
Emerging Glycation-Based Therapeutics—Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors



Abstract
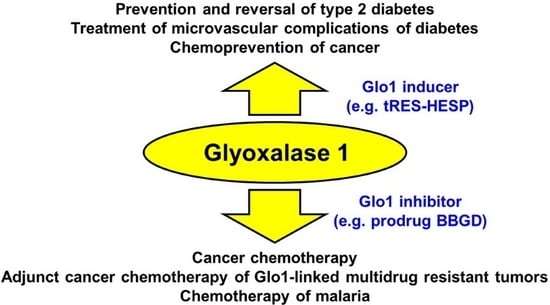
1. Introduction–Dicarbonyl Stress in Health and Disease
2. Development and Applications of Glyoxalase 1 Inducers for Improved Metabolic and Vascular Health
3. Development and Application of Glyoxalase 1 Inhibitors for Cancer Chemotherapy
4. Other Potential Therapeutic Applications and Experimental Applications of Cell Permeable Glyoxalase 1 Inhibitors
5. Concluding Remarks, Future Perspective and Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Babaei-Jadidi, R.; Howell, S.K.; Beisswenger, P.J.; Thornalley, P.J. Degradation products of proteins damaged by glycation, oxidation and nitration in clinical type 1 diabetes. Diabetologia 2005, 48, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Waris, S.; Fleming, T.; Santarius, T.; Larkin, S.J.; Winklhofer-Roob, B.M.; Stratton, M.R.; Rabbani, N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010, 38, 5432–5442. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Activity, regulation, copy number and function in the glyoxalase system. Biochem. Soc. Trans. 2014, 42, 419–424. [Google Scholar] [CrossRef]
- Larsen, K.; Aronsson, A.C.; Marmstal, E.; Mannervik, B. Immunological comparison of glyoxalase I from yeast and mammals with quantitative determination of the enzyme in human tissues by radioimmunoassay. Comp. Biochem. Physiol. B 1985, 82, 625–638. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–334. [Google Scholar] [CrossRef]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 9, 7889. [Google Scholar] [CrossRef]
- Glass, O.; Henao, R.; Patel, K.; Guy, C.D.; Gruss, H.J.; Syn, W.-K.; Moylan, C.A.; Streilein, R.; Hall, R.; Mae Diehl, A.; et al. Serum Interleukin-8, Osteopontin, and Monocyte Chemoattractant Protein 1 Are Associated With Hepatic Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2018, 2, 1344–1355. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef]
- Ajmera, V.; Perito, E.R.; Bass, N.M.; Terrault, N.A.; Yates, K.P.; Gill, R.; Loomba, R.; Diehl, A.M.; Aouizerat, B.E. Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology 2017, 65, 65–77. [Google Scholar] [CrossRef]
- Spoto, B.; Pisano, A.; Zoccali, C. Insulin resistance in chronic kidney disease: A systematic review. Am. J. Physiol.-Ren. Physiol. 2016, 311, F1087–F1108. [Google Scholar] [CrossRef] [PubMed]
- Haller, H.; Bertram, A.; Nadrowitz, F.; Menne, J. Monocyte chemoattractant protein-1 and the kidney. Curr. Opin. Nephrol. Hypertens. 2016, 25, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.J.; Quigley, K.; Cheng, X.; Suresh, A.; Tahir, S.; Ahmed-Jushuf, F.; Nawab, K.; Choy, K.; Walker, S.A.; Mathie, S.A.; et al. Lung Defense through IL-8 Carries a Cost of Chronic Lung Remodeling and Impaired Function. Am. J. Respir. Cell Mol. Biol. 2018, 59, 557–571. [Google Scholar] [CrossRef]
- Valentine, M.S.; Link, P.A.; Herbert, J.A.; Kamga Gninzeko, F.J.; Schneck, M.B.; Shankar, K.; Nkwocha, J.; Reynolds, A.M.; Heise, R.L. Inflammation and Monocyte Recruitment Due to Aging and Mechanical Stretch in Alveolar Epithelium are Inhibited by the Molecular Chaperone 4-Phenylbutyrate. Cell. Mol. Bioeng. 2018, 11, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Kim, Y.S.; Lee, D.-H.; Kim, H.Y.; Lee, J.-I.; Ahn, H.-S.; Sohn, T.S.; Lee, T.-K.; Song, J.Y.; Yeo, C.D.; et al. Association between HOMA-IR and Lung Function in Korean Young Adults based on the Korea National Health and Nutrition Examination Survey. Sci. Rep. 2017, 7, 11726. [Google Scholar] [CrossRef] [PubMed]
- Moreno Velásquez, I.; Gajulapuri, A.; Leander, K.; Berglund, A.; de Faire, U.; Gigante, B. Serum IL8 is not associated with cardiovascular events but with all-cause mortality. BMC Cardiovasc. Disord. 2019, 19, 34. [Google Scholar] [CrossRef]
- Piemonti, L.; Calori, G.; Lattuada, G.; Mercalli, A.; Ragogna, F.; Garancini, M.P.; Ruotolo, G.; Luzi, L.; Perseghin, G. Association Between Plasma Monocyte Chemoattractant Protein-1 Concentration and Cardiovascular Disease Mortality in Middle-Aged Diabetic and Nondiabetic Individuals. Diabetes Care 2009, 32, 2105–2110. [Google Scholar] [CrossRef]
- Ausk, K.J.; Boyko, E.J.; Ioannou, G.N. Insulin resistance predicts mortality in nondiabetic individuals in the U.S. Diabetes Care 2010, 33, 1179–1185. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Multiple roles of glyoxalase 1-mediated suppression of methylglyoxal glycation in cancer biology—Involvement in tumour suppression, tumour growth, multidrug resistance and target for chemotherapy. Semin. Cancer Biol. 2018, 49, 83–93. [Google Scholar] [CrossRef]
- Alhujaily, M.; Abbas, H.; Xue, M.; de la Fuente, A.; Rabbani, N.; Thornalley, P.J. Studies of Glyoxalase 1-Linked Multidrug Resistance Reveal Glycolysis-Derived Reactive Metabolite, Methylglyoxal, Is a Common Contributor in Cancer Chemotherapy Targeting the Spliceosome. Front. Oncol. 2021, 11, 4413. [Google Scholar] [CrossRef]
- Dutertre, M.; Sanchez, G.; Barbier, J.; Corcos, L.; Auboeuf, D. The emerging role of pre-messenger RNA splicing in stress responses: Sending alternative messages and silent messengers. RNA Biol. 2011, 8, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.-C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.; Brouwers, O.; Miyata, T.; Stehouwer, C.; Schalkwijk, C. Glyoxalase-1 overexpression reduces body weight and adipokine expression, and improves insulin sensitivity in high-fat diet-induced obese mice. Diabetologia 2014, 57 (Suppl. S1), 713. [Google Scholar]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of Glyoxalase 1 Mimics Diabetic Nephropathy in Nondiabetic Mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef]
- Berner, A.K.; Brouwers, O.; Pringle, R.; Klaassen, I.; Colhoun, L.; McVicar, C.; Brockbank, S.; Curry, J.W.; Miyata, T.; Brownlee, M.; et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia 2012, 55, 845–854. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnolzer, M.; Lasischka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Zender, L.; Xue, W.; Zuber, J.; Semighini, C.P.; Krasnitz, A.; Ma, B.; Zender, P.; Kubicka, S.; Luk, J.M.; Schirmacher, P.; et al. An Oncogenomics-Based In Vivo RNAi Screen Identifies Tumor Suppressors in Liver Cancer. Cell 2008, 135, 852–864. [Google Scholar] [CrossRef]
- Ashour, A.; Xue, M.; Al-Motawa, M.; Thornalley, P.J.; Rabbani, N. Glycolytic overload-driven dysfunction of periodontal ligament fibroblasts in high glucose concentration, corrected by glyoxalase 1 inducer. BMJ Open Diabetes Res. Care 2020, 8, e001458. [Google Scholar] [CrossRef]
- Dobler, D.; Ahmed, N.; Thornalley, P.J. Peptide mapping of type IV collagen modified minimally by methylglyoxal in vitro reveals mechanism of cell-matrix disruption. Int. J. Exp. Pathol. 2005, 86, A83–A84. [Google Scholar]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Kang, Y.; Edwards, L.G.; Thornalley, P.J. Effect of methylglyoxal on human leukaemia 60 cell growth: Modification of DNA, G1 growth arrest and induction of apoptosis. Leuk. Res. 1996, 20, 397–405. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Edwards, L.G.; Kang, Y.; Wyatt, C.; Davies, N.; Ladan, M.J.; Double, J. Antitumour activity of S-p- bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996, 51, 1365–1372. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Strath, M.; Wilson, R.J.M. Anti-malarial activity in vitro of the glyoxalase I inhibitor diester, S-p-bromobenzylglutathione diethyl ester. Biochem. Pharmacol. 1994, 268, 14189–14825. [Google Scholar]
- Tanner, L.B.; Goglia, A.G.; Wei, M.H.; Sehgal, T.; Parsons, L.R.; Park, J.O.; White, E.; Toettcher, J.E.; Rabinowitz, J.D. Four Key Steps Control Glycolytic Flux in Mammalian Cells. Cell Syst. 2018, 7, 49–62.e8. [Google Scholar] [CrossRef] [PubMed]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. The glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. 1994, 87, 21–29. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Conversion of low density lipoprotein to the pro-atherogenic form by methylglyoxal with increased arterial proteoglycan binding and aortal retention. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.A.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef]
- Mäkinen, V.-P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segrè, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative Genomics Reveals Novel Molecular Pathways and Gene Networks for Coronary Artery Disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Minhas, H.S. Rapid hydrolysis and slow alpha,beta-dicarbonyl cleavage of an agent proposed to cleave glucose-derived protein cross-links. Biochem. Pharmacol. 1999, 57, 303–307. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.R.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Ngianga-Bakwin, K.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved glycemic control and vascular function in overweight and obese subjects by glyoxalase 1 inducer formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Boocock, D.J.; Faust, G.E.S.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I Dose Escalation Pharmacokinetic Study in Healthy Volunteers of Resveratrol, a Potential Cancer Chemopreventive Agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glyoxalase 1 modulation in obesity and diabetes. Antioxid. Redox Signal. 2018, 30, 354–374. [Google Scholar] [CrossRef]
- Li, H.; O’Meara, M.; Zhang, X.; Zhang, K.; Seyoum, B.; Yi, Z.; Kaufman, R.J.; Monks, T.J.; Wang, J.-M. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef]
- Mari, A.; Pacini, G.; Brazzale, A.R.; Ahrén, B. Comparative evaluation of simple insulin sensitivity methods based on the oral glucose tolerance test. Diabetologia 2005, 48, 748–751. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Tura, A.; Winzer, C.; Wagner, O.F.; Ludvik, B.; Hanusch-Enserer, U.; Prager, R.; Pacini, G. Insulin sensitivity during oral glucose tolerance test and its relations to parameters of glucose metabolism and endothelial function in type 2 diabetic subjects under metformin and thiazolidinedione. Diabetes Obes. Metab. 2006, 8, 561–567. [Google Scholar] [CrossRef]
- Hanusch-Enserer, U.; Cauza, E.; Spak, M.; Endler, G.; Dunky, A.; Tura, A.; Wagner, O.; Rosen, H.R.; Pacini, G.; Prager, R. Improvement of Insulin Resistance and Early Atherosclerosis in Patients after Gastric Banding. Obes. Res. 2004, 12, 284–291. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Weickert, M.O.; Thornalley, P.J. Reversal of Insulin Resistance in Overweight and Obese Subjects by trans-Resveratrol and Hesperetin Combination—Link to Dysglycemia, Blood Pressure, Dyslipidemia, and Low-Grade Inflammation. Nutrients 2021, 13, 2374. [Google Scholar] [CrossRef]
- Jo, S.H.; Kim, M.Y.; Park, J.M.; Kim, T.H.; Ahn, Y.H. Txnip contributes to impaired glucose tolerance by upregulating the expression of genes involved in hepatic gluconeogenesis in mice. Diabetologia 2013, 56, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP Regulates Peripheral Glucose Metabolism in Humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Murray, D.L.; Choy, L.N.; Spiegelman, B.M. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 4854–4858. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Pipek, R.; Mandarino, L.J.; DeFronzo, R.A. Tumor necrosis factor-alpha and insulin resistance in obese type 2 diabetic patients. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 88–94. [Google Scholar] [CrossRef]
- Plomgaard, P.; Bouzakri, K.; Krogh-Madsen, R.; Mittendorfer, B.; Zierath, J.R.; Pedersen, B.K. Tumor Necrosis Factor-α Induces Skeletal Muscle Insulin Resistance in Healthy Human Subjects via Inhibition of Akt Substrate 160 Phosphorylation. Diabetes 2005, 54, 2939–2945. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. S2), S157–S163. [Google Scholar] [CrossRef]
- Hallberg, S.J.; Gershuni, V.M.; Hazbun, T.L.; Athinarayanan, S.J. Reversing Type 2 Diabetes: A Narrative Review of the Evidence. Nutrients 2019, 11, 766. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie Restriction-like Effects of 30 Days of Resveratrol Supplementation on Energy Metabolism and Metabolic Profile in Obese Humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef]
- Poulsen, M.M.; Vestergaard, P.F.; Clasen, B.F.; Radko, Y.; Christensen, L.P.; Stødkilde-Jørgensen, H.; Møller, N.; Jessen, N.; Pedersen, S.B.; Jørgensen, J.O.L. High-dose resveratrol supplementation in obese men: An investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013, 62, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.-P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1alpha Cleaves Select microRNAs During ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef]
- Waldhart, A.N.; Dykstra, H.; Peck, A.S.; Boguslawski, E.A.; Madaj, Z.B.; Wen, J.; Veldkamp, K.; Hollowell, M.; Zheng, B.; Cantley, L.C.; et al. Phosphorylation of TXNIP by AKT Mediates Acute Influx of Glucose in Response to Insulin. Cell Rep. 2017, 19, 2005–2013. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.-K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-κB-dependent Inflammatory Genes. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef]
- Chen, J.; Guo, Y.; Zeng, W.; Huang, L.; Pang, Q.; Nie, L.; Mu, J.; Yuan, F.; Feng, B. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am. J. Physiol.-Ren. Physiol. 2014, 306, F916–F925. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Mechanisms of TNF-alpha-induced insulin resistance. Exp. Clin. Endocrinol. Diabetes 1999, 107, 119–125. [Google Scholar] [CrossRef]
- Liu, K.; Zhou, R.; Wang, B.; Mi, M.-T. Effect of resveratrol on glucose control and insulin sensitivity: A meta-analysis of 11 randomized controlled trials. Am. J. Clin. Nutr. 2014, 99, 1510–1519. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Rizza, S.; Muniyappa, R.; Iantorno, M.; Kim, J.-a.; Chen, H.; Pullikotil, P.; Senese, N.; Tesauro, M.; Lauro, D.; Cardillo, C.; et al. Citrus Polyphenol Hesperidin Stimulates Production of Nitric Oxide in Endothelial Cells while Improving Endothelial Function and Reducing Inflammatory Markers in Patients with Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2011, 96, E782–E792. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Momiji, H.; Rabbani, N.; Barker, G.; Bretschneider, T.; Shmygol, A.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of Nrf2 mediate the ARE cytoprotective transcriptional response Antioxid. Redox Signal. 2015, 23, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.-L.; Lin, J.-A.; Shih, P.-H.; Yeh, C.-T.; Yen, G.-C. Pro-cellular survival and neuroprotection of citrus flavonoid: The actions of hesperetin in PC12 cells. Food Funct. 2012, 3, 1082–1090. [Google Scholar] [CrossRef]
- Xue, M.; Momiji, H.; Rabbani, N.; Bretschneider, T.; Rand, D.A.; Thornalley, P.J. Frequency modulated translocational oscillations of Nrf2, a transcription factor functioning like a wireless sensor. Biochem. Soc. Trans. 2015, 43, 669–673. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting cAMP Phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Hexokinase-2-linked glycolytic overload and unscheduled glycolysis—Driver of insulin resistance and development of vascular complications of diabetes. Int. J. Mol. Sci. 2022, 23, 2165. [Google Scholar] [CrossRef]
- Yang, Y.; Li, W.; Liu, Y.; Sun, Y.; Li, Y.; Yao, Q.; Li, J.; Zhang, Q.; Gao, Y.; Gao, L.; et al. Alpha-lipoic acid improves high-fat diet-induced hepatic steatosis by modulating the transcription factors SREBP-1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J. Nutr. Biochem. 2014, 25, 1207–1217. [Google Scholar] [CrossRef]
- Chen, M.-C.; Ye, Y.-Y.; Ji, G.; Liu, J.-W. Hesperidin Upregulates Heme Oxygenase-1 To Attenuate Hydrogen Peroxide-Induced Cell Damage in Hepatic L02 Cells. J. Agric. Food Chem. 2010, 58, 3330–3335. [Google Scholar] [CrossRef]
- Nielsen, I.L.F.; Chee, W.S.S.; Poulsen, L.; Offord-Cavin, E.; Rasmussen, S.E.; Frederiksen, H.; Enslen, M.; Barron, D.; Horcajada, M.N.; Williamson, G. Bioavailability is improved by enzymatic modification of the citrus flavonoid hesperidin in humans: A randomized, double-blind, crossover trial. J. Nutr. 2006, 136, 404–408. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Hexokinase-2 Glycolytic Overload in Diabetes and Ischemia–Reperfusion Injury. Trends Endocrinol. Metab. 2019, 30, 419–431. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Katsuoka, F.; Kitajima, Y.; Nunomiya, A.; Nagatomi, R.; Pi, J.; Biswal, S.S.; Yamamoto, M. Nrf2-Mediated Regulation of Skeletal Muscle Glycogen Metabolism. Mol. Cell. Biol. 2016, 36, 1655–1672. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.; Wadd, W.B. Glyoxalase inhibitors as potential anticancer agents. Biochem. Biophys. Res. Commun. 1969, 35, 593–598. [Google Scholar] [CrossRef]
- Dobler, D.; Ahmed, N.; Song, L.J.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 2006, 55, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Vince, R.; Daluge, S.; Wadd, W.B. Studies on the inhibition of glyoxalase I by S-substituted glutathione. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.E.; Lo, T.W.C.; Thornalley, P.J. Inhibitors of glyoxalase I: Design, synthesis, inhibitory characteristics and biological evaluation. Biochem. Soc. Trans. 1993, 21, 535–540. [Google Scholar] [CrossRef]
- Allen, R.E.; Lo, T.W.C.; Thornalley, P.J. A simplified method for the purification of human red blood cell glyoxalase I. Characteristics, immunoblotting and inhibitor studies. J. Prot. Chem. 1993, 12, 111–119. [Google Scholar] [CrossRef]
- Lo, T.W.C.; Thornalley, P.J. Inhibition of proliferation of human leukemia 60 cells by diethyl esters of glyoxalase inhibitors in vitro. Biochem. Pharmacol. 1992, 44, 2357–2363. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Ladan, M.J.; Ridgway, S.J.S.; Kang, Y. Antitumour activity of S-p-bromobenzylglutathione diesters in vitro: A structure activity study. J. Med. Chem. 1996, 39, 3409–3411. [Google Scholar] [CrossRef]
- Thornalley, P.J. Advances in glyoxalase research. Glyoxalase expression in malignancy, anti-proliferative effects of methylglyoxal, glyoxalase I inhibitor diesters and S-D-lactoylglutathione, and methylglyoxal-modified protein binding and endocytosis by the advanced glycation endproduct receptor. Crit. Rev. Oncol. Haematol. 1995, 20, 99–128. [Google Scholar]
- Creighton, D.J.; Zheng, Z.B.; Holewinski, R.; Hamilton, D.S.; Eiseman, J.L. Glyoxalase I inhibitors in cancer chemotherapy. Biochem. Soc. Trans. 2003, 31, 1378–1382. [Google Scholar] [CrossRef]
- Kavarana, M.J.; Kovaleva, E.G.; Creighton, D.J.; Wollman, M.B.; Eiseman, J.L. Mechanism-Based Competitive Inhibitors of Glyoxalase I: Intracellular Delivery, in Vitro Antitumor Activities, and Stabilities in Human Serum and Mouse Serum. J. Med. Chem. 1999, 42, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Sato, S.; Hashimoto, Y.; Yamori, T.; Tsuruo, T. Selective activation of apoptosis program by S-p-bromobenzylglutathione cyclopentyl diester in glyoxalase I-overexpressing human lung cancer cells. Clin. Cancer Res. 2001, 7, 2513–2518. [Google Scholar]
- Murthy, N.S.R.K.; Bakeris, T.; Kavarana, M.J.; Hamilton, D.S.; Lan, Y.; Creighton, D.J. S-(N-Aryl-N-Hydroxycarbamoyl)Glutathione Derivatives Are Tight-Binding Inhibitors of Glyoxalase-I and Slow Substrates for Glyoxalase-Ii. J. Med. Chem. 1994, 37, 2161–2166. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, E.M.; O’Neill, H.B.; Kavarana, M.J.; Wang, H.B.; Creighton, D.J.; Sentz, D.L.; Eiseman, J.L. Pharmacokinetics and antitumor properties in tumor-bearing mice of an enediol analogue inhibitor of glyoxalase I. Cancer Chemother. Pharmacol. 2000, 46, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Jandial, R.; Neman, J.; Lim, P.P.; Tamae, D.; Kowolik, C.M.; Wuenschell, G.E.; Shuck, S.C.; Ciminera, A.K.; De Jesus, L.R.; Ouyang, C.; et al. Inhibition of GLO1 in Glioblastoma Multiforme Increases DNA-AGEs, Stimulates RAGE Expression, and Inhibits Brain Tumor Growth in Orthotopic Mouse Models. Int. J. Mol. Sci. 2018, 19, 406. [Google Scholar] [CrossRef]
- Hosoda, F.; Arai, Y.; Okada, N.; Shimizu, H.; Miyamoto, M.; Kitagawa, N.; Katai, H.; Taniguchi, H.; Yanagihara, K.; Imoto, I.; et al. Integrated genomic and functional analyses reveal glyoxalase I as a novel metabolic oncogene in human gastric cancer. Oncogene 2015, 34, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Hollenbach, M.; Pohl, S.; Ripoll, C.; Zipprich, A. Inhibition of Glyoxalase-I Leads to Reduced Proliferation, Migration and Colony Formation, and Enhanced Susceptibility to Sorafenib in Hepatocellular Carcinoma. Front. Oncol. 2019, 9, 785. [Google Scholar] [CrossRef]
- Santarius, T.; Bignell, G.R.; Greenan, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. GLO1—A novel amplified gene in human cancer. Genes Chromosomes Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef]
- Kreycy, N.; Gotzian, C.; Fleming, T.; Flechtenmacher, C.; Grabe, N.; Plinkert, P.; Hess, J.; Zaoui, K. Glyoxalase 1 expression is associated with an unfavorable prognosis of oropharyngeal squamous cell carcinoma. BMC Cancer 2017, 17, 382. [Google Scholar] [CrossRef]
- Hamada, H.; Tsuruo, T. Functional role for the 170- to 180-kDa glycoprotein specific to drug-resistant tumor cells as revealed by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1986, 83, 7785–7789. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Kazaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumour agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Shafie, A.; Xue, M.Z.; Thornalley, P.J.; Rabbani, N. Copy number variation of glyoxalase I. Biochem. Soc. Trans. 2014, 42, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Job, B.; Dessen, P.; Tordai, A.; Michiels, S.; Liedtke, C.; Richon, C.; Yan, K.; Wang, B.; Vassal, G.; et al. Molecular Characterization of Breast Cancer with High-Resolution Oligonucleotide Comparative Genomic Hybridization Array. Clin. Cancer Res. 2009, 15, 441–451. [Google Scholar] [CrossRef]
- Chiu, C.G.; Nakamura, Y.; Chong, K.K.; Huang, S.K.; Kawas, N.P.; Triche, T.; Elashoff, D.; Kiyohara, E.; Irie, R.F.; Morton, D.L.; et al. Genome-Wide Characterization of Circulating Tumor Cells Identifies Novel Prognostic Genomic Alterations in Systemic Melanoma Metastasis. Clin. Chem. 2014, 60, 873–885. [Google Scholar] [CrossRef]
- Xue, M.; Shafie, A.; Qaiser, T.; Rajpoot, N.M.; Kaltsas, G.; Gopalakrishnan, K.; Fisk, A.; Dimitriadis, G.K.; Grammatopoulos, D.K.; Rabbani, N.; et al. Glyoxalase 1 copy number variation in patients with well differentiated gastroentero-pancreatic neuroendocrine tumours (GEP-NET). Oncotarget 2017, 8, 76961–76973. [Google Scholar] [CrossRef][Green Version]
- Shafie, A.; Xue, M.; Barker, G.; Zehnder, D.; Thornalley, P.J.; Rabbani, N. Re-appraisal of putative glyoxalase 1 deficient mouse and dicarbonyl stress on embryonic stem cells in vitro. Biochem. J. 2016, 473, 4255–4270. [Google Scholar] [CrossRef]
- Black, J.C.; Atabakhsh, E.; Kim, J.; Biette, K.M.; Van Rechem, C.; Ladd, B.; Burrowes, P.D.; Donado, C.; Mattoo, H.; Kleinstiver, B.P.; et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015, 29, 1018–1031. [Google Scholar] [CrossRef]
- Black, J.C.; Manning, A.L.; Van Rechem, C.; Kim, J.; Ladd, B.; Cho, J.; Pineda, C.M.; Murphy, N.; Daniels, D.L.; Montagna, C.; et al. KDM4A Lysine Demethylase Induces Site-Specific Copy Gain and Rereplication of Regions Amplified in Tumors. Cell 2013, 154, 541–555. [Google Scholar] [CrossRef]
- Demel, H.R.; Feuerecker, B.; Piontek, G.; Seidl, C.; Blechert, B.; Pickhard, A.; Essler, M. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. Am. J. Cancer Res. 2015, 5, 1649–1664. [Google Scholar] [PubMed]
- Maldonado, E.N.; Patnaik, J.; Mullins, M.R.; Lemasters, J.J. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010, 70, 10192–10201. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via AMPK activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Ronen, S.M.; DiStefano, F.; McCoy, C.L.; Robertson, D.; Smith, T.A.D.; Al-Saffar, N.M.; Titley, J.; Cunningham, D.C.; Griffiths, J.R.; Leach, M.O.; et al. Magnetic resonance detects metabolic changes associated with chemotherapy-induced apoptosis. Br. J. Cancer 1999, 80, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Heiden, M.G.V. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Lang-Unnasch, N.; Murphy, A.D. Metabolic changes of the malaria parasite during the transition from the human to the mosquito host. Annu. Rev. Microbiol. 1998, 52, 561–590. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Hunsaker, L.A.; Campos, N.M.; Baack, B.R. d-Lactate production in erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 1990, 42, 277–284. [Google Scholar] [CrossRef]
- Wezena, C.A.; Urscher, M.; Vince, R.; More, S.S.; Deponte, M. Hemolytic and antimalarial effects of tight-binding glyoxalase 1 inhibitors on the host-parasite unit of erythrocytes infected with Plasmodium falciparum. Redox Biol. 2016, 8, 348–353. [Google Scholar] [CrossRef][Green Version]
- Al-Motawa, M.S.; Abbas, H.; Wijten, P.; de la Fuente, A.; Xue, M.; Rabbani, N.; Thornalley, P.J. Vulnerabilities of the SARS-CoV-2 Virus to Proteotoxicity—Opportunity for Repurposed Chemotherapy of COVID-19 Infection. Front. Pharmacol. 2020, 11, 585408. [Google Scholar] [CrossRef]
- De Bock, C.A.; Brug, J.; Walop, J.N. Antiviral Activity of Glyoxals. Nature 1957, 179, 706–707. [Google Scholar] [CrossRef] [PubMed]
- Charyasriwong, S.; Haruyama, T.; Kobayashi, N. In vitro evaluation of the antiviral activity of methylglyoxal against influenza B virus infection. Drug Discov. Ther. 2016, 10, 201–210. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tikellis, C.; Pickering, R.J.; Tsorotes, D.; Huet, O.; Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes 2014, 63, 3915–3925. [Google Scholar] [CrossRef] [PubMed]
- Pun, P.B.L.; Logan, A.; Darley-Usmar, V.; Chacko, B.; Johnson, M.S.; Huang, G.W.; Rogatti, S.; Prime, T.A.; Methner, C.; Krieg, T.; et al. A mitochondria-targeted mass spectrometry probe to detect glyoxals: Implications for diabetes. Free Radic. Biol. Med. 2014, 67, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [Google Scholar] [CrossRef]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef] [PubMed]
- Kawatani, M.; Okumura, H.; Honda, K.; Kanoh, N.; Muroi, M.; Dohmae, N.; Takami, M.; Kitagawa, M.; Futamura, Y.; Imoto, M.; et al. The identification of an osteoclastogenesis inhibitor through the inhibition of glyoxalase I. Proc. Natl. Acad. Sci. USA 2008, 105, 11691–11696. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Minguet, M.; Bou-Teen, D.; Miro-Casas, E.; Castans, C.; Castellano, J.; Bonzon-Kulichenko, E.; Igual, A.; Rodriguez-Lecoq, R.; Vázquez, J.; et al. Ryanodine Receptor Glycation Favors Mitochondrial Damage in the Senescent Heart. Circulation 2019, 139, 949–964. [Google Scholar] [CrossRef]
- Forouhi, N.G.; Misra, A.; Mohan, V.; Taylor, R.; Yancy, W. Dietary and nutritional approaches for prevention and management of type 2 diabetes. BMJ 2018, 361, k2234. [Google Scholar] [CrossRef]
- Taylor, R.; Ramachandran, A.; Yancy, W.S.; Forouhi, N.G. Nutritional basis of type 2 diabetes remission. BMJ 2021, 374, n1449. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Hendler, R.; Simonson, D. Insulin Resistance is a Prominent Feature of Insulin-dependent Diabetes. Diabetes 1982, 31, 795–801. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas; IDF: Brussels, Belgium, 2021. [Google Scholar]
- Feldman, E.L.; Callaghan, B.C.; Pop-Busui, R.; Zochodne, D.W.; Wright, D.E.; Bennett, D.L.; Bril, V.; Russell, J.W.; Viswanathan, V. Diabetic neuropathy. Nat. Rev. Dis. Prim. 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Ashley, D.M.; López, G.Y.; Malinzak, M.; Friedman, H.S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA A Cancer J. Clin. 2020, 70, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Lowenthal, R.M.; Eaton, K. Toxicity of chemotherapy. Hematol./Oncol. Clin. 1996, 10, 967–990. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Evaluation Model | Main Outcome | Reference |
|---|---|---|---|
| GLO1-ARE transcriptional activity | Stable transfectant luciferase reporter cell lines with GLO1-ARE or functionally inactive mutant as negative control | tRES: EC50 = 2.52 ± 0.19 µM and Emax = 100 ± 2% HESP: EC50 = 0.59 ± 0.01 µM and Emax = 24.4 ± 0.1% With 5 µM HESP, for tRES: EC50 = 1.46 ± 0.10 µM and Emax = 95.0 ± 0.1% | [43] |
| Cell vitality markers | Human aortal endothelial cells (HAECs) in primary culture | Decreased glucose metabolism, RAGE, ICAM-1, VCAM-1, E-selectin, and IL8 secretion. | [7,43] |
| Human fibroblasts in primary culture | Increased cellular GSH and decreased RAGE and MMP3 | [43] | |
| Human hepatocyte-like HepG2 cells in vitro | Increased cellular GSH | [43] | |
| Endothelial cell dysfunction in diabetes | Human aortal endothelial cells (HAECs) in primary culture | In high glucose concentration, tRES-HESP (10 μM) corrected HK2-linked glycolytic overload, metabolic dysfunction and IL8 secretion | [7] |
| Fibroblast dysfunction in diabetes | Human periodontal ligament | In high glucose concentration, tRES-HESP (10 μM) corrected HK2-linked glycolytic overload, metabolic dysfunction and adhesion to extracellular matrix | [28] |
| Wound healing in diabetes | Dermal wound healing by topical application of tRES-HESP on alternate days for 6 days in db/db mice | tRES-HESP (5 μM) accelerated wound healing, compared to vehicle control | [46] |
| Metabolic and vascular health | Randomized, double-blind placebo-controlled crossover study in overweight and obese subjects (n = 29). Treatment was for 8 weeks, once-daily by oral capsule containing 90 mg tRES and 120 mg HESP or placebo, with 6 weeks washout (HATFF study; NCT02095873) | Effect of tRES-HESP: target pharmacology—PBMC activity of Glo1 (+27%), and plasma MG concentration (−37%); clinical endpoint-related variables—FPG (−5%), AUCg (−8%) and OGIS (+54 mL min−1m−2); and other—decreased expression of MCP-1, IL-8, COX-2 and RAGE in PBMCs. Urinary excretion of tRES and HESP metabolites increased >2000-fold and >100 fold, respectively. Placebo had no effect. | [43] |
| Cell Lines | Main Outcome | Reference |
|---|---|---|
| Human leukemia 60 (HL60) | GC50 = 4.2 µM; cf. GC50 = 8.3 µM for ethyl diester and GC50 of 4.2–29.2 µM for series of BBG alkyl and cycloalkyl diesters with BBGD fund to be most potent | [32,88,89] |
| National Cancer Institute anticancer screen of 60 human tumor cell lines | GC50 = 5–20 µM for leukemia, NSCLC, colon, CNS, melanoma, ovarian, renal, prostate and breast cancer cell lines—most potent for glioblastoma SNB-19 (data for ethyl diester) | [90] |
| A549, DMS114, DMS273, NCI-H23, NCI-H226, NCI-H460 and NCI-H522 cell lines | GC50 range 4.4–29.7 µM | [93] |
| Sixteen gastric tumor cell lines | GC50 range 3–10 µM | [97] |
| Hepatocellular carcinoma HUH7 | Inhibition of cell growth at 1–10 µM | [98] |
| Glioblastoma multiforme T98 and U87 | GC50: T98, 100.6 µM; and U87, 9.9 µM | [96] |
| Osteosarcoma MG63; lung adenocarcinoma A549, NCI-H522 and NCI-H460; pancreatic carcinoma YAPC; squamous cell carcinoma LB771; and brain astrocytoma CCF-STTG-1 | GC50: MG63, 3.8 µM; A549 23.5 µM; NCI-H522, 7 µM; NCI-H460, 19.8 µM; YAPC, 10 µM; LB771, 9.5 µM; and CCF-STTG-1, 1 µM | [99] |
| FaDu hypopharyngeal carcinoma and CAL27 oral adenosquamous carcinoma | GC50 ca. 3 µM | [100] |
| Tumor Bearing Mouse Model | Main Outcome | Reference |
|---|---|---|
| Xenografts of lung cancer DMS114 and prostate cancer DU-145 s.c. in nude mice. Dosing: BBGD (100 mg/kg/day) i.p. from day 0 to 8. | 40–50% inhibition of tumor growth. | [93] |
| Adenocarcinoma 15A cells s.c. in mice. Dosing: BBGD (50–200 mg/kg) administered i.p. at day 4 post-implant. Tumor mass recorded at day 7 post-treatment | 50–200 mg/kg BBGD decreased tumor volume by 30–42% | [32] |
| Glioblastoma multiforme (GMB) orthotopic xenograft mouse model U87 glioma cells expressing ZsGreen1-firefly luciferase brain tumor xenograft implants in non-obese diabetic/severe combined immunodeficiency mice. Dosing: BBGD (50 mg/kg), i.p. on days 13 and 15 post-implant | Profound decrease in tumor volume at day 17 post-implant. Total tumor volume: vehicle, 4.6 × 1011 µm3; and BBDG-treated, 1.33 × 102 µm3 (>>99.9% decrease; p < 0.0001) | [96] |
| cf. Treatment with S-(N-p-chlorophenyl-N-hydroxycarbamoyl)glutathione ethyl diester (CHGD) | ||
| C57BL/6 (Es-1e) esterase deficient mice with murine B16 melanoma, human prostate PC3 and human colon HT-29 adenocarcinoma. Dosing: i.v. bolus of 80 or 120 mg/kg CHGD, 5 days for 2 weeks or continuous infusion. | Potency was achieved similar to clinical antitumor drugs: Doxorubicin for B16, cisplatin for PC3 and vincristine for HT-29. | [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabbani, N.; Thornalley, P.J. Emerging Glycation-Based Therapeutics—Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. Int. J. Mol. Sci. 2022, 23, 2453. https://doi.org/10.3390/ijms23052453
Rabbani N, Thornalley PJ. Emerging Glycation-Based Therapeutics—Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. International Journal of Molecular Sciences. 2022; 23(5):2453. https://doi.org/10.3390/ijms23052453
Chicago/Turabian StyleRabbani, Naila, and Paul J. Thornalley. 2022. "Emerging Glycation-Based Therapeutics—Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors" International Journal of Molecular Sciences 23, no. 5: 2453. https://doi.org/10.3390/ijms23052453
APA StyleRabbani, N., & Thornalley, P. J. (2022). Emerging Glycation-Based Therapeutics—Glyoxalase 1 Inducers and Glyoxalase 1 Inhibitors. International Journal of Molecular Sciences, 23(5), 2453. https://doi.org/10.3390/ijms23052453