
Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: Focus on Molecular Mediators

 , ,
, , 
Abstract
:- Table of Contents
- Abstract
- 1. Introduction
- 1.1. Blood Pressure Variability and Brain Damage
- 1.2. Hypertension and Stroke
- 1.3. Hypertension and Cerebral Small Vessel Diseases
- 1.4. Hypertension, Dementia and Progression of Brain Damage
- 2. Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: Traditional Mechanisms
- 2.1. Cerebral Blood Flow Autoregulation
- 2.2. Endothelial Dysfunction and Oxidative Stress
- 2.3. Mitochondrial Dysfunction
- 2.4. Microcirculation
- 2.5. Endothelial Activation and BBD Involvement
- 3. Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: New Factors
- 3.1. Role of Neuroinflammation
- 3.2. Role of the Innate Immune System
- 3.2.1. TRLs: Discovery, Structure and Function
- 3.2.2. Mechanisms of DAMPs Presentation
- 3.2.3. TLRs and Brain Damage-Related Hypertension
- 3.2.4. The Potential Therapeutic Role of TLRs in Cardiovascular Disorders
- 4. Conclusions
1. Introduction
1.1. Blood Pressure Variability and Brain Damage
1.2. Hypertension and Stroke
1.3. Hypertension and cSVD
1.4. Hypertension, Dementia and Progression of Brain Damage
2. Pathogenetic Mechanisms of Hypertension–Brain Induced Complications: Traditional Mechanisms
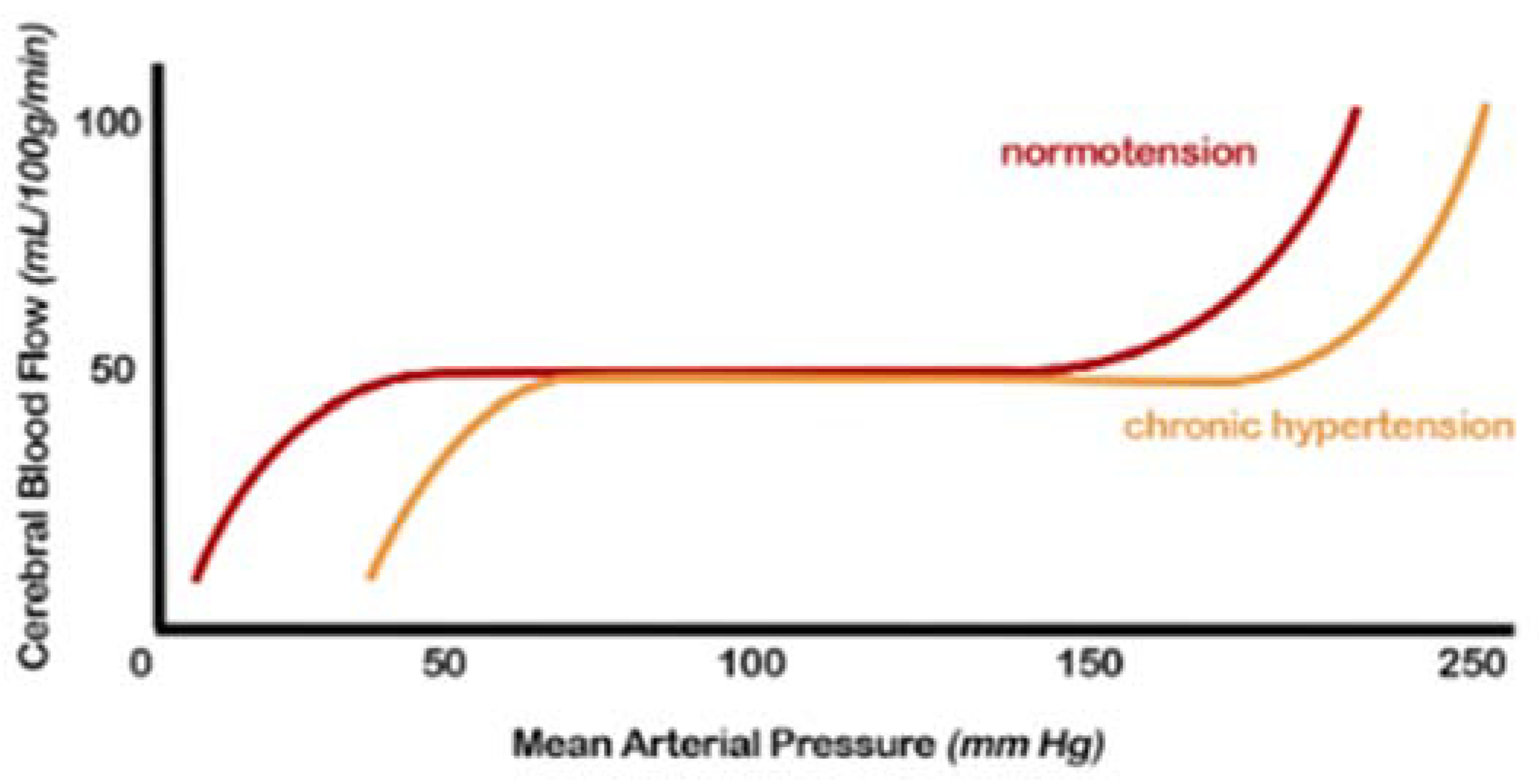
2.1. Cerebral Blood Flow (CBF) Autoregulation
2.2. Endothelial Dysfunction and Oxidative Stress
2.3. Mitochondrial Dysfunction
2.4. Microcirculation
2.5. Endothelial Activation and BBB Involvement
3. Pathogenetic Mechanisms of Hypertension—Brain Induced Complications: New Factors
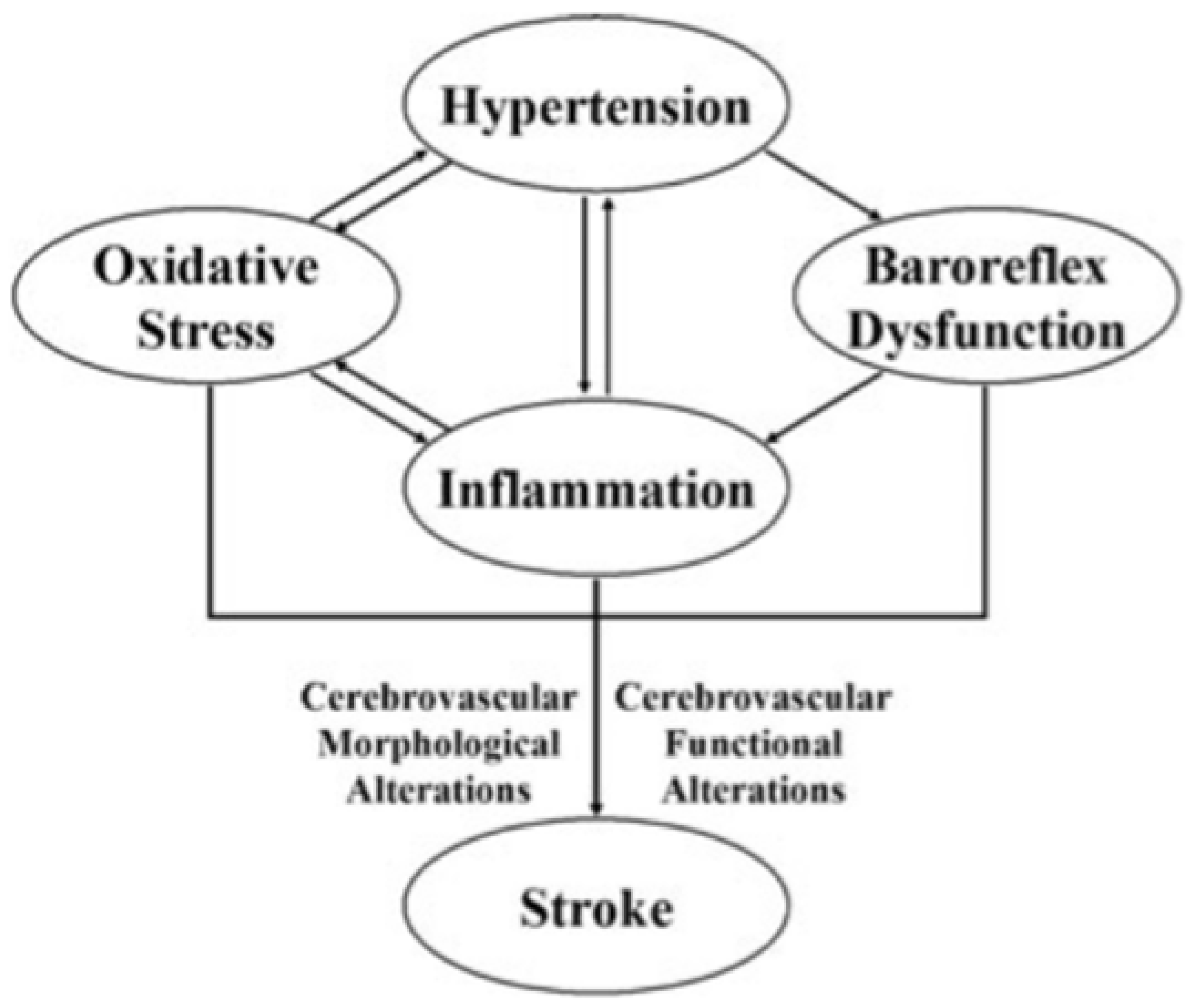
3.1. Role of Neuroinflammation
- The activation of microglia and perivascular and parenchymal resident macrophages;
- The infiltration of peripheral inflammatory cells into the brain.
- The NLRP3 inflammasome is responsible for the activation of IL-1β and the release of IL-18, which phosphorylate insulin receptor substrate 1 (IRS-1), worsening insulin resistance and causing neuronal death. The NLRP3 inflammasome is one of the primary mediators contributing to the neuroinflammation process and consequent brain damage [88].
- DKK-3 concentrations are associated with endothelial dysfunction and atherosclerosis. High or low levels of DKK-3 are able of inducing a worsening of outcomes after ischemic stroke [89].
- The interaction between Dectin-1 and damage-associated molecular patterns (DAMPs) determines the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) and, subsequently, of spleen tyrosine kinase (SYK9), a kinase able to mediate the neuroinflammatory cascade through the release of some cytokines. Therefore, the inflammatory pathway mediated by Dectin-1/SYK plays a fundamental role in postictal neuroinflammation [90].
- The heterodimer CXCL4-CCL5 plays a crucial role in developing brain damage [91].
- MKEY, a cyclic peptide synthesized in mice, can avoid the formation of the heterodimer CXCL4-CCL5, thereby limiting ischemic brain injury and improving neurological deficits [92].
- The expression of some miRNAs, such as 126, 124-3p, 30a and 16, is considerably elevated in patients with acute ischemic brain injury, even if they still cannot be successfully used in clinical routine for obvious reasons, above all being the high cost and their execution in highly specialized laboratories [93].
3.2. Role of the Innate Immune System
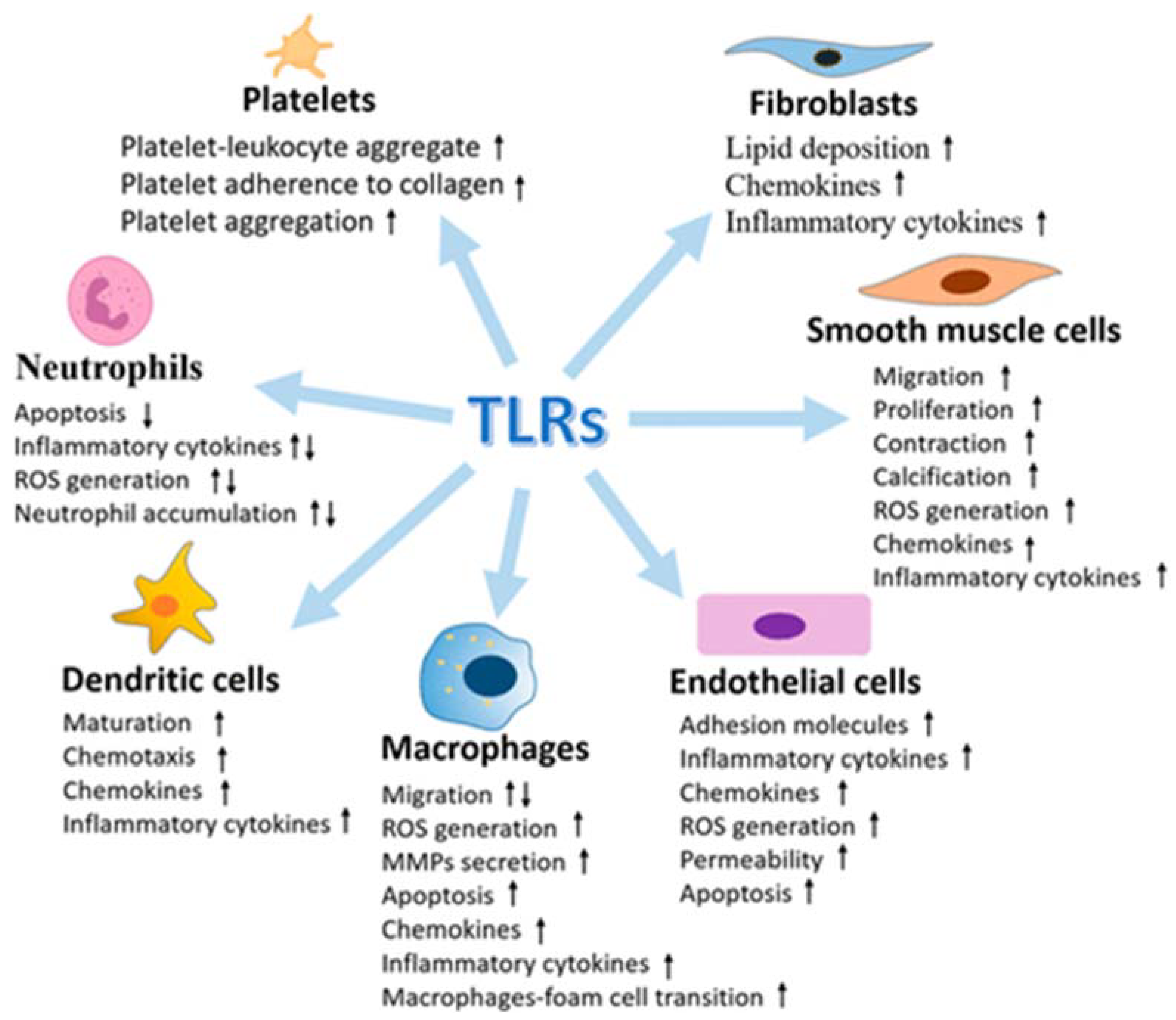
3.2.1. TRLs: Discovery, Structure and Function
- (1)
- An amino (N)-terminal ectodomain that contains leucine-rich repeats and mediates ligand recognition;
- (2)
- A single transmembrane domain that determines cellular localization;
- (3)
- A carboxyl (C)-terminal cytoplasmic domain of the Toll/interleukin-1 receptor (TIR) that mediates downstream signalling.
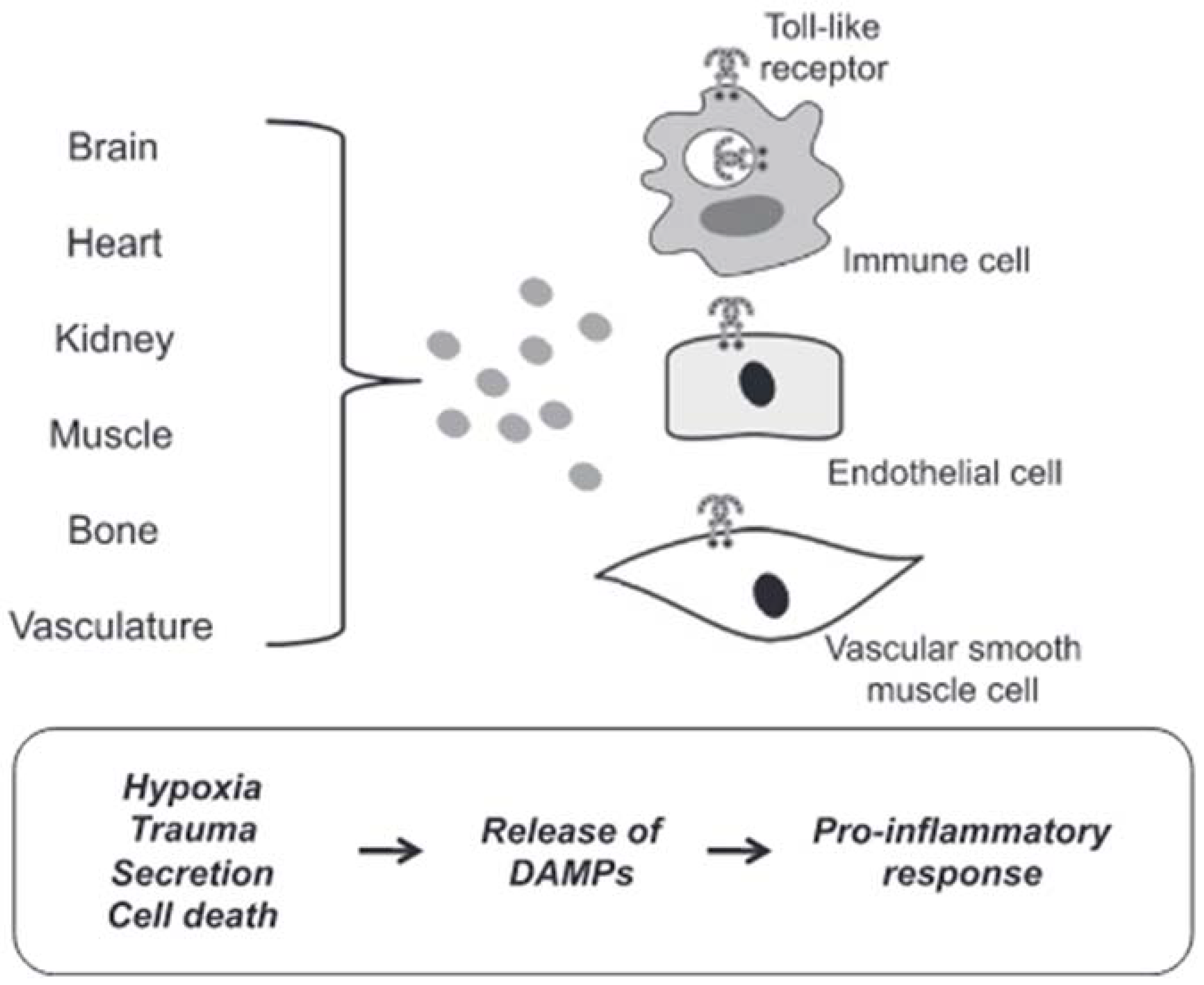
3.2.2. Mechanisms of DAMPs Presentation
3.2.3. TLRs and Brain Damage-Related Hypertension
- Ischemia causes an increase in the expression of TLR2 in neurons (118) and microglia associated with the lesion [119];
- The neurological damage and deficits caused by a stroke were significantly lower in TLR2-deficient mice compared to wild-type controls [118];
- Although acute ischemic lesions (24 to 72 h) have been observed to be smaller in TLR2-deficient mice, the subsequent innate immune response has been reported to be more pronounced, causing progression of the ischemic injury [120].
- TLR3 induces neuroprotection against ischemia through preconditioning [127];
- The expression of TLR7 and TLR8 is associated with a negative outcome with increased inflammatory responses in patients with acute ischemic stroke [128];
- TLR8 agonist induces increased neuronal cell death during oxygen or glucose deprivation, neurological deficit and T cell infiltration after stroke [129];
- TLR9 activation induces neuroprotection against ischemic damage by increasing serum TNF-α by activating PI3K [132].
3.2.4. The Potential Therapeutic Role of TLRs in Cardiovascular Disorders
4. Conclusions
- How can the TLRs signalling alter the different compartments of the blood vessels and the interaction between the layers of the vascular wall?
- How can the activation of TLRs modify the intracellular signal induced by the second messenger in vascular cells?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE | angiotensin-converting enzyme |
| Ang II | angiotensin II |
| ADP | adenosine diphosphate |
| ATP | adenosine triphosphate |
| BBB | brain–blood barrier |
| CBF | cerebral blood flow |
| CCL5 | chemokine (C-C motif) ligand 5 |
| cGMP | cyclic guanosine monophosphate |
| CRP | C-reactive protein |
| cSVD | cerebral small vessel disease |
| CX3CL1 | chemokine (C-X3-C motif) ligand 1 |
| CXCL4 | chemokine (C-X-C motif) ligand 4 |
| CXCL7 | chemokine (C-X-C motif) ligand 7 |
| CXCL8 | chemokine (C-X-C motif) ligand 8 |
| COX-2 | cyclooxygenase-2 |
| DAMPs | damage-associated molecular patterns |
| DKK-3 | Dickkopf WNT Signalling Pathway Inhibitor 3 |
| ECs | endothelial cells |
| ERF | endothelial releasing factors |
| IL | interleukine |
| iNOS | inducible nitric oxide synthase |
| IFN-α | interferone-α |
| IRS-1 | insulin receptor substrate 1 |
| ITAMs | immunoreceptor tyrosine-based activation motifs |
| miRNAs | microRNAs |
| MyD88 | myeloid differentiation primary response gene88-dependent pathway |
| mtDNA | mitochondrial DNA |
| mtNOS | mitochondrial isoform of nitric oxide synthase |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NLR | neutrophil-to-lymphocyte ratio |
| NLRP3 | NLR family pyrin domain-containing 3 |
| NO | nitric oxide |
| OXPHOS | mitochondria mediate oxidative phosphorylation |
| PAMPs | pathogen associated molecular patterns |
| P13K | protein kinase B mechanism-dependent signalling |
| PRRs | family of pattern recognition receptors |
| ROS | reactive oxygen species |
| SHRs | spontaneously hypertensive rats |
| SYK | spleen tyrosine kinase |
| TIR | Toll/interleukin-1 receptor |
| TIRAP | Toll-interleukin 1 receptor domain-containing adapter protein |
| TLRs | Toll-like receptors |
| TNF-α | tumour necrosis factor-alfa |
| TGF-β | transforming growth factor-beta |
| sICAM-1 | soluble intercellular adhesion molecule-1 |
| sVCAM-1 | soluble vascular cell adhesion molecules-1 |
| VSMCs | vascular smooth muscle cells |
References
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.; Coca, A.; De Simone, G.; Dominiczak, A.; et al. Authors/Task Force Members. 2018 Practice Guidelines for the management of arterial hypertension of the European Society of Hypertension and the European Society of Cardiology: ESH/ESC Task Force for the Management of Arterial Hypertension. J. Hypertens. 2018, 36, 2284–2309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, A. Hypertension and the brain: A risk factor for more than heart disease. Cerebrovasc. Dis. 2016, 42, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Barodka, V.M.; Joshi, B.L.; Berkowitz, D.E.; Hogue, C.W.; Nyhan, D. Review article: Implications of vascular aging. Anesth. Analg. 2011, 112, 1048–1060. [Google Scholar] [CrossRef] [Green Version]
- Kai, H.; Kudo, H.; Takayama, N.; Yasuoka, S.; Aoki, Y.; Imaizumi, T. Molecular mechanism of aggravation of hypertensive organ damages by short-term blood pressure variability. Curr. Hypert. Rev. 2014, 10, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Howard, S.C.; Dolan, E.; O’Brien, E.; Dobson, J.E.; Dahlöf, B.; Sever, P.S.; Poulter, N.R. Prognostic significance of visit- to- visit variability, maximum systolic blood pressure, and episodic hypertension. Lancet 2010, 375, 895–905. [Google Scholar] [CrossRef]
- Matsumoto, A.; Satoh, M.; Kikuya, M.; Ohkubo, T.; Hirano, M.; Inoue, R.; Hashimoto, T.; Hara, A.; Hirose, T.; Obara, T.; et al. Day-to-day variability in home blood pressure is associated with cognitive decline: The Ohasama study. Hypertension 2014, 63, 1333–1338. [Google Scholar] [CrossRef] [Green Version]
- Kelly, D.M.; Rothwell, P.M.M. Blood pressure and the brain: The neurology of hypertension. Pract. Neurol. 2020, 20, 100–111. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular smooth muscle cells and arterial stiffening: Relevance in development, aging, and disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Imai, Y.; Hozawa, A.; Ohkubo, T.; Tsuji, I.; Yamaguchi, J.; Matsubara, M.; Michimata, M.; Hashimoto, J.; Fujiwara, T.; Nagai, K.; et al. Predictive values of automated blood pressure measurement: What can we learn from the Japanese population—The Ohasama study. Blood. Pres. Monit. 2001, 6, 335–359. [Google Scholar] [CrossRef]
- Lawes, C.M.; Vander Hoorn, S.; Rodgers, A. Global burden of blood-pressure-related disease, 2001. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Sacco, R.L.; Adams, R.; Albers, G.; Alberts, M.J.; Benavente, O.; Furie, K.; Goldstein, L.B.; Gorelick, P.; Halperin, J.; Harbaugh, R.; et al. Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: A statement for healthcare professionals from the American Heart Association/American Stroke Association Council on Stroke: Co-sponsored by the Council on Cardiovascular Radiology and Intervention: The American Academy of Neurology affirms the value of this guideline. Circulation 2006, 113, e409–e449. [Google Scholar] [PubMed]
- Yu, J.G.; Zhou, R.R.; Cai, G.J. From Hypertension to Stroke: Mechanisms and potential prevention strategies. CNS Neurosci. Ther. 2011, 17, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Dahlof, B. Prevention of stroke in patients with hypertension. Am. J. Cardiol. 2007, 100, S17–S24. [Google Scholar] [CrossRef]
- Jennings, J.R.; Muldoon, M.F.; Ryan, C.; Price, J.C.; Greer, P.; Sutton-Tyrrell, K.; van der Veen, F.M.; Meltzer, C.C. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology 2005, 64, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Rodriguez-Iturbe, B. Mechanisms of disease: Oxidative stress and inflammation in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 582–593. [Google Scholar] [CrossRef]
- Colomba, D.; Duro, G.; Corrao, S.; Argano, C.; Di Chiara, T.; Nuzzo, D.; Pizzo, F.; Parrinello, G.; Scaglione, R.; Licata, G. Endothelial nitric oxide synthase gene polymorphisms and cardiovascular damage in hypertensive subjects: An Italian case-control study. Immun. Ageing 2008, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Tzoulaki, I.; Murray, G.D.; Lee, A.J.; Rumley, A.; Lowe, G.D.; Fowkes, F.G. Relative value of inflammatory, hemostatic, and rheological factors for incident myocardial infarction and stroke: The Edinburgh Artery Study. Circulation 2007, 115, 2119–2127. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Puleo, M.G.; Velardo, M.C.; Corpora, F.; Daidone, M.; Pinto, A. Molecular biology of atherosclerotic ischemic strokes. Int. J. Mol. Sci. 2020, 21, 9372. [Google Scholar] [CrossRef]
- Maida, C.M.; Norrito, R.L.; Daidone, M.; Tuttolomondo, A.; Pinto, A. Neuroinflammatory mechanisms in ischemic stroke: Focus on cardioembolic stroke, background, and therapeutic approaches. Int. J. Mol. Sci. 2020, 21, 6454. [Google Scholar] [CrossRef]
- Persson, P.B. Baroreflexes in hypertension: A mystery revisited. Hypertension 2005, 46, 1095–1096. [Google Scholar] [CrossRef]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Mechanisms of sporadic cerebral small vessel disease: Insights from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Gąsecki, D.; Kwarciany, M.; Nyka, W.; Narkiewicz, K. Hypertension, brain damage and cognitive decline. Curr. Hypertens. Rep. 2013, 15, 547–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehab, A.; Abdulle, A. Cognitive and autonomic dysfunction measures in normal controls, white coat and borderline hypertension. BMC Cardiovasc. Disord. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richmond, R.; Law, J.; Kay-Lambkin, F. Higher blood pressure associated with higher cognition and functionality among centenarians in Australia. Am. J. Hypertens. 2011, 24, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, F.C.; Levey, A.I.; Steenland, N.K. High blood pressure and cognitive decline in mild cognitive impairment. J. Am. Geriatr. Soc. 2013, 61, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Ronnemaa, E.; Zethelius, B.; Lannfelt, L.; Kilander, L. Vascular risk factors and dementia: 40-year follow-up of a population based cohort. Dement. Geriatr. Cogn. Disord. 2011, 31, 460–466. [Google Scholar] [CrossRef]
- Maillard, P.; Seshadri, S.; Beiser, A.; Himali, J.J.; Au, R.; Fletcher, E.; Carmichael, O.; Wolf, P.A.; De Carli, C. Effects of systolic blood pressure on white-matter integrity in young adults in the Framingham Heart Study: A cross-sectional study. Lancet Neurol. 2012, 11, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Pistoia, F.; Sacco, S.; Degan, D.; Tiseo, C.; Ornello, R.; Carole, A. Hypertension and stroke: Epidemiological aspects and clinical evaluation. High Blood Press. Cardiovasc. Prev. 2016, 23, 9–18. [Google Scholar]
- McGrath, E.R.; Beiser, A.S.; DeCarli, C.; Plourde, K.L.; Vasan, R.S.; Greenberg, S.M.; Seshadri, S. Blood pressure from mid to late life and risk of incident dementia. Neurology 2017, 89, 2447–2454. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Nyenhuis, D. On behalf of the American Society of Hypertension Writing Group. Blood pressure and treatment of persons with hypertension as it relates to cognitive outcomes including executive function. J. Am. Soc. Hypertens. 2012, 6, 309–315. [Google Scholar] [CrossRef]
- Scuteri, A. Brain injury as end-organ damage in hypertension. Lancet Neurol. 2012, 11, 1015–1017. [Google Scholar] [CrossRef]
- Au, R.; Massaro, J.M.; Wolf, P.A.; Young, M.E.; Beiser, A.; Seshadri, S.; D’Agostino, R.B.; De Carli, C. Association of white matter hyperintensity volume with decreased cognitive functioning: The Framingham Heart Study. Arch. Neurol. 2006, 63, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Winblad, B.; Fratiglioni, L. The age-dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005, 4, 487–489. [Google Scholar] [CrossRef]
- Iadecola, C.; Gottesman, R.F. Neurovascular and cognitive dysfunction in hypertension: Epidemiology, pathobiology and treatment. Circ. Res. 2019, 124, 1025–1044. [Google Scholar] [CrossRef]
- Nadar, S.K.; Tayebjee, M.H.; Messerli, F.; Lip, G.Y. Target organ damage in hypertension: Pathophysiology and implications for drug theraphy. Curr. Pharm. Des. 2006, 12, 1581–1592. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Daidone, M.; Pinto, A. Endothelial dysfunction and inflammation in ischemic stroke pathogenesis. Curr. Pharm. Des. 2020, 26, 4209–4219. [Google Scholar] [CrossRef]
- Feldmann, E.; Skolnick, B.E. Cerebral hemodynamics, autoregulation, and blood pressure management. J. Stroke Cerebrovasc. Dis. 1999, 8, 176–182. [Google Scholar] [CrossRef]
- Veglio, F.; Paglieri, C.; Rabbia, F.; Bisbocci, D.; Bergui, M.; Cerrato, P. Hypertension and cerebrovascular damage. Atherosclerosis 2009, 205, 331–341. [Google Scholar] [CrossRef]
- Talman, W.T.; Dragon, D.N. Neuronal nitric oxide mediates cerebral vasodilatation during acute hypertension. Brain Res. 2007, 1139, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Hata, R.; Bader, M.; Walther, T.; Hossmann, K.A. Larger anastomoses in angiotensinogen-knockout mice attenuate early metabolic disturbances after middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1999, 19, 1092–1098. [Google Scholar] [CrossRef] [Green Version]
- Dupuis, F.; Atkinson, J.; Limiñana, P.; Chillonet, J.M. Captopril improves cerebrovascular structure and function in old hypertensive rats. Br. J. Pharmacol. 2005, 144, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Mayhan, W.G.; Faraci, F.M.; Heistad, D.D. Disruption of the blood-brain barrier in cerebrum and brain stem during acute hypertension. Am. J. Physiol. Heart Circ. Physiol. 1986, 251, 1171–1175. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 2, 511–540. [Google Scholar]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Pries, A.R.; Secomb, T.W.; Gaehtgens, P. The endothelial surface layer. Pflug. Arch. 2000, 440, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Maida, C.D.; Vasto, S.; Di Raimondo, D.; Casuccio, A.; Vassallo, V.; Daidone, M.; Del Cuore, A.; Pacinella, G.; Cirrincione, A.; Simonetta, I.; et al. Inflammatory activation and endothelial dysfunction markers in patients with permanent atrial fibrillation: A cross-sectional study. Aging 2020, 12, 8423–8433. [Google Scholar] [CrossRef] [PubMed]
- Poggesi, A.; Pasi, M.; Pescini, F.; Pantoni, L.; Inzitari, D. Circulating biologic markers of endothelial dysfunction in cerebral small vessel disease: A review. J. Cereb. Blood Flow Metab. 2016, 36, 72–94. [Google Scholar] [CrossRef] [Green Version]
- Licata, G.; Di Chiara, T.; Licata, A.; Triolo, G.; Argano, C.; Parrinello, G.; Pinto, A.; Corrao, S.; Duro, G.; Scaglione, R. Relationship between circulating E-selectin, DD genotype of angiotensin converting enzyme, and cardiovascular damage in central obese subjects. Metabolism 2003, 52, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- González, J.; Valls, N.; Brito, R.; Rodrigo, R. Essential hypertension and oxidative stress: New insights. World J. Cardiol. 2014, 6, 353–366. [Google Scholar] [CrossRef]
- Puddu, P.; Puddu, G.M.; Galletti, L.; Cravero, E.; Muscari, A. Mitochondrial dysfunction as an initiating event in atherogenesis: A plausible hypothesis. Cardiology 2005, 103, 137–141. [Google Scholar] [CrossRef]
- Ballinger, S.W.; Patterson, C.; Yan, C.N.; Doan, R.; Burow, D.L.; Young, C.G.; Yakes, F.M.; van Houten, B.; Ballinger, C.A.; Freeman, B.A.; et al. Hydrogen peroxide and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ. Res. 2000, 86, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Tostes, R.C.; Webb, R.C. Mitochondrial aldehyde dehydrogenase prevents ROS-induced vascular contraction in angiotensin-II hypertensive mice. J. Am. Soc. Hypertens. 2011, 5, 154–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondria l oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsuki, T.; Matsumoto, M.; Suzuki, K.; Taniguchi, N.; Kamada, T. Mitochondrial lipid peroxidation and superoxide dismutase in rat hypertensive target organs. Am. J. Physiol. 1995, 268, H1418–H1421. [Google Scholar] [CrossRef]
- Patten, D.A.; Germain, M.; Kelly, M.A.; Slack, R.S. Reactive oxygen species: Stuck in the middle of neurodegeneration. J. Alzheimers Dis. 2010, 20, S357–S367. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Campistrous, A.; Hao, L.; Xiang, W.; Ton, D.; Semchuk, P.; Sander, J.; Ellison, M.J.; Fernandez-Patron, C. Mitochondrial dysfunction in the hypertensive rat brain: Respiratory complexes exhibit assembly defects in hypertension. Hypertension 2008, 51, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional modules and structural basis of conformational coupling in mitochondrial complex I. Science 2010, 329, 448–451. [Google Scholar] [CrossRef]
- Calderón-Cortés, E.; Cortés-Rojo, C.; Clemente-Guerrero, M.; Manzo-Avalos, S.; Villalobos-Molina, R.; Boldogh, I.; Saavedra-Molina, A. Changes in mitochondrial functionality and calcium uptake in hypertensive rats as a function of age. Mitochondrion 2008, 8, 262–272. [Google Scholar] [CrossRef] [Green Version]
- Aguilera-Aguirre, L.; González-Hernández, J.C.; Pérez-Vázquez, V.; Ramírez, J.; Clemente-Guerrero, M.; Villalobos-Molina, R.; Saavedra-Molina, A. Role of intramitochondrial nitric oxide in rat heart and kidney during hypertension. Mitochondrion 2002, 1, 413–423. [Google Scholar] [CrossRef]
- Feihl, F.; Liaudet, L.; Waeber, B.; Levy, B.I. Hypertension: A disease of the microcirculation? Hypertension 2006, 48, 1012–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, R.S. Effects of altered cerebral hemodynamics on cognitive function. J. Alzheimers Dis. 2012, 32, 633–642. [Google Scholar] [CrossRef]
- Tian, G.H.; Sun, K.; Huang, P.; Zhou, C.M.; Yao, H.J.; Huo, Z.J.; Hao, H.F.; Yang, L.; Pan, C.S.; He, K.; et al. Long-term stimulation with electroacupuncture at DU20 and ST36 rescues hippocampal neuron through attenuating cerebral blood flow in spontaneously hypertensive rats. Evid.-Based Complement. Altern. Med. 2013, 2013, 482947. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Al-Shahi Salman, R.; Warach, S.; Launer, L.J.; Van Buchem, M.A.; Breteler, M.M. Microbleed Study Group: Cerebral micro bleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Chrissobolis, S.; Miller, A.A.; Drummond, G.R.; Kemp-Harper, B.K.; Sobey, C.G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733–1745. [Google Scholar] [CrossRef] [PubMed]
- Deplanque, D.; Lavallee, P.C.; Labreuche, J.; Gongora-Rivera, F.; Jaramillo, A.; Brenner, D.; Abboud, H.; Klein, I.F.; Touboul, P.J.; Vicaut, E.; et al. Lacunar-BICHAT Investigators: Cerebral and extracerebral vasoreactivity in symptomatic lacunar stroke patients: A case-control study. Int. J. Stroke 2013, 8, 413–421. [Google Scholar] [CrossRef]
- Markus, H.S.; Hunt, B.; Palmer, K.; Enzinger, C.; Schmidt, H.; Schmidt, R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: Longitudinal results of the Austrian stroke prevention study. Stroke 2005, 36, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Kazama, K.; Anrather, J.; Zhou, P.; Girouard, H.; Frys, K.; Milner, T.A.; Iadecola, C. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ. Res. 2004, 95, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Didion, S.P.; Faraci, F.M. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke 2003, 34, 2038–2042. [Google Scholar] [CrossRef] [Green Version]
- Pires, P.W.; Jackson, W.F.; Dorrance, A.M. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H127–H136. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, A.H.; Oommen, A.T.; Bridges, L.R. Endothelial cells and human cerebral small vessel disease. Brain Pathol. 2015, 25, 44–50. [Google Scholar] [CrossRef] [PubMed]
- del Zoppo, G.J. Aging and the neurovascular unit. Ann. N. Y. Acad. Sci. 2012, 1268, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- van Hooren, K.W.; Spijkers, L.J.; van Breevoort, D.; Fernandez-Borja, M.; Bierings, R.; van Buul, J.D.; Alewijnse, A.E.; Peters, S.L.; Voorberg, J. Sphingosine-1-phosphate receptor 3 mediates sphingosine-1-phosphate induced release of Weibel-Palade bodies from endothelial cells. PLoS ONE 2014, 9, e91346. [Google Scholar] [CrossRef]
- Shoamanesh, A.; Preis, S.R.; Beiser, A.S.; Vasan, R.S.; Benjamin, E.J.; Kase, C.S.; Wolf, P.A.; DeCarli, C.; Romero, J.R.; Seshadri, S. Inflammatory biomarkers, cerebral microbleeds, and small vessel disease: Framingham heart study. Neurology 2015, 84, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Rouhl, R.P.; Damoiseaux, J.G.; Lodder, J.; Theunissen, R.O.; Knottnerus, I.L.; Staals, J.; Henskens, L.H.; Kroon, A.A.; de Leeuw, P.W.; Tervaert, J.W.; et al. Vascular inflammation in cerebral small vessel disease. Neurobiol. Aging 2012, 33, 1800–1806. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Spizzo, I.; Diep, H.; Drummond, G.R.; Widdop, R.E.; Vinh, A. Differential phenotypes of tissue-infiltrating T cells during angiotensin II-induced hypertension in mice. PLoS ONE 2014, 9, e114895. [Google Scholar] [CrossRef]
- Trott, D.W.; Harrison, D.G. The immune system in hypertension. Adv. Physiol. Educ. 2014, 38, 20–24. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.; Hoshide, S.; Kario, K. Association of prothrombotic status with markers of cerebral small vessel disease in elderly hypertensive patients. Am. J. Hypertens. 2012, 25, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- Marques, F.; Sousa, J.C.; Sousa, N.; Palha, J.A. Blood-brain-barriers in aging and in Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Wardlaw, J.M.; Doubal, F.; Armitage, P.; Chappell, F.; Carpenter, T.; Muñoz Maniega, S.; Farrall, A.; Sudlow, C.; Dennis, M.; Dhillon, B. Lacunar stroke is associated with diffuse blood-brain barrier dysfunction. Ann. Neurol. 2009, 65, 194–202. [Google Scholar] [CrossRef]
- Weiner, H.L.; Selkoe, D.J. Inflammation and therapeutic vaccination in CNS diseases. Nature 2002, 420, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Maida, C.; Pinto, A. Inflammation and inflammatory cell recruitment in acute cerebrovascular diseases. Curr. Immunol. Rev. 2015, 11, 24–32. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Casuccio, A.; Di Raimondo, D.; Buttà, C.; Clemente, G.; Della Corte, V.; Guggino, G.; Arnao, V.; Maida, C.; et al. Peripheral frequency of CD4+ CD28− cells in acute ischemic stroke. Medicine 2015, 94, e813. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Arnao, V.; Pinto, A.; Licata, G. Inflammation in ischemic stroke subtypes. Curr. Pharm. Des. 2012, 18, 4289–4310. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Sciacca, R.; Di Raimondo, D.; Serio, A.; D’Aguanno, G.; La Placa, S.; Pecoraro, R.; Arnao, V.; Marino, L.; Monaco, S.; et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes: Relationship with TOAST subtype, outcome and infarct site. J. Neuroimmunol. 2009, 215, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Pétrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Monaghan, A.; Kioschis, P.; Wu, W.; Zuniga, A.; Bock, D.; Poustka, A.; Delius, H.; Niehrs, C. Dickkopf genes are coordinately expressed in mesodermal lineages. Mech. Dev. 1999, 87, 45–56. [Google Scholar] [CrossRef]
- Ye, X.; Hao, Q.; Ma, W.-J.; Zhao, Q.-C.; Wang, W.-W.; Yin, H.-H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.-X.; et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflamm. 2020, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Koenen, R.R.; Von Hundelshausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.M.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef]
- Terao, S.; Yilmaz, G.; Stokes, K.Y.; Russell, J.; Ishikawa, M.; Kawase, T.; Granger, D.N. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia–reperfusion. Stroke 2008, 39, 2560–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.; Wang, F.; Li, H.; Yin, Z.; Sandip, C.; Lou, Y.; Wang, Y.; Chen, C.; Wang, D.W. Circulating miR-30a, miR-126 and let-7b as biomarker for ischemic stroke in humans. BMC Neurol. 2013, 13, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goulopoulou, S.; McCarthy, C.G.; Webb, R.C. Toll-like receptors in the vascular system: Sensing the dangers within. Pharmacol. Rev. 2016, 68, 142–167. [Google Scholar] [CrossRef]
- Lazaridis, A.; Gavriilaki, L.; Douma, S.; Gkaliagkousi, E. Toll-like receptors in the pathogenesis of essential hypertension. A forthcoming immune-driven theory in full effect. Int. J. Mol. Sci. 2021, 22, 3451. [Google Scholar] [CrossRef]
- Zhou, Y.; Little, P.J.; Downey, L.; Afroz, R.; Wu, Y.; Ta, H.T.; Xu, S.; Kamato, D. The role of toll-like receptors in atherothrombotic cardiovascular disease. ACS Pharmacol. Transl. Sci. 2020, 3, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.V.; Jürgens, G.; Nüsslein-Volhard, C. Establishment of dorsal-ventral polarity in the Drosophila embryo: Genetic studies on the role of the Toll gene product. Cell 1985, 42, 779–789. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Rifkin, I.R.; Leadbetter, E.A.; Busconi, L.; Viglianti, G.; Marshak-Rothstein, A. Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol. Rev. 2005, 204, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.M.; Ketelhuth, D.F.; Johansson, M.E.; Gerdes, N.; Liu, S.; Yamamoto, M.; Akira, S.; Hansson, G.K. Toll-like receptor 3 and 4 signalling through the TRIF and TRAM adaptors in haematopoietic cells promotes atherosclerosis. Cardiovasc. Res. 2013, 99, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Yamasaki, S. Sensing necrotic cells. Adv. Exp. Med. Biol. 2012, 738, 144–152. [Google Scholar]
- Davidovich, P.; Kearney, C.J.; Martin, S.J. Inflammatory outcomes of apoptosis, necrosis and necroptosis. Biol. Chem. 2014, 395, 1163–1171. [Google Scholar] [CrossRef]
- Erridge, C.; Burdess, A.; Jackson, A.J.; Murray, C.; Riggio, M.; Lappin, D.; Milligan, S.; Spickett, C.M.; Webb, D.J. Vascular cell responsiveness to Toll-like receptor ligands in carotid atheroma. Eur. J. Clin. Investig. 2008, 38, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Bomfim, G.F.; Echem, C.; Martins, C.B.; Costa, T.J.; Sartoretto, S.M.; Dos Santos, R.A.; Oliveira, M.A.; Akamine, E.H.; Fortes, Z.B.; Tostes, R.C.; et al. Toll-like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sci. 2015, 122, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Majumder, S.; Pushpakumar, S.; Juin, S.K.; Jala, V.R.; Sen, U. Toll-like receptor 4 mutation protects the kidney from Ang-II-induced hypertensive injury. Pharmacol. Res. 2022, 175, 106030. [Google Scholar] [CrossRef]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [Green Version]
- Vijay-Kumar, M.; Aitken, J.D.; Carvalho, F.A.; Cullender, T.C.; Mwangi, S.; Srinivasan, S.; Sitaraman, S.V.; Knight, R.; Ley, R.E.; Gewirtz, A.T. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 2010, 328, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Harwani, S.C.; Chapleau, M.W.; Legge, K.L.; Ballas, Z.K.; Abboud, F.M. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ. Res. 2012, 111, 1190–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, C.G.; Wenceslau, C.F.; Goulopoulou, S.; Ogbi, S.; Baban, B.; Sullivan, J.C.; Matsumoto, T.; Webb, R.C. Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res. 2015, 107, 119–130. [Google Scholar] [CrossRef]
- Rodrigues, F.L.; Silva, L.E.; Hott, S.C.; Bomfim, G.F.; da Silva, C.A.; Fazan, R., Jr.; Resstel, L.B.; Tostes, R.C.; Carneiro, F.S. Toll-like receptor 9 plays a key role in the autonomic cardiac and baroreflex control of arterial pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R714–R723. [Google Scholar] [CrossRef] [Green Version]
- Shichita, T.; Ito, M.; Yoshimura, A. Post-ischemic inflammation regulates neural damage and protection. Front. Cell Neurosci. 2014, 8, 319. [Google Scholar] [CrossRef]
- Li, L.; Acioglu, C.; Heary, R.F.; Elkabes, S. Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav. Immun. 2021, 91, 740–755. [Google Scholar] [CrossRef]
- Fadakar, K.; Dadkhahfar, S.; Esmaeili, A.; Rezaei, N. The role of Toll-like receptors (TLRs) in stroke. Rev. Neurosci. 2014, 25, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Arumugam, T.V.; Xu, X.; Cheng, A.; Mughal, M.R.; Jo, D.G.; Lathia, J.D.; Siler, D.A.; Chigurupati, S.; Ouyang, X.; et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 13798–13803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnardt, S.; Lehmann, S.; Kaul, D.; Tschimmel, K.; Hoffmann, O.; Cho, S.; Krueger, C.; Nitsch, R.; Meisel, A.; Weber, J.R. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. J. Neuroimmunol. 2007, 190, 28–33. [Google Scholar] [CrossRef]
- Bohacek, I.; Cordeau, P.; Lalancette-Hébert, M.; Gorup, D.; Weng, Y.C.; Gajovic, S.; Kriz, J. Toll-like receptor 2 deficiency leads to delayed exacerbation of ischemic injury. J. Neuroinflamm. 2012, 9, 191. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Ma, J.; Ha, T.; Kelley, J.L.; Kao, R.L.; Schweitzer, J.B.; Kalbfleisch, J.H.; Williams, D.L.; Li, C. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res. 2009, 1262, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Liu, L.; Chen, Y.; Ha, T.; Kelley, J.; Schweitzer, J.; Kalbfleisch, J.H.; Kao, R.L.; Williams, D.L.; Li, C. TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J. Immunol. 2011, 187, 1458–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansing, L.H.; Harris, T.H.; Welsh, F.A.; Kasner, S.E.; Hunter, C.A.; Kariko, K. Toll- like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 2011, 70, 646–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.C.; Wang, P.F.; Fang, H.; Chen, J.; Xiong, X.Y.; Yang, Q.W. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke 2013, 44, 2545–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradillo, J.M.; Fernández-López, D.; García-Yébenes, I.; Sobrado, M.; Hurtado, O.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J. Neurochem. 2009, 109, 287–294. [Google Scholar] [CrossRef]
- Moraga, A.; Pradillo, J.M.; Cuartero, M.I.; Hernández-Jiménez, M.; Oses, M.; Moro, M.A.; Lizasoain, I. Toll-like receptor 4 modulates cell migration and cortical neurogenesis after focal cerebral ischemia. FASEB J. 2014, 28, 4710–4718. [Google Scholar] [CrossRef]
- Shi, H.; Gabarin, N.; Hickey, E.; Askalan, R. TLR-3 receptor activation protects the very immature brain from ischemic injury. J. Neuroinflamm. 2013, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Brea, D.; Sobrino, T.; Rodríguez-Yáñez, M.; Ramos-Cabrer, P.; Agulla, J.; Rodríguez-González, R.; Campos, F.; Blanco, M.; Castillo, J. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clin. Immunol. 2011, 139, 193–198. [Google Scholar] [CrossRef]
- Tang, S.C.; Yeh, S.J.; Li, Y.I.; Wang, Y.C.; Baik, S.H.; Santro, T.; Widiapradja, A.; Manzanero, S.; Sobey, C.G.; Jo, D.G.; et al. Evidence for a detrimental role of TLR8 in ischemic stroke. Exp. Neurol. 2013, 250, 341–347. [Google Scholar] [CrossRef]
- Hyakkoku, K.; Hamanaka, J.; Tsuruma, K.; Shimazawa, M.; Tanaka, H.; Uematsu, S.; Akira, S.; Inagaki, N.; Nagai, H.; Hara, H. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience 2010, 171, 258–267. [Google Scholar] [CrossRef]
- Stevens, S.L.; Ciesielski, T.M.; Marsh, B.J.; Yang, T.; Homen, D.S.; Boule, J.L.; Lessov, N.S.; Simon, R.P.; Stenzel-Poore, M.P. Toll-like receptor 9: A new target of ischemic preconditioning in the brain. J. Cereb. Blood Flow Metab. 2008, 28, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Ha, T.; Wang, X.; Liu, L.; Zhang, X.; Kimbrough, E.O.; Sha, Z.; Guan, M.; Schweitzer, J.; Kalbfleisch, J.; et al. The TLR9 ligand, CpG-ODN, induces protection against cerebral ischemia/reperfusion injury via activation of PI3K/Akt signaling. J. Am. Heart Assoc. 2014, 3, e000629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avbelj, M.; Horvat, S.; Jerala, R. The role of intermediary domain of MyD88 in cell activation and therapeutic inhibition of TLRs. J. Immunol. 2011, 187, 2394–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.N.; Mann, E.; Behrens, M.M.; Gaidarova, S.; Rebek, M.; Rebek, J., Jr.; Bartfai, T. MyD88-dependent and -independent signaling by IL-1 in neurons probed by bifunctional Toll/IL-1 receptor domain/BB-loop mimetics. Proc. Natl. Acad. Sci. USA 2006, 103, 2953–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassell, B.W.; Seropian, I.M.; Toldo, S.; Salloum, F.N.; Smithson, L.; Varma, A.; Hoke, N.N.; Gelwix, C.; Chau, V.; Abbate, A. Pharmacologic inhibition of myeloid differentiation factor 88 (MyD88) prevents left ventricular dilation and hypertrophy after experimental acute myocardial infarction in the mouse. J. Cardiovasc. Pharmacol. 2010, 55, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murgueitio, M.S.; Rakers, C.; Frank, A.; Wolber, G. Balancing inflammation: Computational design of small-molecule toll-like receptor modulators. Trends Pharmacol. Sci. 2017, 38, 155–168. [Google Scholar] [CrossRef]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-like receptor signaling as a promising therapy for inflammatory diseases: A journey from molecular to nano therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Albanese, A.; Tuttolomondo, A.; Anile, C.; Sabatino, G.; Pompucci, A.; Pinto, A.; Licata, G.; Mangiola, A. Spontaneous chronic subdural hematomas in young adults with a deficiency in coagulation factor XIII. Report of three cases. J. Neurosurg. 2005, 102, 1130–1132. [Google Scholar] [CrossRef]
- Corte, V.D.; Tuttolomondo, A.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Pinto, A. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 2016, 22, 4658–4668. [Google Scholar] [CrossRef]
- Zanoli, L.; Boutouyrie, P.; Fatuzzo, P.; Granata, A.; Lentini, P.; Oztürk, K.; Cappello, M.; Theocharidou, E.; Tuttolomondo, A.; Pinto, A.; et al. Inflammation and Aortic Stiffness: An Individual Participant Data Meta-Analysis in Patients With Inflammatory Bowel Disease. J. Am. Heart Assoc. 2017, 6, e007003. [Google Scholar] [CrossRef] [Green Version]
- Basili, S.; Raparelli, V.; Napoleone, L.; Talerico, G.; Corazza, G.R.; Perticone, F.; Sacerdoti, D.; Andriulli, A.; Licata, A.; Pietrangelo, A.; et al. Platelet Count Does Not Predict Bleeding in Cirrhotic Patients: Results from the PRO-LIVER Study. Am. J. Gastroenterol. 2018, 113, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, S.; Malato, A.; Saccullo, G.; Iorio, A.; Di Ianni, M.; Caracciolo, C.; Coco, L.L.; Raso, S.; Santoro, M.; Guarneri, F.P.; et al. Residual vein thrombosis for assessing duration of anticoagulation after unprovoked deep vein thrombosis of the lower limbs: The extended DACUS study. Am. J. Hematol. 2011, 86, 914–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Raimondo, D.; Tuttolomondo, A.; Buttà, C.; Casuccio, A.; Giarrusso, L.; Miceli, G.; Licata, G.; Pinto, A. Metabolic and anti-inflammatory effects of a home-based programme of aerobic physical exercise. Int. J. Clin. Pract. 2013, 67, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Pinto, A.; Tuttolomondo, A.; Di Raimondo, D.; Fernandez, P.; Licata, G. Risk factors profile and clinical outcome of ischemic stroke patients admitted in a Department of Internal Medicine and classified by TOAST classification. Int. Angiol. 2006, 25, 261–267. [Google Scholar]
- Tuttolomondo, A.; Pinto, A.; Corrao, S.; Di Raimondo, D.; Fernandez, P.; Di Sciacca, R.; Arnao, V.; Licata, G. Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis 2009, 203, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Simonetta, I.; Daidone, M.; Mogavero, A.; Ortello, A.; Pinto, A. Metabolic and Vascular Effect of the Mediterranean Diet. Int. J. Mol. Sci. 2019, 20, 4716. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vascular Factors | Stiffness of arteries; compliance of resistant artery; vascular remodelling |
| Neural Factors | Sympathetic, parasympathetic and baroreflex system |
| Humoral Factors | Renin–angiotensin–aldosterone system; endothelin |
| Others | Stress, emotion, exercise, circadian rhythm, climate, environment |
| Regulation of Vascular Tone |
|
| |
| Balanced Blood Fluidity/Thrombosis |
|
| |
| Vascular Inflammation and Immunological Process Control |
|
| |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Chiara, T.; Del Cuore, A.; Daidone, M.; Scaglione, S.; Norrito, R.L.; Puleo, M.G.; Scaglione, R.; Pinto, A.; Tuttolomondo, A. Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: Focus on Molecular Mediators. Int. J. Mol. Sci. 2022, 23, 2445. https://doi.org/10.3390/ijms23052445
Di Chiara T, Del Cuore A, Daidone M, Scaglione S, Norrito RL, Puleo MG, Scaglione R, Pinto A, Tuttolomondo A. Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: Focus on Molecular Mediators. International Journal of Molecular Sciences. 2022; 23(5):2445. https://doi.org/10.3390/ijms23052445
Chicago/Turabian StyleDi Chiara, Tiziana, Alessandro Del Cuore, Mario Daidone, Stefania Scaglione, Rosario Luca Norrito, Maria Grazia Puleo, Rosario Scaglione, Antonio Pinto, and Antonino Tuttolomondo. 2022. "Pathogenetic Mechanisms of Hypertension–Brain-Induced Complications: Focus on Molecular Mediators" International Journal of Molecular Sciences 23, no. 5: 2445. https://doi.org/10.3390/ijms23052445