
Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MiT Transcription Factors Control Lysosome Biogenesis
3. The Lysosome as a Signaling Hub
4. The Lysosome as a Platform for the Nutrient-Sensing Machinery
5. Lysosomes and Cell Cycle Regulation
5.1. Control of the Cell Cycle by the Lysosomal Machinery
5.2. Regulation of Lysosomes by the Cell Cycle Machinery
6. Role of Lysosomes in Stem Cell Metabolism and Fate
6.1. Role of the Lysosomal Machinery in Embryonic Stem Cells (ESCs)
6.2. Role of the Lysosomal Machinery in Adult Stem Cells
6.3. Role of the Lysosomal Machinery in Cancer Stem Cells
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Feng, W.; Rong, Y.; Liu, R. Anatomy of autophagy: From the beginning to the end. Cell. Mol. Life Sci. 2018, 75, 815–831. [Google Scholar] [CrossRef]
- Savini, M.; Zhao, Q.; Wang, M.C. Lysosomes: Signaling Hubs for Metabolic Sensing and Longevity. Trends Cell Biol. 2019, 29, 876–887. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; W. W. Norton & Company: New York, NY, USA, 2015. [Google Scholar]
- Cooper, G.M. The Cell. A Molecular Approach; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Ohkuma, S.; Moriyama, Y.; Takano, T. Identification and characterization of a proton pump on lysosomes by fluorescein-isothiocyanate-dextran fluorescence. Proc. Natl. Acad. Sci. USA 1982, 79, 2758–2762. [Google Scholar] [CrossRef] [Green Version]
- Schwake, M.; Schroder, B.; Saftig, P. Lysosomal membrane proteins and their central role in physiology. Traffic 2013, 14, 739–748. [Google Scholar] [CrossRef]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. A shortcut to the lysosome: The mannose-6-phosphate-independent pathway. Mol. Genet. Metab. 2012, 107, 257–266. [Google Scholar] [CrossRef]
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12 (Suppl. 2), 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy in protein and organelle turnover. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [Green Version]
- Boya, P. Lysosomal function and dysfunction: Mechanism and disease. Antioxid. Redox. Signal. 2012, 17, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Wyant, G.A.; Abu-Remaileh, M.; Wolfson, R.L.; Chen, W.W.; Freinkman, E.; Danai, L.V.; Vander Heiden, M.G.; Sabatini, D.M. mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 2017, 171, 642–654.e12. [Google Scholar] [CrossRef]
- Fan, X.; Jin, W.Y.; Lu, J.; Wang, J.; Wang, Y.T. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat. Neurosci. 2014, 17, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Settembre, C.; Medina, D.L. TFEB and the CLEAR network. Methods Cell Biol. 2015, 126, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.Q.; Zhao, Q.; Zhou, X.; Mattei, M.G.; de Crombrugghe, B. TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol. Cell. Biol. 1993, 13, 4505–4512. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Jian, Y.; Xu, M.; Huang, X.; Wang, N.; Liu, Z.; Li, Q.; Li, J.; Zhou, H.; Xu, L.; et al. CDK4/6 regulate lysosome biogenesis through TFEB/TFE3. J. Cell Biol. 2020, 219, e201911036. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Diab, H.I.; Lishu, L.; Jeong, A.L.; Patange, S.; Raben, N.; Puertollano, R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014, 7, ra9. [Google Scholar] [CrossRef] [Green Version]
- Ploper, D.; Taelman, V.F.; Robert, L.; Perez, B.S.; Titz, B.; Chen, H.W.; Graeber, T.G.; von Euw, E.; Ribas, A.; De Robertis, E.M. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E420–E429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Friedrichsen, H.J.; Andrews, S.; Picaud, S.; Volpon, L.; Ngeow, K.; Berridge, G.; Fischer, R.; Borden, K.L.B.; Filippakopoulos, P.; et al. A TFEB nuclear export signal integrates amino acid supply and glucose availability. Nat. Commun. 2018, 9, 2685. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- Hsu, C.L.; Lee, E.X.; Gordon, K.L.; Paz, E.A.; Shen, W.C.; Ohnishi, K.; Meisenhelder, J.; Hunter, T.; La Spada, A.R. MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat. Commun. 2018, 9, 942. [Google Scholar] [CrossRef]
- Odle, R.I.; Walker, S.A.; Oxley, D.; Kidger, A.M.; Balmanno, K.; Gilley, R.; Okkenhaug, H.; Florey, O.; Ktistakis, N.T.; Cook, S.J. An mTORC1-to-CDK1 Switch Maintains Autophagy Suppression during Mitosis. Mol. Cell 2020, 77, 228–240.e7. [Google Scholar] [CrossRef] [Green Version]
- Ferron, M.; Settembre, C.; Shimazu, J.; Lacombe, J.; Kato, S.; Rawlings, D.J.; Ballabio, A.; Karsenty, G. A RANKL-PKCbeta-TFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Genes Dev. 2013, 27, 955–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Puertollano, R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 2018, 293, 12525–12534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, H.J.; Kim, H.; Oh, S.; Lee, J.G.; Kee, M.; Ko, H.J.; Kweon, M.N.; Won, K.J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. Eur. Mol. Biol. Organ. J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W.; Bar-Peled, L. Lysosome: The metabolic signaling hub. Traffic 2019, 20, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2013, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Brunn, G.J.; Hudson, C.C.; Sekulic, A.; Williams, J.M.; Hosoi, H.; Houghton, P.J.; Abraham, R.T. Phosphorylation of the Translational Repressor PHAS-I by the Mammalian Target of Rapamycin. Science 2012, 99, 10–13. [Google Scholar] [CrossRef]
- Brown, E.J.; Beal, P.A.; Keith, C.T.; Chen, J.; Shin, T.B.; Schreiber, S.L. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature 1995, 377, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the Human mTOR Complex I and Its Implications for Rapamycin Inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Bonifacino, J.S. Lysosome Positioning Influences mTORC2 and AKT Signaling. Mol. Cell 2019, 75, 26–38.e3. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef]
- Zheng, X.F.; Fiorentino, D.; Chen, J.; Crabtree, G.R.; Schreiber, S.L. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 1995, 82, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Garami, A.; Zwartkruis, F.J.T.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 2003, 11, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Buerger, C.; DeVries, B.; Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem. Biophys. Res. Commun. 2006, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kim, E. Rag GTPase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef]
- Shen, K.; Choe, A.; Sabatini, D.M. Intersubunit Crosstalk in the Rag GTPase Heterodimer Enables mTORC1 to Respond Rapidly to Amino Acid Availability. Mol. Cell 2017, 68, 552–565.e8. [Google Scholar] [CrossRef]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef] [Green Version]
- Villegas, F.; Lehalle, D.; Mayer, D.; Rittirsch, M.; Stadler, M.B.; Zinner, M.; Olivieri, D.; Vabres, P.; Duplomb-Jego, L.; De Bont, E.; et al. Lysosomal Signaling Licenses Embryonic Stem Cell Differentiation via Inactivation of Tfe3. Cell Stem Cell 2019, 24, 257–270.e8. [Google Scholar] [CrossRef] [Green Version]
- Meireles, A.M.; Shen, K.; Zoupi, L.; Iyer, H.; Bouchard, E.L.; Williams, A.; Talbot, W.S. The Lysosomal Transcription Factor TFEB Represses Myelination Downstream of the Rag-Ragulator Complex. Dev. Cell 2018, 47, 319–330.e5. [Google Scholar] [CrossRef] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, N.; Lima, T.B.; Mercaldi, G.F.; Nascimento, A.F.Z.; Silva, A.L.S.; Righetto, G.L.; Bar-Peled, L.; Shen, K.; Sabatini, D.M.; Gozzo, F.C.; et al. C7orf59/LAMTOR4 phosphorylation and structural flexibility modulate Ragulator assembly. FEBS Open Bio. 2019, 9, 1589–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, Z.; Wang, L.; Deng, W.; Wang, J.; Wu, G. Structural insight into the Ragulator complex which anchors mTORC1 to the lysosomal membrane. Cell Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, R.E.; Cho, K.F.; Rappold, R.; Thrun, A.; Tofaute, M.; Kim, D.J.; Moldavski, O.; Hurley, J.H.; Zoncu, R. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat. Cell Biol. 2018, 20, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, X.; Saucedo, L.J.; Ru, B.; Edgar, B.A.; Pan, D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003, 5, 578–581. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccio, M.; Bos, J.L.; Zwartkruis, F.J.T. Regulation of the small GTPase Rheb by amino acids. Oncogene 2006, 25, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Corradetti, M.N.; Inoki, K.; Guan, K.-L. TSC2: Filling the GAP in the mTOR signaling pathway. Trends Biochem. Sci. 2004, 29, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Efeyan, A.; Sabatini, D.M. Nutrients and growth factors in mTORC1 activation. Biochem. Soc. Trans. 2013, 41, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Liang, Y.; He, Q.; Yao, R.; Bao, W.; Bao, L.; Wang, Y.; Wang, Z. Current models of mammalian target of rapamycin complex 1 (mTORC1) activation by growth factors and amino acids. Int. J. Mol. Sci. 2014, 15, 20753–20769. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef] [Green Version]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mtorc1 at the lysosome. Cell 2014, 156, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Prentzell, M.T.; Rehbein, U.; Cadena Sandoval, M.; De Meulemeester, A.S.; Baumeister, R.; Brohee, L.; Berdel, B.; Bockwoldt, M.; Carroll, B.; Chowdhury, S.R.; et al. G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell 2021, 184, 655–674.e27. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.H.; Wu, X.N.; Wu, S.Q.; Lu, B.J.; Dong, M.Q.; Zhang, H.; Sun, P.; Lin, S.C.; Guan, K.L.; et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef]
- Deng, L.; Jiang, C.; Chen, L.; Jin, J.; Wei, J.; Zhao, L.; Chen, M.; Pan, W.; Xu, Y.; Chu, H.; et al. The Ubiquitination of RagA GTPase by RNF152 Negatively Regulates mTORC1 Activation. Mol. Cell 2015, 58, 804–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Hong, S.; Ikeda, T.; Mori, H.; MacDougald, O.A.; Nada, S.; Okada, M.; Inoki, K. Amino Acids Enhance Polyubiquitination of Rheb and Its Binding to mTORC1 by Blocking Lysosomal ATXN3 Deubiquitinase Activity. Mol. Cell 2020, 80, 437–451.e6. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Kondo, K.; Itoh, T.; Ikari, S.; Nada, S.; Okada, M.; Noda, T. Rheb localized on the Golgi membrane activates lysosome-localized mTORC1 at the Golgi-lysosome contact site. J. Cell Sci. 2018, 131, jcs208017. [Google Scholar] [CrossRef] [Green Version]
- Angarola, B.; Ferguson, S.M. Weak membrane interactions allow Rheb to activate mTORC1 signaling without major lysosome enrichment. Mol. Biol Cell 2019, 30, 2750–2760. [Google Scholar] [CrossRef]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid Suspends the Circadian Clock in Hypoxia through Inhibition of mTOR. Cell 2018, 174, 72–87.e32. [Google Scholar] [CrossRef] [Green Version]
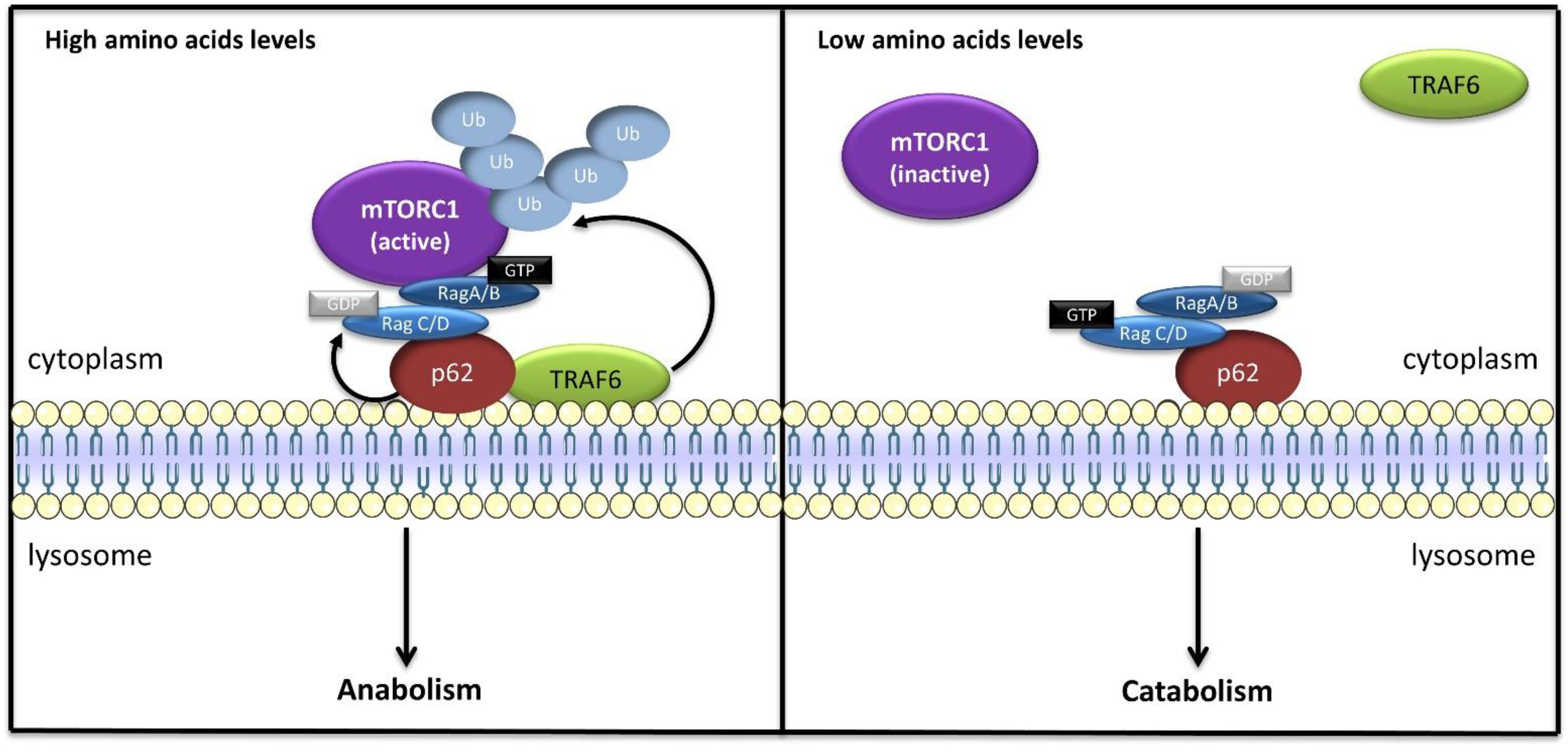
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 Is a Key Regulator of Nutrient Sensing in the mTORC1 Pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. Feedback on Fat: p62-mTORC1-Autophagy Connections. Cell 2011, 147, 724–727. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araújo, M.E.G.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Heublein, S.; Kazi, S.; Ögmundsdóttir, M.H.; Attwood, E.V.; Kala, S.; Boyd, C.A.R.; Wilson, C.; Goberdhan, D.C.I. Proton-assisted amino-acid transporters are conserved regulators of proliferation and amino-acid-dependent mtorc1 activation. Oncogene 2010, 29, 4068–4079. [Google Scholar] [CrossRef] [Green Version]
- Goberdhan, D.C.I.; Wilson, C.; Harris, A.L. Amino Acid Sensing by mTORC1: Intracellular Transporters Mark the Spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef] [Green Version]
- Jewell, J.L.; Russell, R.C.; Guan, K.-L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stransky, L.A.; Forgac, M. Amino acid availability modulates vacuolar H+-ATPase assembly. J. Biol. Chem. 2015, 290, 27360–27369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, S.; Brown, D. Regulation of luminal acidification by the V-ATPase. Physiology 2013, 28, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Myers, M.; Forgac, M. Characterization of the V0 domain of the coated vesicle (H+)-ATPase. J. Biol. Chem. 1992, 267, 9773–9778. [Google Scholar] [CrossRef]
- Puopolo, K.; Forgac, M. Functional reassembly of the coated vesicle proton pump. J. Biol. Chem. 1990, 265, 14836–14841. [Google Scholar] [CrossRef]
- Parra, K.J.; Kane, P.M. Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol. Cell. Biol. 1998, 18, 7064–7074. [Google Scholar] [CrossRef] [Green Version]
- Sautin, Y.Y.; Lu, M.; Gaugler, A.; Zhang, L.; Gluck, S.L. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol. Cell. Biol. 2005, 25, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Ögmundsdóttir, M.H.; Heublein, S.; Kazi, S.; Reynolds, B.; Visvalingam, S.M.; Shaw, M.K.; Goberdhan, D.C.I. Proton-assisted Amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PLoS ONE 2012, 7, e36616. [Google Scholar] [CrossRef] [Green Version]
- Beaumatin, F.; O’Prey, J.; Barthet, V.J.A.; Zunino, B.; Parvy, J.P.; Bachmann, A.M.; O’Prey, M.; Kania, E.; Gonzalez, P.S.; Macintosh, R.; et al. mTORC1 Activation Requires DRAM-1 by Facilitating Lysosomal Amino Acid Efflux. Mol. Cell 2019, 76, 163–176.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Tsun, Z.; Wolfson, R.; Shen, K.; Wyant, G.; Plovanich, M.; Yuan, E.; Jones, T.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing 1–4. Am. J. Clin. Nutr. 2014, 99, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhang, W.; Zhou, Y.; Li, F.; Wei, H.; Peng, J. Recent Advances in Understanding Amino Acid Sensing Mechanisms that Regulate mTORC1. Int. J. Mol. Sci. 2016, 17, 1636. [Google Scholar] [CrossRef] [Green Version]
- Hundal, H.S.; Taylor, P.M. Amino acid transceptors: Gate keepers of nutrient exchange and regulators of nutrient signaling. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E603–E613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal v-ATPase-Ragulator Complex Is a Common Activator for AMPK and mTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Orozco, J.M.; Krawczyk, P.A.; Scaria, S.M.; Cangelosi, A.L.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. Dihydroxyacetone phosphate signals glucose availability to mTORC1. Nat. Metab. 2020, 2, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Almacellas, E.; Pelletier, J.; Manzano, A.; Gentilella, A.; Ambrosio, S.; Mauvezin, C.; Tauler, A. Phosphofructokinases Axis Controls Glucose-Dependent mTORC1 Activation Driven by E2F1. iScience 2019, 20, 434–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
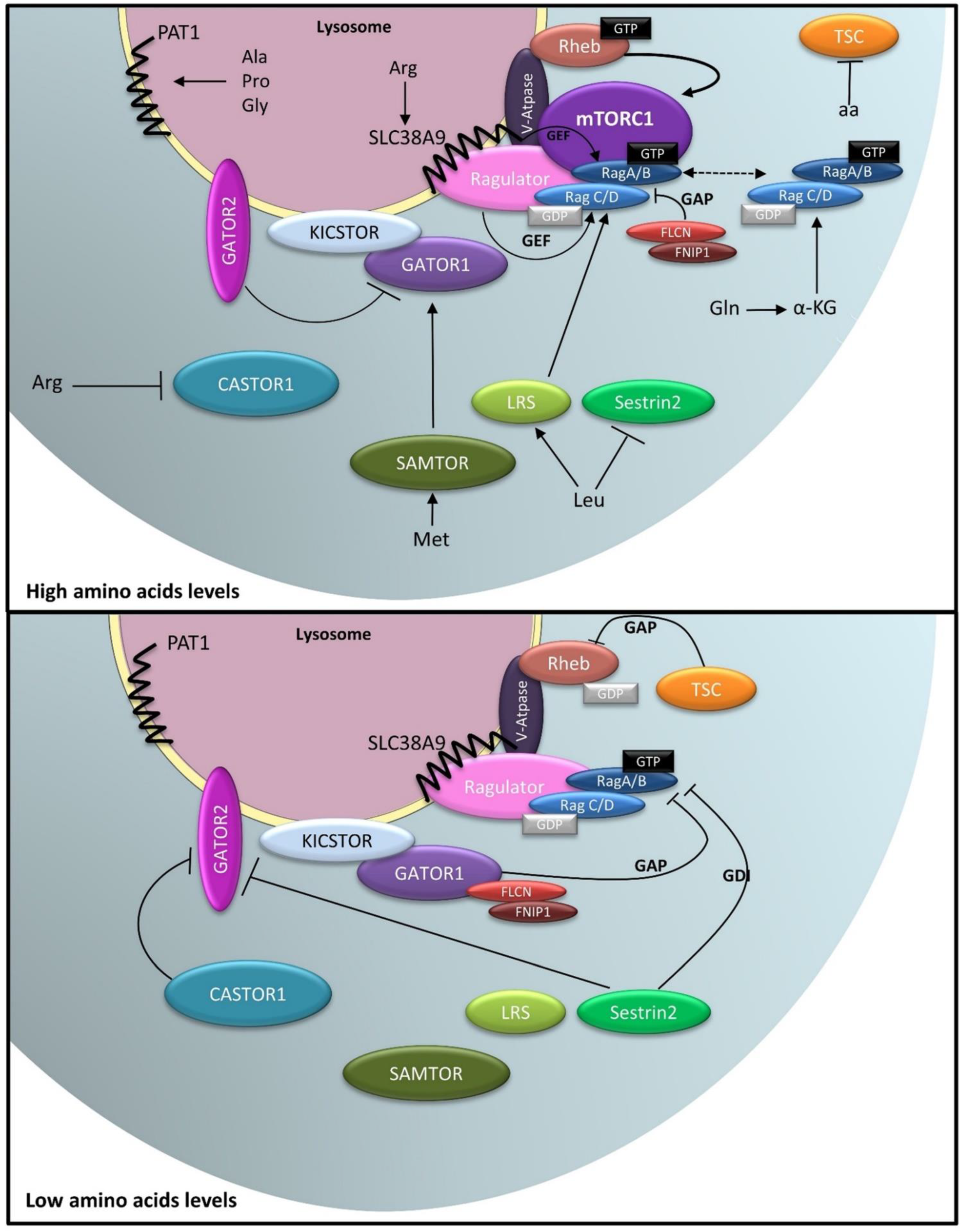
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, K.; Huang, R.K.; Brignole, E.J.; Condon, K.J.; Valenstein, M.L.; Chantranupong, L.; Bomaliyamu, A.; Choe, A.; Hong, C.; Yu, Z.; et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 2018, 556, 64–69. [Google Scholar] [CrossRef]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Wolfson, R.L.; Orozco, J.M.; Saxton, R.A.; Scaria, S.M.; Bar-Peled, L.; Spooner, E.; Isasa, M.; Gygi, S.P.; Sabatini, D.M. The sestrins interact with gator2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Ro, S.-H.; Kim, M.; Park, H.-W.; Semple, I.A.; Park, H.; Cho, U.-S.; Wang, W.; Guan, K.-L.; Karin, M.; et al. Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci. Rep. 2015, 5, 9502. [Google Scholar] [CrossRef]
- Shen, K.; Valenstein, M.L.; Gu, X.; Sabatini, D.M. Arg-78 of Nprl2 catalyzes GATOR1-stimulated GTP hydrolysis by the Rag GTPases. J. Biol. Chem. 2019, 294, 2970–2975. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2015, 351, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Knockenhauer, K.E.; Wolfson, R.L.; Chantranupong, L.; Pacold, M.E.; Wang, T.; Schwartz, T.U.; Sabatini, D.M. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 2016, 351, 53–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Yin, N.; Li, M.O. Sestrins function as guanine nucleotide dissociation inhibitors for rag GTPases to control mTORC1 signaling. Cell 2014, 159, 122–133. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Palm, W.; Peng, M.; King, B.; Lindsten, T.; Li, M.O.; Koumenis, C.; Thompson, C.B. GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev. 2015, 29, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Ferguson, S.M. GATOR1-dependent recruitment of FLCN-FNIP to lysosomes coordinates Rag GTPase heterodimer nucleotide status in response to amino acids. J. Cell Biol. 2018, 217, 2765–2776. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Rogala, K.B.; Chou, H.T.; Huang, R.K.; Yu, Z.; Sabatini, D.M. Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 2019, 179, 1319–1329.e8. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Kim, J.H.; Yoon, I.; Lee, C.; Fallahi Sichani, M.; Kang, J.S.; Kang, J.; Guo, M.; Lee, K.Y.; Han, G.; et al. Coordination of the leucine-sensing Rag GTPase cycle by leucyl-tRNA synthetase in the mTORC1 signaling pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E5279–E5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.S.; Son, K.; Arauz, E.; Han, J.M.; Kim, S.; Chen, J. Leucyl-tRNA Synthetase Activates Vps34 in Amino Acid-Sensing mTORC1 Signaling. Cell Rep. 2016, 16, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.S.; Du, G.; Backer, J.M.; Frohman, M.A.; Chen, J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol. 2011, 195, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.; Salloum, D.; Bernfeld, E.; Gorodetsky, E.; Akselrod, A.; Frias, M.A.; Sudderth, J.; Chen, P.H.; DeBerardinis, R.; Foster, D.A. Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J. Biol. Chem. 2017, 292, 6303–6311. [Google Scholar] [CrossRef] [Green Version]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar] [CrossRef] [Green Version]
- Byfield, M.P.; Murray, J.T.; Backer, J.M. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J. Biol. Chem. 2005, 280, 33076–33082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Z.; Pedersen, N.M.; Wang, L.; Torgersen, M.L.; Stenmark, H.; Raiborg, C. PtdIns3P controls mTORC1 signaling through lysosomal positioning. J. Cell Biol 2017, 216, 4217–4233. [Google Scholar] [CrossRef] [Green Version]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernfeld, E.; Menon, D.; Vaghela, V.; Zerin, I.; Faruque, P.; Frias, M.A.; Foster, D.A. Phospholipase D-dependent mTOR complex 1 (mTORC1) activation by glutamine. J. Biol. Chem. 2018, 293, 16390–16401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, G.G.; Papazotos, F.; Pawling, J.; Rajendran, D.; Knight, J.D.R.; Martinez, S.; Taipale, M.; Schramek, D.; Dennis, J.W.; Gingras, A.C. The GATOR-Rag GTPase pathway inhibits mTORC1 activation by lysosome-derived amino acids. Science 2020, 370, 351–356. [Google Scholar] [CrossRef]
- Kwon, J.S.; Everetts, N.J.; Wang, X.; Wang, W.; Della Croce, K.; Xing, J.; Yao, G. Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep. 2017, 20, 3223–3235. [Google Scholar] [CrossRef] [Green Version]
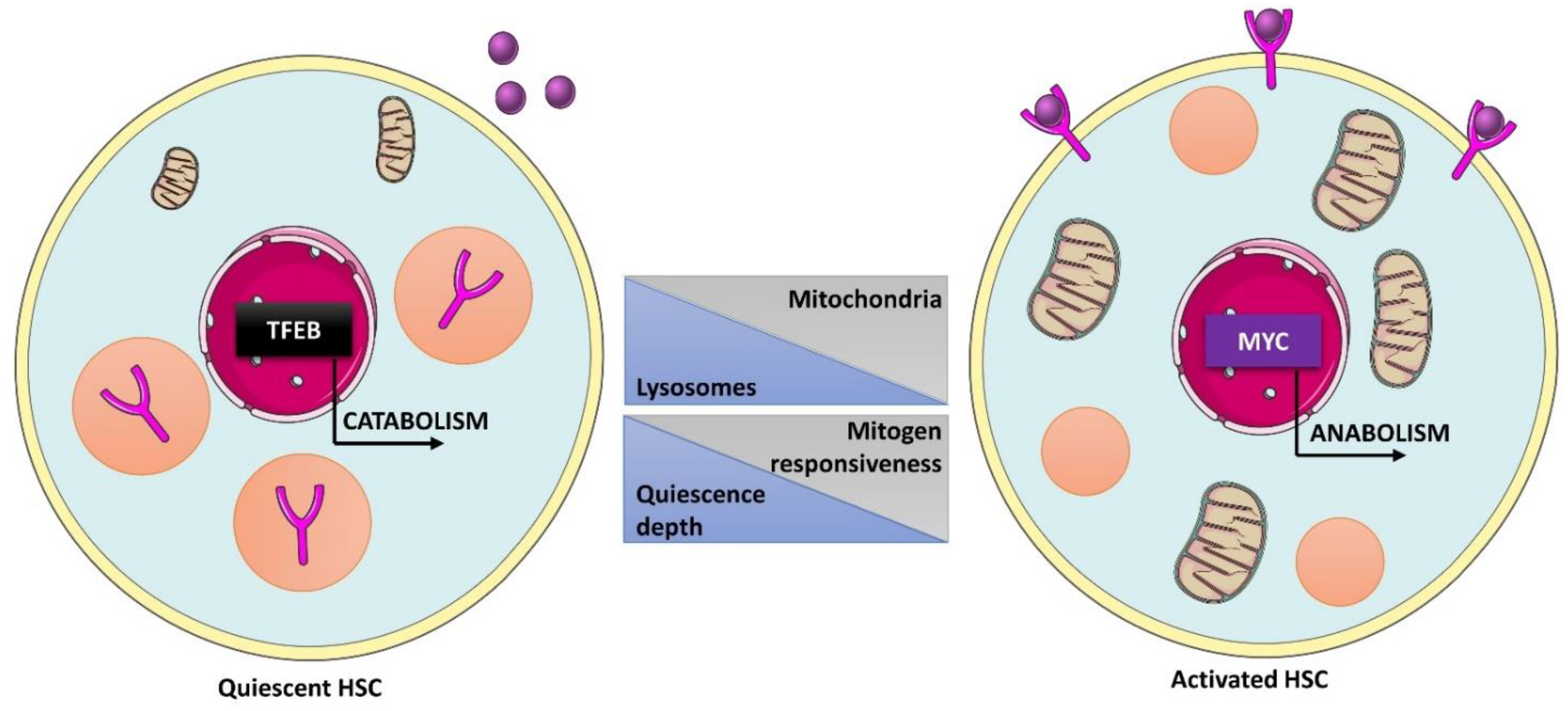
- Fujimaki, K.; Li, R.; Chen, H.; Della Croce, K.; Zhang, H.H.; Xing, J.; Bai, F.; Yao, G. Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. USA 2019, 116, 22624–22634. [Google Scholar] [CrossRef]
- Ianniciello, A.; Rattigan, K.M.; Helgason, G.V. The Ins and Outs of Autophagy and Metabolism in Hematopoietic and Leukemic Stem Cells: Food for Thought. Front. Cell Dev. Biol. 2018, 6, 120. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [Green Version]
- Julian, L.M.; Stanford, W.L. Organelle Cooperation in Stem Cell Fate: Lysosomes as Emerging Regulators of Cell Identity. Front. Cell Dev. Biol. 2020, 8, 591. [Google Scholar] [CrossRef]
- Doménech, E.; Maestre, C.; Esteban-Martínez, L.; Partida, D.; Pascual, R.; Fernández-Miranda, G.; Seco, E.; Campos-Olivas, R.; Pérez, M.; Megias, D.; et al. AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat. Cell Biol. 2015, 17, 1304–1316. [Google Scholar] [CrossRef] [PubMed]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of Mitochondrial Function Impairs Lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
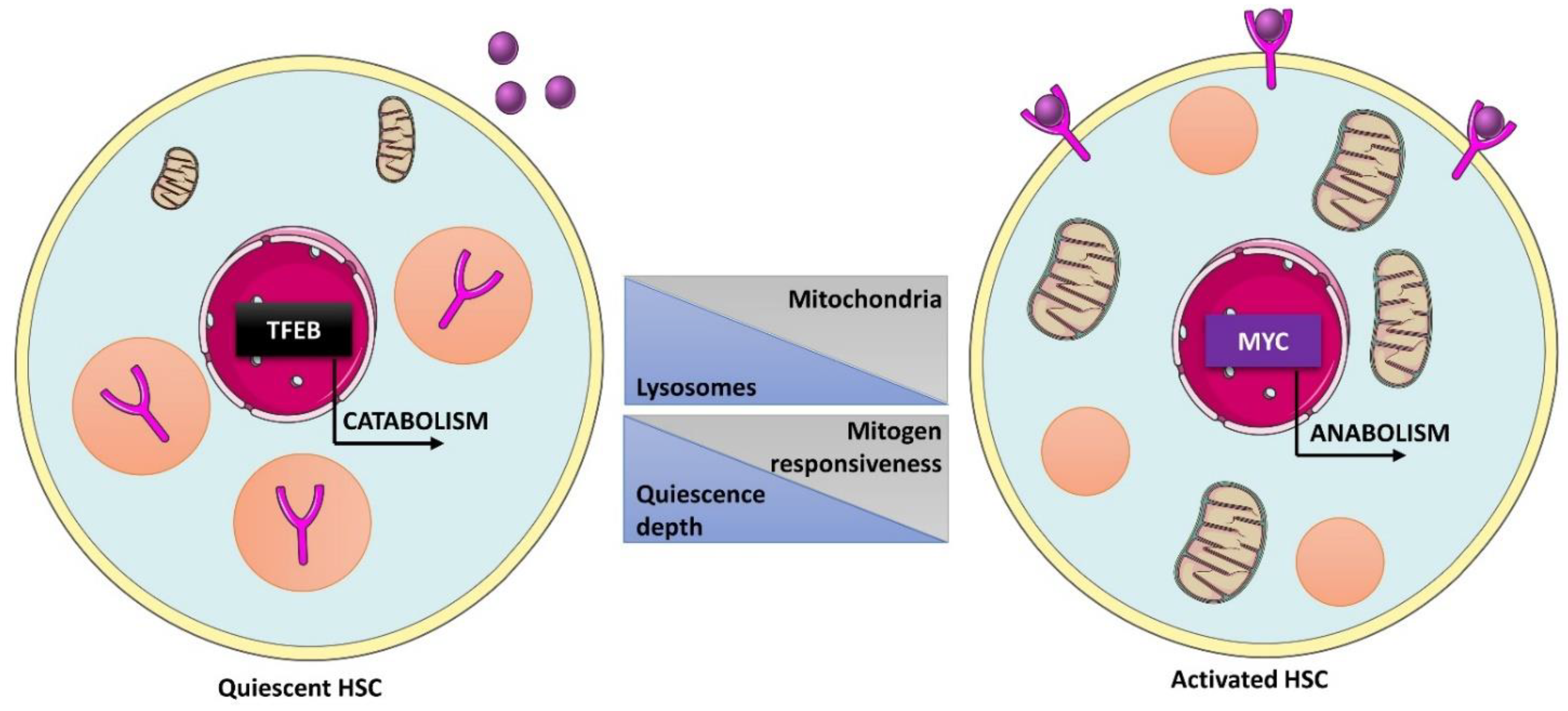
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 2014, 510, 393–396. [Google Scholar] [CrossRef]
- Condon, K.J.; Orozco, J.M.; Adelmann, C.H.; Spinelli, J.B.; van der Helm, P.W.; Roberts, J.M.; Kunchok, T.; Sabatini, D.M. Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2021, 118, e2022120118. [Google Scholar] [CrossRef]
- Brady, O.A.; Jeong, E.; Martina, J.A.; Pirooznia, M.; Tunc, I.; Puertollano, R. The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. eLife 2018, 7, e40856. [Google Scholar] [CrossRef]
- Fassl, A.; Brain, C.; Abu-Remaileh, M.; Stukan, I.; Butter, D.; Stepien, P.; Feit, A.S.; Bergholz, J.; Michowski, W.; Otto, T.; et al. Increased lysosomal biomass is responsible for the resistance of triple-negative breast cancers to CDK4/6 inhibition. Sci. Adv. 2020, 6, eabb2210. [Google Scholar] [CrossRef]
- Martinez-Carreres, L.; Puyal, J.; Leal-Esteban, L.C.; Orpinell, M.; Castillo-Armengol, J.; Giralt, A.; Dergai, O.; Moret, C.; Barquissau, V.; Nasrallah, A.; et al. CDK4 Regulates Lysosomal Function and mTORC1 Activation to Promote Cancer Cell Survival. Cancer Res. 2019, 79, 5245–5259. [Google Scholar] [CrossRef] [Green Version]
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182. [Google Scholar] [CrossRef] [Green Version]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar] [CrossRef]
- Annunziata, I.; van de Vlekkert, D.; Wolf, E.; Finkelstein, D.; Neale, G.; Machado, E.; Mosca, R.; Campos, Y.; Tillman, H.; Roussel, M.F.; et al. MYC competes with MiT/TFE in regulating lysosomal biogenesis and autophagy through an epigenetic rheostat. Nat. Commun. 2019, 10, 3623. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Prat, L.; Kaufmann, K.B.; Schneiter, F.; Voisin, V.; Murison, A.; Chen, J.; Chan-Seng-Yue, M.; Gan, O.I.; McLeod, J.L.; Smith, S.A.; et al. TFEB-mediated endolysosomal activity controls human hematopoietic stem cell fate. Cell Stem Cell 2021, 28, 1838–1850.e10. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Tajeddine, N.; Vitale, I.; Criollo, A.; Vicencio, J.M.; Hickman, J.A.; Geneste, O.; Kroemer, G. Cell Cycle-Dependent Induction of Autophagy, Mitophagy and Reticulophagy. Cell Cycle 2007, 6, 2263–2267. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Nowosad, A.; Jeannot, P.; Callot, C.; Creff, J.; Perchey, R.T.; Joffre, C.; Codogno, P.; Manenti, S.; Besson, A. p27 controls Ragulator and mTOR activity in amino acid-deprived cells to regulate the autophagy-lysosomal pathway and coordinate cell cycle and cell growth. Nat. Cell Biol. 2020, 22, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Nowosad, A.; Creff, J.; Jeannot, P.; Culerrier, R.; Codogno, P.; Manenti, S.; Nguyen, L.; Besson, A. p27 controls autophagic vesicle trafficking in glucose-deprived cells via the regulation of ATAT1-mediated microtubule acetylation. Cell Death Dis. 2021, 12, 481. [Google Scholar] [CrossRef]
- Su, M.; Wang, J.; Wang, C.; Wang, X.; Dong, W.; Qiu, W.; Wang, Y.; Zhao, X.; Zou, Y.; Song, L.; et al. MicroRNA-221 inhibits autophagy and promotes heart failure by modulating the p27/CDK2/mTOR axis. Cell Death Differ. 2014, 22, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Momen, A.; Wu, J.; Noyan, H.; Li, R.; von Harsdorf, R.; Husain, M. p27 protein protects metabolically stressed cardiomyocytes from apoptosis by promoting autophagy. J. Biol. Chem. 2014, 289, 16924–16935. [Google Scholar] [CrossRef] [Green Version]
- Guiley, K.Z.; Stevenson, J.W.; Lou, K.; Barkovich, K.J.; Kumarasamy, V.; Wijeratne, T.U.; Bunch, K.L.; Tripathi, S.; Knudsen, E.S.; Witkiewicz, A.K.; et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science 2019, 366, eaaw2106. [Google Scholar] [CrossRef]
- Furuya, T.; Kim, M.; Lipinski, M.; Li, J.; Kim, D.; Lu, T.; Shen, Y.; Rameh, L.; Yankner, B.; Tsai, L.H.; et al. Negative Regulation of Vps34 by Cdk Mediated Phosphorylation. Mol. Cell 2010, 38, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Yi, J.; Gubas, A.; Wang, Y.T.; Wu, Y.; Ren, Y.; Wu, M.; Shi, Y.; Ouyang, C.; Tan, H.W.S.; et al. Suppression of autophagy during mitosis via CUL4-RING ubiquitin ligases-mediated WIPI2 polyubiquitination and proteasomal degradation. Autophagy 2019, 15, 1917–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almacellas, E.; Pelletier, J.; Day, C.; Ambrosio, S.; Tauler, A.; Mauvezin, C. Lysosomal degradation ensures accurate chromosomal segregation to prevent chromosomal instability. Autophagy 2021, 17, 796–813. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Xie, R.; Nguyen, S.; Ye, M.; McKeehan, W.L. Robust autophagy/mitophagy persists during mitosis. Cell Cycle 2009, 8, 1616–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Ji, X.; Wang, D.; Liu, J.; Zhang, X. Autophagic flux is highly active in early mitosis and differentially regulated throughout the cell cycle. Oncotarget 2016, 7, 39705–39718. [Google Scholar] [CrossRef] [Green Version]
- Belaid, A.; Cerezo, M.; Chargui, A.; Corcelle-Termeau, E.; Pedeutour, F.; Giuliano, S.; Ilie, M.; Rubera, I.; Tauc, M.; Barale, S.; et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013, 73, 4311–4322. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L.; Prescott, A.R.; Cooper, J.; Brachmann, S.M.; Wang, L.; Tang, X.; Backer, J.M.; Lucocq, J.M. Inhibition of autophagy in mitotic animal cells. Traffic 2002, 3, 878–893. [Google Scholar] [CrossRef]
- Rape, M. Cell cycle: On-time delivery of Plk1 during cytokinesis. Curr. Biol. 2007, 17, R506–R508. [Google Scholar] [CrossRef] [Green Version]
- Saurin, A.T.; Brownlow, N.; Parker, P.J. Protein kinase C epsilon in cell division: Control of abscission. Cell Cycle 2009, 8, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mierzwa, B.; Gerlich, D.W. Cytokinetic abscission: Molecular mechanisms and temporal control. Dev. Cell 2014, 31, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ettinger, A.W.; Wilsch-Brauninger, M.; Marzesco, A.M.; Bickle, M.; Lohmann, A.; Maliga, Z.; Karbanova, J.; Corbeil, D.; Hyman, A.A.; Huttner, W.B. Proliferating versus differentiating stem and cancer cells exhibit distinct midbody-release behaviour. Nat. Commun. 2011, 2, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, C.; Jentsch, S. Midbody ring disposal by autophagy is a post-abscission event of cytokinesis. Nat. Cell Biol. 2009, 11, 65–70. [Google Scholar] [CrossRef]
- Kuo, T.-C.; Chen, C.-T.; Baron, D.; Onder, T.T.; Loewer, S.; Almeida, S.; Weismann, C.M.; Xu, P.; Houghton, J.-M.; Gao, F.-B.; et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat. Cell Biol. 2011, 13, 1214–1223. [Google Scholar] [CrossRef] [Green Version]
- Sardina, F.; Monteonofrio, L.; Ferrara, M.; Magi, F.; Soddu, S.; Rinaldo, C. HIPK2 Is Required for Midbody Remnant Removal Through Autophagy-Mediated Degradation. Front. Cell Dev. Biol. 2020, 8, 572094. [Google Scholar] [CrossRef]
- Dionne, L.K.; Peterman, E.; Schiel, J.; Gibieza, P.; Skeberdis, V.A.; Jimeno, A.; Wang, X.J.; Prekeris, R. FYCO1 regulates accumulation of post-mitotic midbodies by mediating LC3-dependent midbody degradation. J. Cell Sci. 2017, 130, 4051–4062. [Google Scholar] [CrossRef] [Green Version]
- Peterman, E.; Gibieza, P.; Schafer, J.; Skeberdis, V.A.; Kaupinis, A.; Valius, M.; Heiligenstein, X.; Hurbain, I.; Raposo, G.; Prekeris, R. The post-abscission midbody is an intracellular signaling organelle that regulates cell proliferation. Nat. Commun. 2019, 10, 3181. [Google Scholar] [CrossRef]
- Messmer, T.; von Meyenn, F.; Savino, A.; Santos, F.; Mohammed, H.; Lun, A.T.L.; Marioni, J.C.; Reik, W. Transcriptional Heterogeneity in Naive and Primed Human Pluripotent Stem Cells at Single-Cell Resolution. Cell Rep. 2019, 26, 815–824.e4. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Kobayashi, S.; Hiratani, I. Epigenetic differences between naive and primed pluripotent stem cells. Cell. Mol. Life Sci. 2018, 75, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Nichols, J.; Smith, A. Naive and primed pluripotent states. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [Green Version]
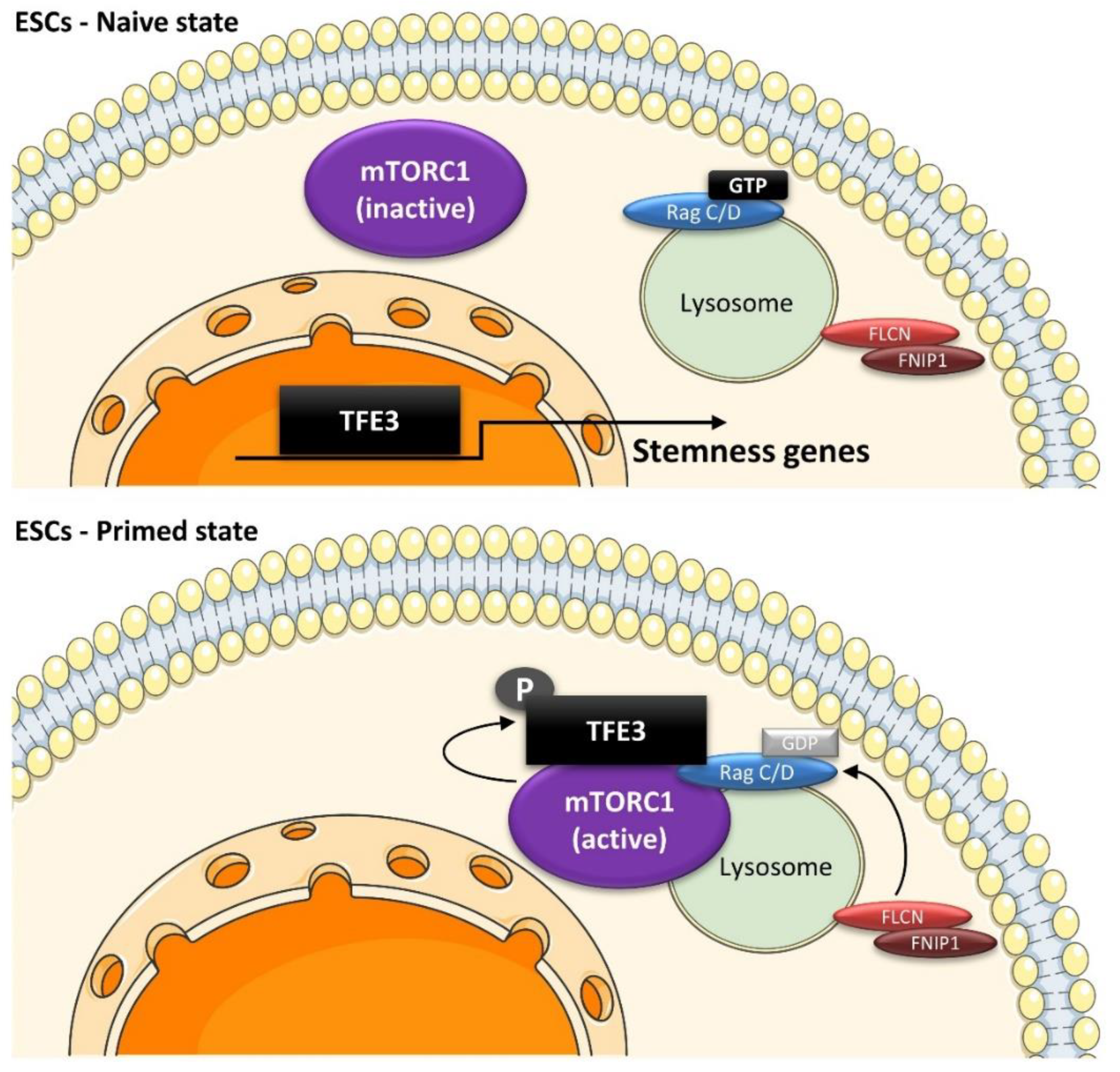
- Betschinger, J.; Nichols, J.; Dietmann, S.; Corrin, P.D.; Paddison, P.J.; Smith, A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription factor Tfe3. Cell 2013, 153, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, J.; Detraux, D.; Kuppers, D.; Wang, Y.; Cavanaugh, C.; Sidhu, S.; Levy, S.; Robitaille, A.M.; Ferreccio, A.; Bottorff, T.; et al. Folliculin regulates mTORC1/2 and WNT pathways in early human pluripotency. Nat. Commun. 2019, 10, 632. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.P.; Kamireddy, A.; Van Nostrand, J.L.; Eichner, L.J.; Shokhirev, M.N.; Dayn, Y.; Shaw, R.J. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016, 30, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDougall, M.S.; Clarke, R.; Merrill, B.J. Intracellular Ca(2+) Homeostasis and Nuclear Export Mediate Exit from Naive Pluripotency. Cell Stem Cell 2019, 25, 210–224.e6. [Google Scholar] [CrossRef]
- Loeffler, D.; Wehling, A.; Schneiter, F.; Zhang, Y.; Muller-Botticher, N.; Hoppe, P.S.; Hilsenbeck, O.; Kokkaliaris, K.D.; Endele, M.; Schroeder, T. Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 2019, 573, 426–429. [Google Scholar] [CrossRef] [Green Version]
- Spevak, C.C.; Elias, H.K.; Kannan, L.; Ali, M.A.E.; Martin, G.H.; Selvaraj, S.; Eng, W.S.; Ernlund, A.; Rajasekhar, V.K.; Woolthuis, C.M.; et al. Hematopoietic Stem and Progenitor Cells Exhibit Stage-Specific Translational Programs via mTOR- and CDK1-Dependent Mechanisms. Cell Stem Cell 2020, 26, 755–765.e7. [Google Scholar] [CrossRef]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Piao, W.; Takamura, T.; Kori, H.; Miyachi, H.; Kitano, S.; Iwamoto, Y.; Yamada, M.; Imayoshi, I.; Shioda, S.; et al. Enhanced lysosomal degradation maintains the quiescent state of neural stem cells. Nat. Commun. 2019, 10, 5446. [Google Scholar] [CrossRef]
- Ibrayeva, A.; Bay, M.; Pu, E.; Jorg, D.J.; Peng, L.; Jun, H.; Zhang, N.; Aaron, D.; Lin, C.; Resler, G.; et al. Early stem cell aging in the mature brain. Cell Stem Cell 2021, 28, 955–966.e7. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Gonzalez, J.L.; Gomez-Sanchez, L.; Mavillard, F.; Linares-Clemente, P.; Rivero, M.C.; Valenzuela-Villatoro, M.; Munoz-Bravo, J.L.; Pardal, R.; Fernandez-Chacon, R. Loss of postnatal quiescence of neural stem cells through mTOR activation upon genetic removal of cysteine string protein-alpha. Proc. Natl. Acad. Sci. USA 2019, 116, 8000–8009. [Google Scholar] [CrossRef] [Green Version]
- Pastore, N.; Huynh, T.; Herz, N.J.; Calcagni, A.; Klisch, T.J.; Brunetti, L.; Kim, K.H.; De Giorgi, M.; Hurley, A.; Carissimo, A.; et al. TFEB regulates murine liver cell fate during development and regeneration. Nat. Commun. 2020, 11, 2461. [Google Scholar] [CrossRef]
- Schaaf, G.J.; van Gestel, T.J.M.; In ‘t Groen, S.L.M.; de Jong, B.; Boomaars, B.; Tarallo, A.; Cardone, M.; Parenti, G.; van der Ploeg, A.T.; Pijnappel, W. Satellite cells maintain regenerative capacity but fail to repair disease-associated muscle damage in mice with Pompe disease. Acta Neuropathol. Commun. 2018, 6, 119. [Google Scholar] [CrossRef] [Green Version]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.A.; Annunziata, F.; Zare, H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Feeney, E.J.; Spampanato, C.; Puertollano, R.; Ballabio, A.; Parenti, G.; Raben, N. What else is in store for autophagy? Exocytosis of autolysosomes as a mechanism of TFEB-mediated cellular clearance in Pompe disease. Autophagy 2013, 9, 1117–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, A.; Pasut, A.; Matsumoto, M.; Yamashita, R.; Fung, J.; Monteleone, E.; Saghatelian, A.; Nakayama, K.I.; Clohessy, J.G.; Pandolfi, P.P. mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 2017, 541, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Wu, X.; Fogarty, S.; Kang, S.A.W.; Dephoure, N.; Gygi, S.P.; Sabatini, D.M.; Rutter, J. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc. Natl. Acad. Sci. USA 2019, 116, 10382–10391. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.G.; Silva, M.T.; da Cunha, F.M.; Moriscot, A.S.; Aoki, M.S.; Miyabara, E.H. Leucine supplementation improves regeneration of skeletal muscles from old rats. Exp. Gerontol. 2015, 72, 269–277. [Google Scholar] [CrossRef]
- Jin, C.L.; Ye, J.L.; Yang, J.; Gao, C.Q.; Yan, H.C.; Li, H.C.; Wang, X.Q. mTORC1 Mediates Lysine-Induced Satellite Cell Activation to Promote Skeletal Muscle Growth. Cells 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyabara, E.H.; Conte, T.C.; Silva, M.T.; Baptista, I.L.; Bueno, C., Jr.; Fiamoncini, J.; Lambertucci, R.H.; Serra, C.S.; Brum, P.C.; Pithon-Curi, T.; et al. Mammalian target of rapamycin complex 1 is involved in differentiation of regenerating myofibers in vivo. Muscle Nerve 2010, 42, 778–787. [Google Scholar] [CrossRef]
- Ge, Y.; Wu, A.L.; Warnes, C.; Liu, J.; Zhang, C.; Kawasome, H.; Terada, N.; Boppart, M.D.; Schoenherr, C.J.; Chen, J. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am. J. Physiol. Cell Physiol. 2009, 297, C1434–C1444. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.H.; Rando, T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef] [Green Version]
- Beumer, J.; Clevers, H. Regulation and plasticity of intestinal stem cells during homeostasis and regeneration. Development 2016, 143, 3639–3649. [Google Scholar] [CrossRef] [Green Version]
- Asano, J.; Sato, T.; Ichinose, S.; Kajita, M.; Onai, N.; Shimizu, S.; Ohteki, T. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017, 20, 1050–1060. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.; Sandor, G.O.; Juhasz, G. Autophagy maintains stem cells and intestinal homeostasis in Drosophila. Sci. Rep. 2018, 8, 4644. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Brauer, C.; Hao, Z.; Elia, A.J.; Fortin, J.M.; Nechanitzky, R.; Brauer, P.M.; Sheng, Y.; Mana, M.D.; Chio, I.I.C.; Haight, J.; et al. Mule Regulates the Intestinal Stem Cell Niche via the Wnt Pathway and Targets EphB3 for Proteasomal and Lysosomal Degradation. Cell Stem Cell 2016, 19, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.; Kovacs, L.; Sandor, G.O.; Juhasz, G. Stem-cell-specific endocytic degradation defects lead to intestinal dysplasia in Drosophila. Dis. Model. Mech. 2016, 9, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Holowatyj, A.N.; Roy, T.; Pronovost, S.M.; Marchetti, M.; Liu, H.; Ulrich, C.M.; Edgar, B.A. An SH3PX1-Dependent Endocytosis-Autophagy Network Restrains Intestinal Stem Cell Proliferation by Counteracting EGFR-ERK Signaling. Dev. Cell 2019, 49, 574–589.e5. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.R.; Zeng, X.; Zhao, J.; Liu, Y.; Hou, G.; Liu, H.; Hou, S.X. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature 2016, 538, 109–113. [Google Scholar] [CrossRef]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [Green Version]
- Turcu, A.L.; Versini, A.; Khene, N.; Gaillet, C.; Caneque, T.; Muller, S.; Rodriguez, R. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chemistry 2020, 26, 7369–7373. [Google Scholar] [CrossRef]
- Balic, A.; Sorensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Jacobs, K.A.; Andre-Gregoire, G.; Maghe, C.; Thys, A.; Li, Y.; Harford-Wright, E.; Trillet, K.; Douanne, T.; Alves Nicolau, C.; Frenel, J.S.; et al. Paracaspase MALT1 regulates glioma cell survival by controlling endo-lysosome homeostasis. EMBO J. 2020, 39, e102030. [Google Scholar] [CrossRef]
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C.; et al. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal disruption preferentially targets acute myeloid leukemia cells and progenitors. J. Clin. Investig. 2013, 123, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.T.; Hamai, A.; Hienzsch, A.; Caneque, T.; Muller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [Green Version]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef]
- Zangrossi, M.; Romani, P.; Chakravarty, P.; Ratcliffe, C.D.H.; Hooper, S.; Dori, M.; Forcato, M.; Bicciato, S.; Dupont, S.; Sahai, E.; et al. EphB6 Regulates TFEB-Lysosomal Pathway and Survival of Disseminated Indolent Breast Cancer Cells. Cancers 2021, 13, 1079. [Google Scholar] [CrossRef] [PubMed]
- Zangrossi, M.; Chakravarty, P.; Romani, P.; Dupont, S.; Hooper, S.; Sahai, E.; Montagner, M. A Lung Organotypic Coculture Reveals a Role for TFEB-Lysosomal Axis in the Survival of Disseminated Dormant Cancer Cells. Cancers 2021, 13, 1007. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharm. 2019, 112, 108690. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Koseki, J.; Takahashi, H.; Miyoshi, N.; Nishida, N.; Nishimura, J.; Hata, T.; Matsuda, C.; Mizushima, T.; Yamamoto, H.; et al. Disruption of Endolysosomal RAB5/7 Efficiently Eliminates Colorectal Cancer Stem Cells. Cancer Res. 2019, 79, 1426–1437. [Google Scholar] [CrossRef] [Green Version]
- Sung, G.J.; Kim, S.H.; Kwak, S.; Park, S.H.; Song, J.H.; Jung, J.H.; Kim, H.; Choi, K.C. Inhibition of TFEB oligomerization by co-treatment of melatonin with vorinostat promotes the therapeutic sensitivity in glioblastoma and glioma stem cells. J. Pineal. Res. 2019, 66, e12556. [Google Scholar] [CrossRef]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef] [Green Version]
- Intlekofer, A.M.; Finley, L.W.S. Metabolic signatures of cancer cells and stem cells. Nat. Metab. 2019, 1, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Semenza, G.L. Serine Synthesis Helps Hypoxic Cancer Stem Cells Regulate Redox. Cancer Res. 2016, 76, 6458–6462. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowosad, A.; Besson, A. Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness. Int. J. Mol. Sci. 2022, 23, 2290. https://doi.org/10.3390/ijms23042290
Nowosad A, Besson A. Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness. International Journal of Molecular Sciences. 2022; 23(4):2290. https://doi.org/10.3390/ijms23042290
Chicago/Turabian StyleNowosad, Ada, and Arnaud Besson. 2022. "Lysosomes at the Crossroads of Cell Metabolism, Cell Cycle, and Stemness" International Journal of Molecular Sciences 23, no. 4: 2290. https://doi.org/10.3390/ijms23042290