SC, Proctor MT, Xia H, Ho E, Kang DS, Schuster K, Bicca MA, Seckler HS, Viola KL, Patrie SM, et al. An Essential Role for Alzheimer’s-Linked Amyloid Beta Oligomers in Neurodevelopment: Transient Expression of Multiple Proteoforms during Retina Histogenesis. International Journal of Molecular Sciences. 2022; 23(4):2208. https://doi.org/10.3390/ijms23042208
Chicago/Turabian StyleBartley, Samuel C., Madison T. Proctor, Hongjie Xia, Evelyn Ho, Dong S. Kang, Kristen Schuster, Maíra A. Bicca, Henrique S. Seckler, Kirsten L. Viola, Steven M. Patrie, and et al. 2022. "An Essential Role for Alzheimer’s-Linked Amyloid Beta Oligomers in Neurodevelopment: Transient Expression of Multiple Proteoforms during Retina Histogenesis" International Journal of Molecular Sciences 23, no. 4: 2208. https://doi.org/10.3390/ijms23042208
APA StyleBartley, S. C., Proctor, M. T., Xia, H., Ho, E., Kang, D. S., Schuster, K., Bicca, M. A., Seckler, H. S., Viola, K. L., Patrie, S. M., Kelleher, N. L., De Mello, F. G., & Klein, W. L. (2022). An Essential Role for Alzheimer’s-Linked Amyloid Beta Oligomers in Neurodevelopment: Transient Expression of Multiple Proteoforms during Retina Histogenesis. International Journal of Molecular Sciences, 23(4), 2208. https://doi.org/10.3390/ijms23042208