
Novel Loss of Function Variant in BCKDK Causes a Treatable Developmental and Epileptic Encephalopathy

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
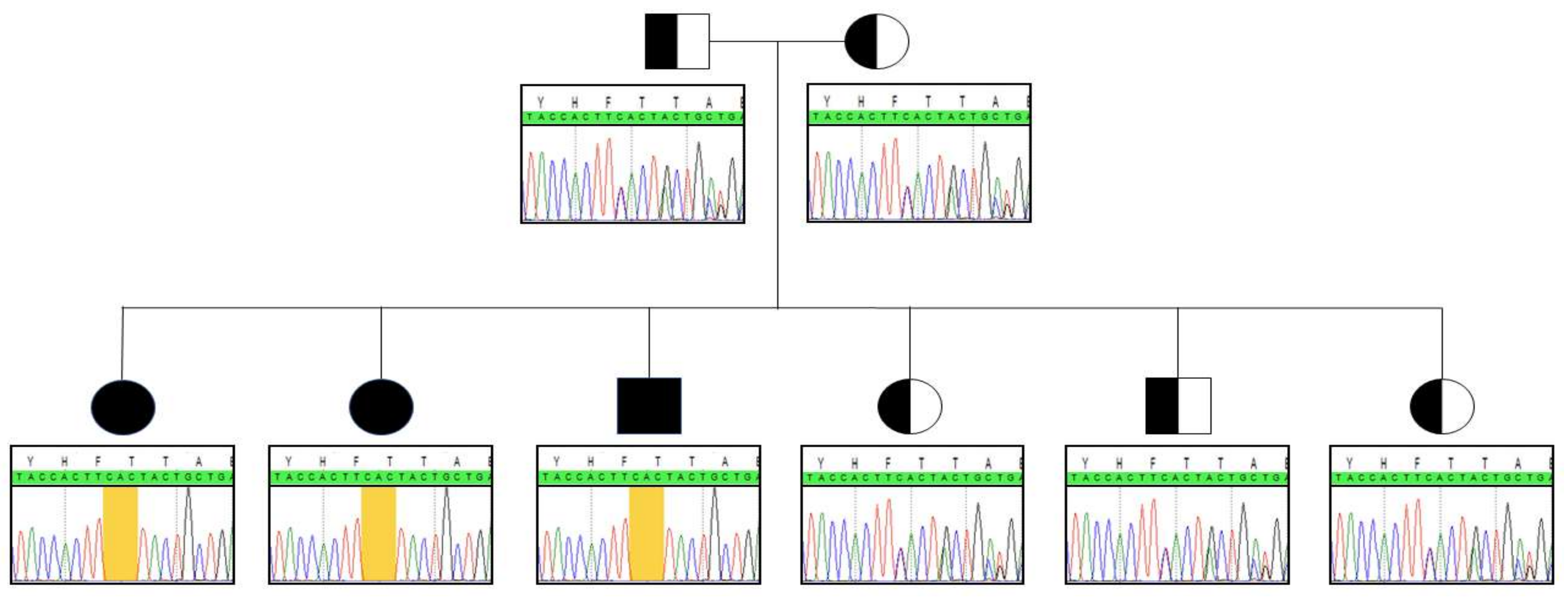
2.1. Novel Mutation
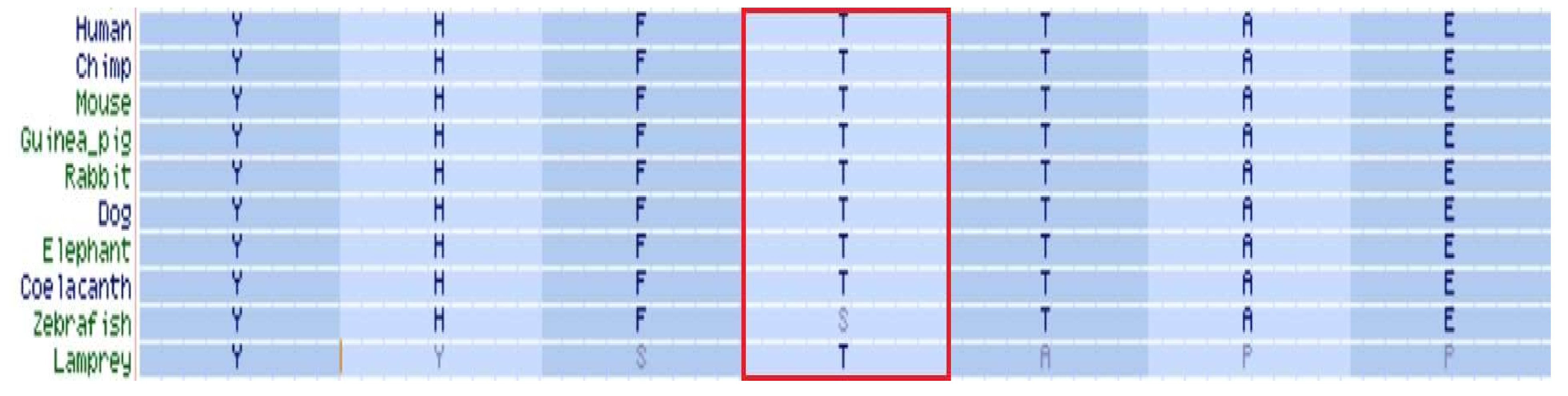
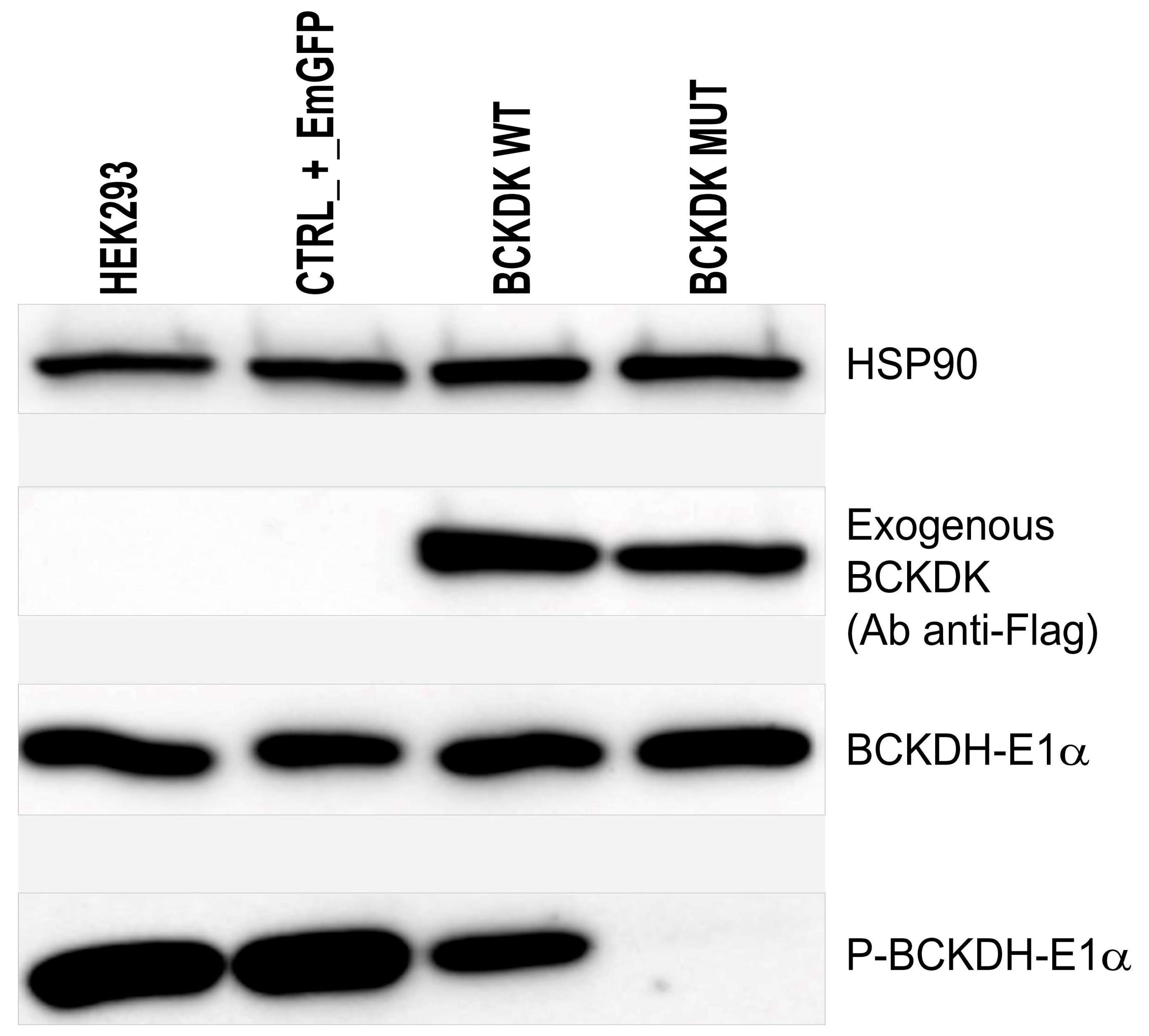
2.2. Protein Conformation and Loss of Function
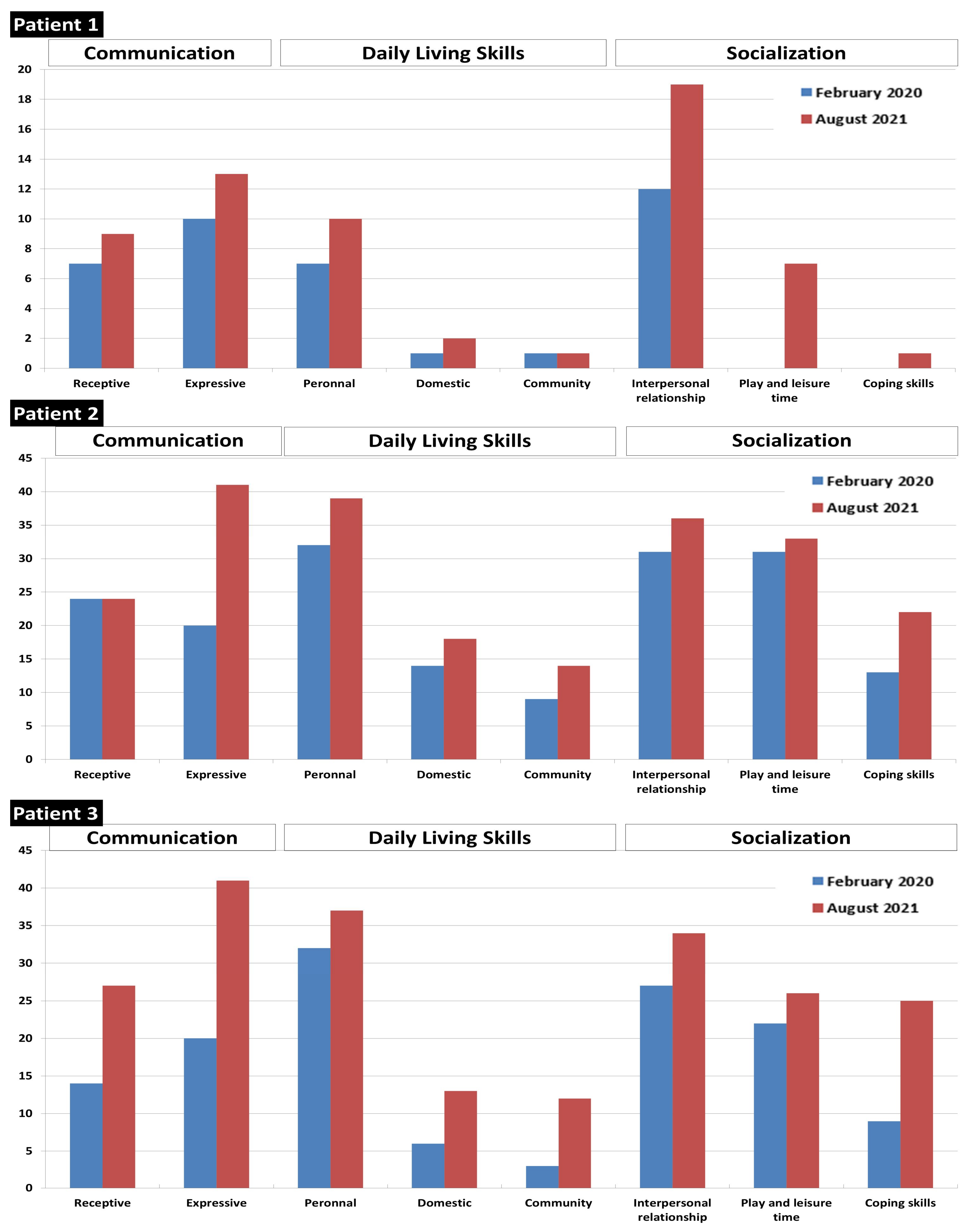
2.3. Patients’ Follow-Up and Treatment
3. Discussion
4. Materials and Methods
4.1. Subjects and Samples
4.2. Sequencing
4.3. Biochemical Analysis
4.4. Lentiviral Constructs
4.5. Cell Culture
4.6. Western Blot
4.7. Computed Tertiary Structure
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BCKDK | Branched-Chain α-Ketoacid Dehydrogenase Kinase |
| BCKDH | Branched-Chain α-Ketoacid Dehydrogenase |
| BCAA | Branched-Chain Amino Acids |
| CSF | Cerebrospinal Fluid |
| DBS | Dried Blood Spots |
| NBS | Newborn Screening |
| WES | Whole Exome Sequencing |
References
- Harris, R.A.; Joshi, M.; Jeoung, N.H. Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem. Biophys. Res. Commun. 2004, 313, 391–396. [Google Scholar]
- Holeček, M. Branched-chain amino acids in health and disease: Metabolism, alterations in blood plasma, and as supplements. Nutr. Metab. 2018, 15, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monirujjaman, M.; Ferdouse, A. Metabolic and Physiological Roles of Branched-Chain Amino Acids. Adv. Mol. Biol. 2014, 2014, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Conway, M.E.; Hutson, S.M. BCAA Metabolism and NH3 Homeostasis. In Advances in Neurobiology; Springer: Cham, Switzerland, 2016; Volume 13, pp. 99–132. [Google Scholar]
- Blackburn, P.R.; Gass, J.M.; Pinto e Vairo, F.; Farnham, K.M.; Atwal, H.K.; Macklin, S.; Klee, E.W.; Atwal, P.S. Maple syrup urine disease: Mechanisms and management. Appl. Clin. Genet. 2017, 10, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.L.; Li, C.J.; Xing, Y.; Yang, Y.H.; Jia, J.P. Hypervalinemia and hyperleucine-isoleucinemia caused by mutations in the branched-chain-amino-acid aminotransferase gene. J. Inherit. Metab. Dis. 2015, 38, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Oyarzabal, A.; Martínez-Pardo, M.; Merinero, B.; Navarrete, R.; Desviat, L.R.; Ugarte, M.; Rodríguez-Pombo, P. A Novel Regulatory Defect in the Branched-Chain α-Keto Acid Dehydrogenase Complex Due to a Mutation in the PPM 1 K Gene Causes a Mild Variant Phenotype of Maple Syrup Urine Disease. Hum. Mutat. 2013, 34, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novarino, G.; El-Fishawy, P.; Kayserili, H.; Meguid, N.A.; Scott, E.M.; Schroth, J.; Silhavy, J.L.; Kara, M.; Khalil, R.O.; Ben-Omran, T.; et al. Mutations in BCKD-kinase Lead to a Potentially Treatable Form of Autism with Epilepsy. Science 2012, 338, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cazorla, A.; Oyarzabal, A.; Fort, J.; Robles, C.; Castejón, E.; Ruiz-Sala, P.; Bodoy, S.; Merinero, B.; Lopez-Sala, A.; Dopazo, J.; et al. Two Novel Mutations in the BCKDK (Branched-Chain Keto-Acid Dehydrogenase Kinase) Gene Are Responsible for a Neurobehavioral Deficit in Two Pediatric Unrelated Patients. Hum. Mutat. 2014, 35, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Jayapalan, S.; Natarajan, J. Classification and domain analysis of protein kinases in hominids. Curr. Sci. 2016, 110, 828–838. [Google Scholar] [CrossRef]
- Oyarzabal, A.; Bravo-Alonso, I.; Sánchez-Aragó, M.; Rejas, M.T.; Merinero, B.; García-Cazorla, A.; Artuch, R.; Ugarte, M.; Rodríguez-Pombo, P. Mitochondrial response to the BCKDK-deficiency: Some clues to understand the positive dietary response in this form of autism. Biochim. Biophys. Acta-Mol. Basis Dis. 2016, 1862, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Oliva, A.; Grassi, S.; Brugada, R.; Campuzano, O. Update on the Genetic Basis of Sudden Unexpected Death in Epilepsy. Int. J. Mol. Sci. 2019, 20, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filee, R.; Schoos, R.; Boemer, F. Evaluation of Physiological Amino Acids Profiling by Tandem Mass Spectrometry. JIMD Rep. 2013, 13, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chace, D.H.; Kalas, T.A.; Naylor, E.W. Use of Tandem Mass Spectrometry for Multianalyte Screening of Dried Blood Specimens from Newborns. Clin. Chem. 2003, 49, 1797–1817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chace, D.H.; Kalas, T.A.; Naylor, E.W. The application of tandem mass spectrometry to neonatal screening for inherited disorders of intermediary metabolism. Annu. Rev. Genom. Hum. Genet. 2002, 3, 17–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emi, N.; Friedmann, T.; Yee, J.K. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J. Virol. 1991, 65, 1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252-8. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Plasma BCAA | CSF BCAA | NBS BCAA | ||||||||||||||
| ID | Gender | Leu * (74–203) | Ile * (42–124) | Val * (145–337) | Leu/Phe * (1.2–3.0) | Ile/Phe * (0.7–1.7) | Val/Phe * (2.0–5.4) | Leu * (7–44) | Ile * (4–24) | Val * (10–41) | Leu/Phe * (0.9–2.2) | Ile/Phe * (0.4–1.7) | Val/Phe * (1.1–2.8) | Xle * (<486) | Val * (<269) | Xle/Phe * (<6.0) | Val/Phe * (<3.1) |
| Patient 1 | Female | 29 | 13 | 62 | 0.4 | 0.2 | 0.9 | 3.3 | 1.4 | 6.4 | 0.20 | 0.08 | 0.38 | 84 | 47 | 1.4 | 0.8 |
| Patient 2 | Female | 15 | 7 | 44 | 0.3 | 0.1 | 0.9 | 1.8 | 1.0 | 4.4 | 0.11 | 0.06 | 0.28 | 89 | 44 | 1.8 | 0.9 |
| Patient 3 | Male | 51 | 21 | 73 | 0.6 | 0.3 | 0.9 | 3.8 | 1.4 | 6 | 0.23 | 0.08 | 0.36 | 210 | 81 | 3.2 | 1.2 |
| Unaffected Siblings | Plasma BCAA | CSF BCAA | NBS BCAA | ||||||||||||||
| ID | Gender | Leu * (74–203) | Ile * (42–124) | Val * (145–337) | Leu/Phe * (1.2–3.0) | Ile/Phe * (0.7–1.7) | Val/Phe * (2.0–5.4) | Leu * (7–44) | Ile * (4–24) | Val * (10–41) | Leu/Phe * (0.9–2.2) | Ile/Phe * (0.4–1.7) | Val/Phe * (1.1–2.8) | Xle * (<486) | Val * (<269) | Xle/Phe * (<6.0) | Val/Phe * (<3.1) |
| Sister 1 | Female | 126 | 62 | 177 | 1.3 | 0.7 | 1.9 | / | / | / | / | / | / | 321 | 145 | 5.7 | 2.6 |
| Brother 1 | Male | 125 | 58 | 136 | 2.0 | 0.9 | 2.2 | / | / | / | / | / | / | 246 | 112 | 4.4 | 2.0 |
| Sister 2 | Female | 112 | 46 | 130 | 2.3 | 1.0 | 2.7 | / | / | / | / | / | / | 253 | 81 | 4.8 | 1.5 |
| Parents | Plasma BCAA | ||||||||||||||||
| ID | Gender | Leu * (74–228) | Ile * (37–132) | Val * (105–352) | Leu/Phe * (0.9–2.6) | Ile/Phe * (0.7–1.5) | Val/Phe * (1.7–4.8) | ||||||||||
| Father | Male | 156 | 66 | 222 | 2.4 | 1.0 | 3.4 | ||||||||||
| Mother | Female | 134 | 64 | 175 | 1.6 | 0.8 | 2.1 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boemer, F.; Josse, C.; Luis, G.; Di Valentin, E.; Thiry, J.; Cello, C.; Caberg, J.-H.; Dadoumont, C.; Harvengt, J.; Lumaka, A.; et al. Novel Loss of Function Variant in BCKDK Causes a Treatable Developmental and Epileptic Encephalopathy. Int. J. Mol. Sci. 2022, 23, 2253. https://doi.org/10.3390/ijms23042253
Boemer F, Josse C, Luis G, Di Valentin E, Thiry J, Cello C, Caberg J-H, Dadoumont C, Harvengt J, Lumaka A, et al. Novel Loss of Function Variant in BCKDK Causes a Treatable Developmental and Epileptic Encephalopathy. International Journal of Molecular Sciences. 2022; 23(4):2253. https://doi.org/10.3390/ijms23042253
Chicago/Turabian StyleBoemer, François, Claire Josse, Géraldine Luis, Emmanuel Di Valentin, Jérôme Thiry, Christophe Cello, Jean-Hubert Caberg, Caroline Dadoumont, Julie Harvengt, Aimé Lumaka, and et al. 2022. "Novel Loss of Function Variant in BCKDK Causes a Treatable Developmental and Epileptic Encephalopathy" International Journal of Molecular Sciences 23, no. 4: 2253. https://doi.org/10.3390/ijms23042253
APA StyleBoemer, F., Josse, C., Luis, G., Di Valentin, E., Thiry, J., Cello, C., Caberg, J.-H., Dadoumont, C., Harvengt, J., Lumaka, A., Bours, V., & Debray, F.-G. (2022). Novel Loss of Function Variant in BCKDK Causes a Treatable Developmental and Epileptic Encephalopathy. International Journal of Molecular Sciences, 23(4), 2253. https://doi.org/10.3390/ijms23042253