
Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
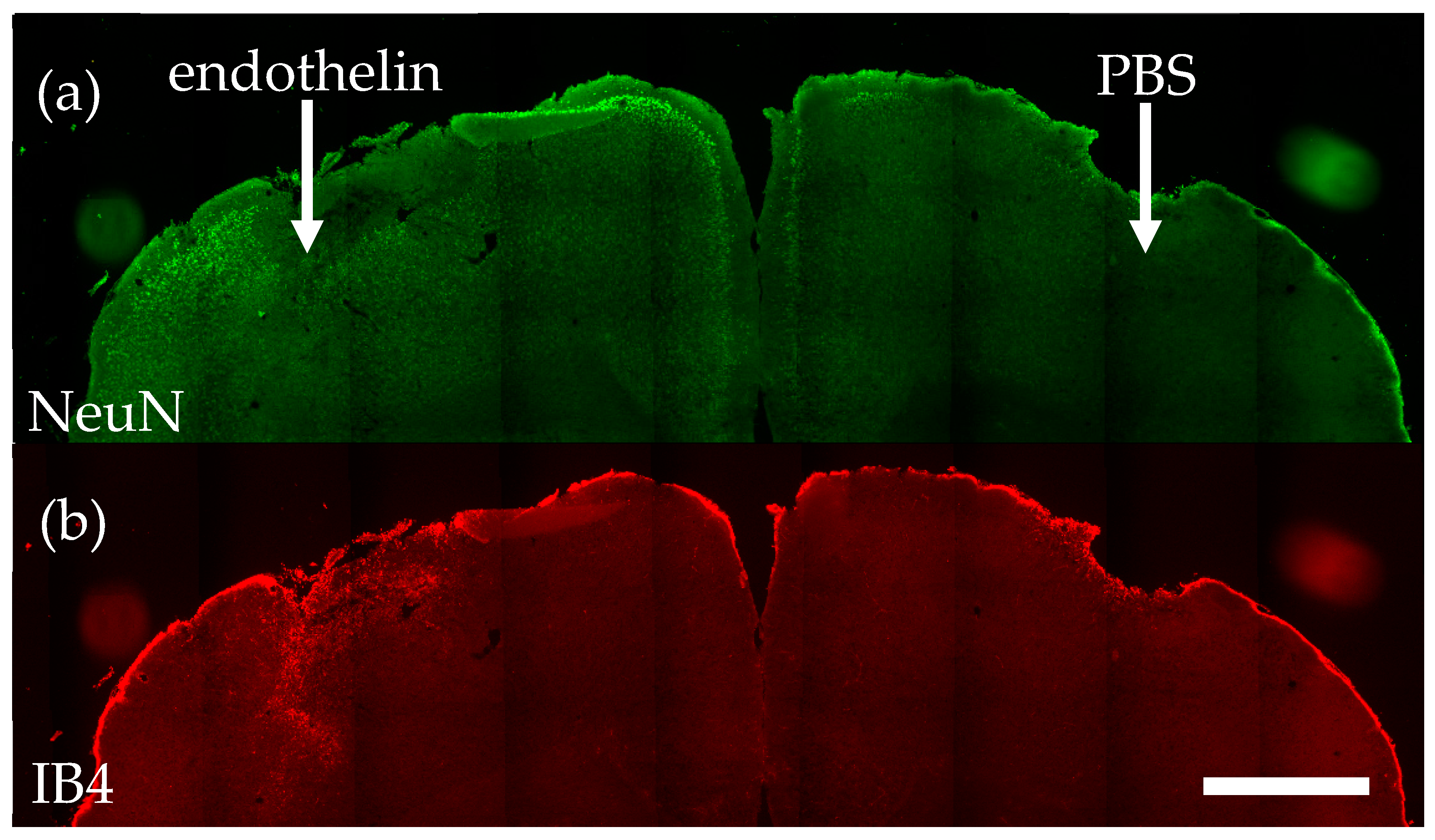
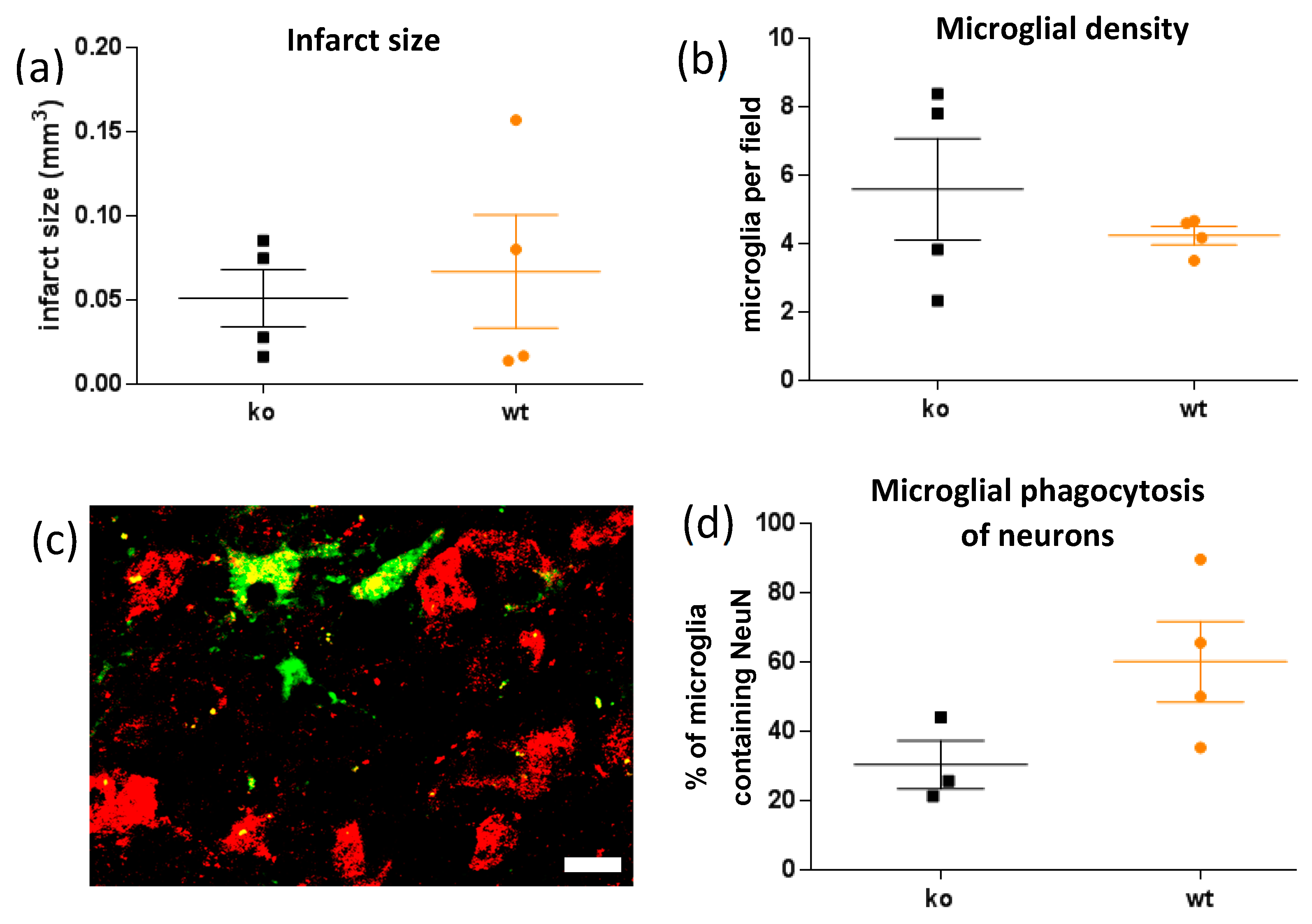
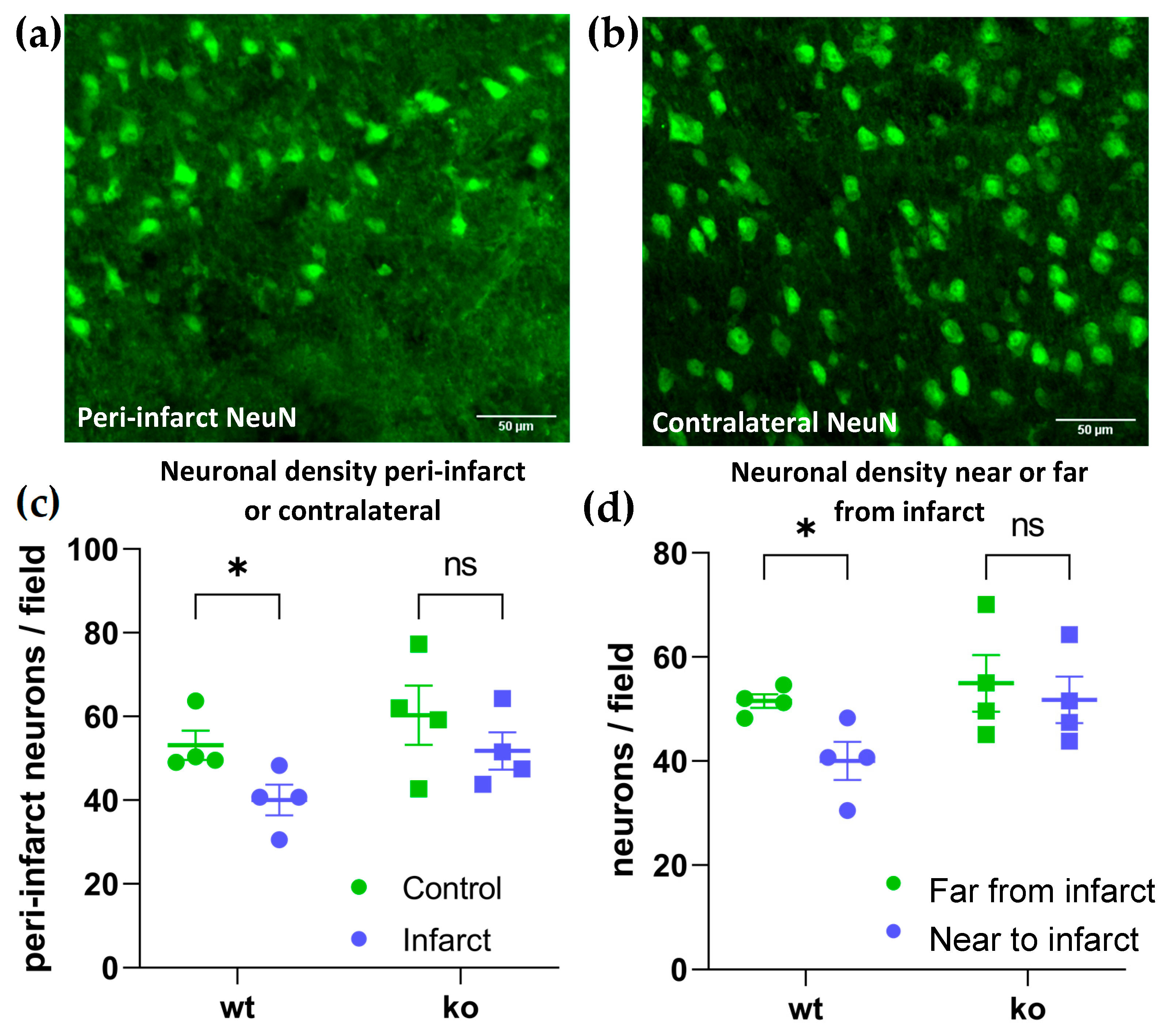
2. Results
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.-C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective Neuronal Loss in Ischemic Stroke and Cerebrovascular Disease. J. Cereb. Blood Flow Metab. 2013, 34, 2–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujioka, M.; Taoka, T.; Matsuo, Y.; Mishima, K.; Ogoshi, K.; Kondo, Y.; Tsuda, M.; Fujiwara, M.; Asano, T.; Sakaki, T.; et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003, 54, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Katchanov, J.; Waeber, C.; Gertz, K.; Gietz, A.; Winter, B.; Brück, W.; Dirnagl, U.; Veh, R.W.; Endres, M. Selective Neuronal Vulnerability Following Mild Focal Brain Ischemia in the Mouse. Brain Pathol. 2006, 13, 452–464. [Google Scholar] [CrossRef]
- Wegener, S.; Weber, R.; Ramos-Cabrer, P.; Uhlenkueken, U.; Sprenger, C.; Wiedermann, D.; Villringer, A.; Hoehn, M. Temporal profile of T2-Weighted MRI Distinguishes between Pannecrosis and Selective Neuronal Death after Transient Focal Cerebral Ischemia in the Rat. J. Cereb. Blood Flow Metab. 2006, 26, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.H.; Liu, K.-F.; Ye, Z.-R.; Gutierrez, J.A. Incomplete Infarct and Delayed Neuronal Death After Transient Middle Cerebral Artery Occlusion in Rats. Stroke 1997, 28, 2303–2310. [Google Scholar] [CrossRef]
- Fricker, M.; Tolkovsky, A.M.; Borutaite, V.; Coleman, M.; Brown, G.C. Neuronal Cell Death. Physiol. Rev. Am. Physiol. Soc. 2018, 98, 813–880. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Brown, G.C. Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. Int. J. Mol. Sci. 2021, 22, 13442. [Google Scholar] [CrossRef]
- Wang, K.; Li, J.; Zhang, Y.; Huang, Y.; Chen, D.; Shi, Z.; Smith, A.D.; Li, W.; Gao, Y. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci. Ther. 2021, 27, 528–539. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2017, 285, 3566–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lee, S.-R.; Choi, S.S.; Yeo, H.-G.; Chang, K.-T.; Lee, H.J. Therapeutically Targeting Neuroinflammation and Microglia after Acute Ischemic Stroke. BioMed Res. Int. 2014, 2014, 297241. [Google Scholar] [CrossRef]
- Kriz, J.; Lalancette-Hebert, M. Inflammation, plasticity and real-time imaging after cerebral ischemia. Acta Neuropathol. 2009, 117, 497–509. [Google Scholar] [CrossRef] [Green Version]
- Miyajima, N.; Ito, M.; Rokugawa, T.; Iimori, H.; Momosaki, S.; Omachi, S.; Shimosegawa, E.; Hatazawa, J.; Abe, K. Detection of neuroinflammation before selective neuronal loss appearance after mild focal ischemia using [18F]DPA-714 imaging. EJNMMI Res. 2018, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Emmrich, J.V.; Ejaz, S.; Williamson, D.J.; Hong, Y.T.; Sitnikov, S.; Fryer, T.D.; Aigbirhio, F.I.; Wulff, H.; Baron, J.-C. Assessing the Effects of Cytoprotectants on Selective Neuronal Loss, Sensorimotor Deficit and Microglial Activation after Temporary Middle Cerebral Occlusion. Brain Sci. 2019, 9, 287. [Google Scholar] [CrossRef] [Green Version]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527. [Google Scholar] [CrossRef]
- Deroide, N.; Li, X.; Lerouet, D.; Van Vré, E.; Baker, L.; Harrison, J.; Poittevin, M.; Masters, L.; Nih, L.; Margaill, I.; et al. MFGE8 inhibits inflammasome-induced IL-1β production and limits postischemic cerebral injury. J. Clin. Investig. 2013, 123, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Davaanyam, D.; Kim, I.-D.; Lee, J.-K. Intranasal Delivery of RGD-Containing Osteopontin Heptamer Peptide Confers Neuroprotection in the Ischemic Brain and Augments Microglia M2 Polarization. Int. J. Mol. Sci. 2021, 22, 9999. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anwar, S.; Pons, V.; Rivest, S. Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells 2020, 9, 1595. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y 6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Milde, S.; van Tartwijk, F.W.; Vilalta, A.; Hornik, T.C.; Dundee, J.M.; Puigdellívol, M.; Brown, G.C. Inflammatory neuronal loss in the substantia nigra induced by systemic lipopolysaccharide is prevented by knockout of the P2Y6 receptor in mice. J. Neuroinflamm. 2021, 18, 225. [Google Scholar] [CrossRef]
- Puigdellívol, M.; Milde, S.; Vilalta, A.; Cockram, T.O.; Allendorf, D.H.; Lee, J.Y.; Dundee, J.M.; Pampuščenko, K.; Borutaite, V.; Nuthall, H.N.; et al. The microglial P2Y6 receptor mediates neuronal loss and memory deficits in neurodegeneration. Cell Rep. 2021, 37, 110148. [Google Scholar] [CrossRef]
- Tennant, K.A.; Jones, T.A. Sensorimotor behavioral effects of endothelin-1 induced small cortical infarcts in C57BL/6 mice. J. Neurosci. Methods 2009, 181, 18–26. [Google Scholar] [CrossRef]
- Sommer, C.J. Ischemic stroke: Experimental models and reality. Acta Neuropathol. 2017, 133, 245–261. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Giacomelli, Á.; Albino, C.M.; de Souza, H.D.N.; Corrêa-Velloso, J.; Santos, A.P.D.J.; Baranova, J.; Ulrich, H. P2Y6 and P2X7 Receptor Antagonism Exerts Neuroprotective/ Neuroregenerative Effects in an Animal Model of Parkinson’s Disease. Front. Cell. Neurosci. 2019, 13, 476. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-D.; Ding, J.-Q.; Xiao, Q.; Chen, S.-D. P2Y6 receptor and immunoinflammation. Neurosci. Bull. 2009, 25, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Wen, R.; Shen, H.; Huang, S.; Wang, L.; Li, Z.; Peng, P.; Mamtilahun, M.; Tang, Y.; Shen, F.; Tian, H.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Gao, Y.; He, C.; Wei, H.; Zhang, J.; Zhang, H.; Hu, L.; Jiang, W. Purinergic Receptor P2Y6 Is a Negative Regulator of NK Cell Maturation and Function. J. Immunol. 2021, 207, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, H.; Li, X.; Wu, J.; Xue, T.; Wu, J.; Shen, H.; Shen, M.; Chen, G. TMEM16F Aggravates Neuronal Loss by Mediating Microglial Phagocytosis of Neurons in a Rat Experimental Cerebral Ischemia and Reperfusion Model. Front. Immunol. 2020, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Alawieh, A.; Langley, E.F.; Tomlinson, S. Targeted complement inhibition salvages stressed neurons and inhibits neuroinflammation after stroke in mice. Sci. Transl. Med. 2018, 10, eaao6459. [Google Scholar] [CrossRef] [Green Version]
- Alawieh, A.M.; Langley, E.F.; Feng, W.; Spiotta, A.M.; Tomlinson, S. Complement-Dependent Synaptic Uptake and Cognitive Decline after Stroke and Reperfusion Therapy. J. Neurosci. 2020, 40, 4042–4058. [Google Scholar] [CrossRef]
- Nishimura, A.; Sunggip, C.; Tozaki-Saitoh, H.; Shimauchi, T.; Numaga-Tomita, T.; Hirano, K.; Ide, T.; Boeynaems, J.-M.; Kurose, H.; Tsuda, M.; et al. Purinergic P2Y6 receptors heterodimerize with angiotensin AT1 receptors to promote angiotensin II–induced hypertension. Sci. Signal. 2016, 9, ra7. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milde, S.; Brown, G.C. Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain. Int. J. Mol. Sci. 2022, 23, 2304. https://doi.org/10.3390/ijms23042304
Milde S, Brown GC. Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain. International Journal of Molecular Sciences. 2022; 23(4):2304. https://doi.org/10.3390/ijms23042304
Chicago/Turabian StyleMilde, Stefan, and Guy C. Brown. 2022. "Knockout of the P2Y6 Receptor Prevents Peri-Infarct Neuronal Loss after Transient, Focal Ischemia in Mouse Brain" International Journal of Molecular Sciences 23, no. 4: 2304. https://doi.org/10.3390/ijms23042304