
Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis

 , ,
, ,  , ,
, ,
Abstract
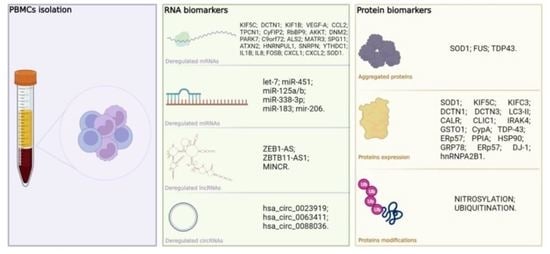
:
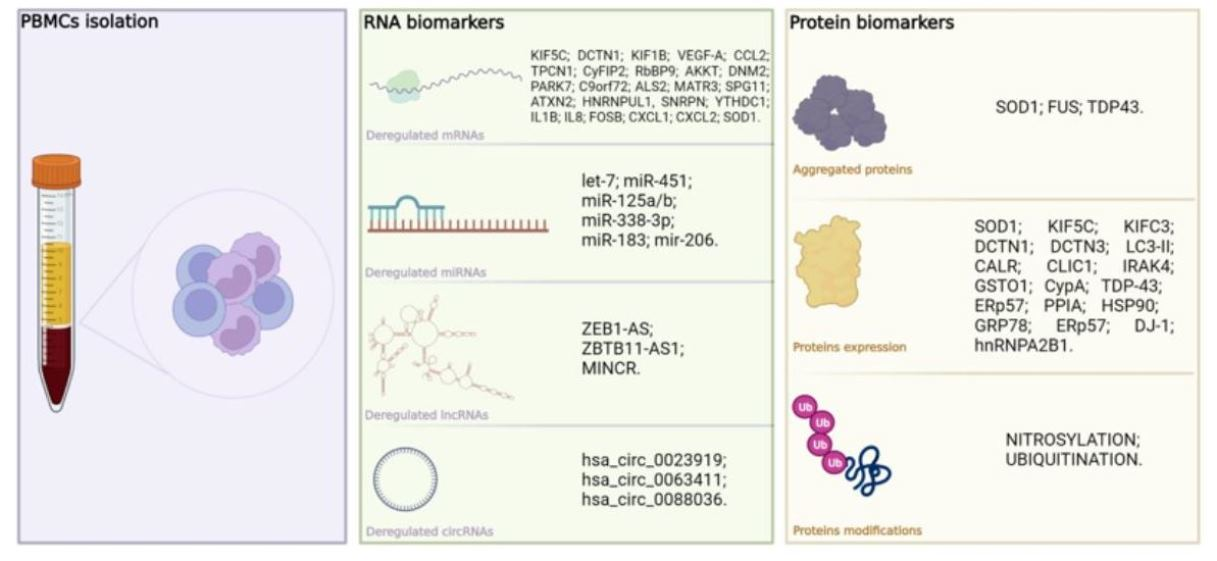{kind=link}
1. Introduction
1.1. Amyotrophic Lateral Sclerosis
1.2. Peripheral Blood Mononuclear Cells Biomarkers
- (i)
- diagnostic biomarkers, used to detect the presence of the disease or to identify subjects with a disease subtype;
- (ii)
- predictive biomarkers, used to identify which treatment a patient or a group of patients will respond to;
- (iii)
- prognostic biomarkers, used to predict disease progression.
1.3. White Cells Subpopulations and ALS
2. Altered RNA Expression in Peripheral Blood of ALS Patients
2.1. RNA Metabolism Involvement in ALS Pathogenesis
2.2. Deregulated mRNA in ALS Patients’ Peripheral Blood
2.3. Deregulated microRNAs in ALS Patients’ Peripheral Blood
2.4. Other Candidate Noncoding RNAs as ALS Biomarker in Peripheral Blood
3. Protein Expression in PBMCs of ALS Patients
Mass Spectometry Analysis in PBMCs of ALS Patients
4. Biomarkers and Clinical Correlation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Erkkinen, M.G.; Kim, M.-O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated Prevalence and Incidence of Amyotrophic Lateral Sclerosis and SOD1 and C9orf72 Genetic Variants. Neuroepidemiology 2021, 55, 342–353. [Google Scholar] [CrossRef]
- Rusina, R.; Vandenberghe, R.; Bruffaerts, R. Cognitive and Behavioral Manifestations in ALS: Beyond Motor System Involvement. Diagnostics 2021, 11, 624. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, M.; Heverin, M.; McLaughlin, R.L.; Hardiman, O. Lifetime Risk and Heritability of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2019, 76, 1367–1374. [Google Scholar] [CrossRef]
- Ingre, C.; Roos, P.M.; Piehl, F.; Kamel, F.; Fang, F. Risk factors for amyotrophic lateral sclerosis. Clin. Epidemiol. 2015, 7, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiman-Patterson, T.D.; Blankenhorn, E.P.; Sher, R.B.; Jiang, J.; Welsh, P.; Dixon, M.C.; Jeffrey, J.I.; Wong, P.; Cox, G.A.; Alexander, G.M. Genetic background effects on disease onset and lifespan of the mutant dynactin p150Glued mouse model of motor neuron disease. PLoS ONE 2015, 10, e0117848. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Cereda, C.; Leoni, E.; Milani, P.; Pansarasa, O.; Mazzini, G.; Guareschi, S.; Alvisi, E.; Ghiroldi, A.; Diamanti, L.; Bernuzzi, S.; et al. Altered Intracellular Localization of SOD1 in Leukocytes from Patients with Sporadic Amyotrophic Lateral Sclerosis. PLoS ONE 2013, 8, e75916. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, M.; Pansarasa, O.; Dell’Orco, M.; Crippa, V.; Gagliardi, S.; Sproviero, D.; Bernuzzi, S.; Diamanti, L.; Ceroni, M.; Tedeschi, G.; et al. Nuclear Phospho-SOD1 Protects DNA from Oxidative Stress Damage in Amyotrophic Lateral Sclerosis. J. Clin. Med. 2019, 8, 729. [Google Scholar] [CrossRef] [Green Version]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, B.M.; Hartmann, H.; Serdaroglu, A.; Schludi, M.H.; Hornburg, D.; Meissner, F.; Orozco, D.; Colombo, A.; Tahirovic, S.; Michaelsen, M.; et al. TDP-43 loss of function inhibits endosomal trafficking and alters trophic signaling in neurons. EMBO J. 2016, 35, 2350–2370. [Google Scholar] [CrossRef]
- Liu, G.; Coyne, A.N.; Pei, F.; Vaughan, S.; Chaung, M.; Zarnescu, D.C.; Buchan, J.R. Endocytosis regulates TDP-43 toxicity and turnover. Nat. Commun. 2017, 8, 2092. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Braun, R.J.; Sommer, C.; Carmona-Gutierrez, D.; Khoury, C.M.; Ring, J.; Büttner, S.; Madeo, F. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem. 2011, 286, 19958–19972. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, T.; Wang, T.; Wang, B. Suppression of lncRNA-ATB prevents amyloid-β-induced neurotoxicity in PC12 cells via regulating miR-200/ZNF217 axis. Biomed. Pharmacother. 2018, 108, 707–715. [Google Scholar] [CrossRef]
- Duan, W.; Li, X.; Shi, J.; Guo, Y.; Li, Z.; Li, C. Mutant TAR DNA-binding protein-43 induces oxidative injury in motor neuron-like cell. Neuroscience 2010, 169, 1621–1629. [Google Scholar] [CrossRef]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef]
- Deng, H.-X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Fogh, I.; Ratti, A.; Gellera, C.; Lin, K.; Tiloca, C.; Moskvina, V.; Corrado, L.; Sorarù, G.; Cereda, C.; Corti, S.; et al. A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Deng, L.; Mei, P.; Zhou, Y.; Wang, B.; Zhang, J.; Lin, J.; Wei, Y.; Zhang, X.; Xu, R. Genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging 2014, 35, 1778.e9–1778.e23. [Google Scholar] [CrossRef]
- Zhang, M.; Xi, Z.; Saez-Atienzar, S.; Chia, R.; Moreno, D.; Sato, C.; Montazer Haghighi, M.; Traynor, B.J.; Zinman, L.; Rogaeva, E. Combined epigenetic/genetic study identified an ALS age of onset modifier. Acta Neuropathol. Commun. 2021, 9, 75. [Google Scholar] [CrossRef]
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Rosado, M.; Silva, R.; G Bexiga, M.; G Jones, J.; Manadas, B.; Anjo, S.I. Advances in biomarker detection: Alternative approaches for blood-based biomarker detection. Adv. Clin. Chem. 2019, 92, 141–199. [Google Scholar] [CrossRef]
- Mosallaei, M.; Ehtesham, N.; Rahimirad, S.; Saghi, M.; Vatandoost, N.; Khosravi, S. PBMCs: A new source of diagnostic and prognostic biomarkers. Arch. Physiol. Biochem. 2020. [Google Scholar] [CrossRef]
- Zondler, L.; Müller, K.; Khalaji, S.; Bliederhäuser, C.; Ruf, W.P.; Grozdanov, V.; Thiemann, M.; Fundel-Clemes, K.; Freischmidt, A.; Holzmann, K.; et al. Peripheral monocytes are functionally altered and invade the CNS in ALS patients. Acta Neuropathol. 2016, 132, 391–411. [Google Scholar] [CrossRef]
- Verber, N.S.; Shepheard, S.R.; Sassani, M.; McDonough, H.E.; Moore, S.A.; Alix, J.J.P.; Wilkinson, I.D.; Jenkins, T.M.; Shaw, P.J. Biomarkers in Motor Neuron Disease: A State of the Art Review. Front. Neurol. 2019, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Can. J. Neurol. Sci. Le J. Can. des Sci. Neurol. 2017, 44, 90–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardo, G.; Pozzi, S.; Pignataro, M.; Lauranzano, E.; Spano, G.; Garbelli, S.; Mantovani, S.; Marinou, K.; Papetti, L.; Monteforte, M.; et al. Amyotrophic lateral sclerosis multiprotein biomarkers in peripheral blood mononuclear cells. PLoS ONE 2011, 6, e25545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luotti, S.; Pasetto, L.; Porcu, L.; Torri, V.; Elezgarai, S.R.; Pantalone, S.; Filareti, M.; Corbo, M.; Lunetta, C.; Mora, G.; et al. Diagnostic and prognostic values of PBMC proteins in amyotrophic lateral sclerosis. Neurobiol. Dis. 2020, 139, 104815. [Google Scholar] [CrossRef] [PubMed]
- Leoni, E.; Bremang, M.; Mitra, V.; Zubiri, I.; Jung, S.; Lu, C.-H.; Adiutori, R.; Lombardi, V.; Russell, C.; Koncarevic, S.; et al. Combined Tissue-Fluid Proteomics to Unravel Phenotypic Variability in Amyotrophic Lateral Sclerosis. Sci. Rep. 2019, 9, 4478. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Feng, W.; Huang, R.; Guo, X.; Chen, Y.; Zheng, Z.; Shang, H. Evidence for peripheral immune activation in amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 347, 90–95. [Google Scholar] [CrossRef]
- Murdock, B.J.; Zhou, T.; Kashlan, S.R.; Little, R.J.; Goutman, S.A.; Feldman, E.L. Correlation of Peripheral Immunity With Rapid Amyotrophic Lateral Sclerosis Progression. JAMA Neurol. 2017, 74, 1446–1454. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Staff, N.P.; Bornschlegl, S.; Butler, G.W.; Maas, M.L.; Kazamel, M.; Zubair, A.; Gastineau, D.A.; Windebank, A.J.; Dietz, A.B. Comprehensive immune profiling reveals substantial immune system alterations in a subset of patients with amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0182002. [Google Scholar] [CrossRef]
- McGill, R.B.; Steyn, F.J.; Ngo, S.T.; Thorpe, K.A.; Heggie, S.; Ruitenberg, M.J.; Henderson, R.D.; McCombe, P.A.; Woodruff, T.M. Monocytes and neutrophils are associated with clinical features in amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa013. [Google Scholar] [CrossRef] [Green Version]
- Rolfes, L.; Schulte-Mecklenbeck, A.; Schreiber, S.; Vielhaber, S.; Herty, M.; Marten, A.; Pfeuffer, S.; Ruck, T.; Wiendl, H.; Gross, C.C.; et al. Amyotrophic lateral sclerosis patients show increased peripheral and intrathecal T-cell activation. Brain Commun. 2021, 3, fcab157. [Google Scholar] [CrossRef]
- Giovannelli, I.; Heath, P.; Shaw, P.J.; Kirby, J. The involvement of regulatory T cells in amyotrophic lateral sclerosis and their therapeutic potential. Amyotroph. Lateral Scler. Frontotemporal Degener. 2020, 21, 435–444. [Google Scholar] [CrossRef]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8(+) T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef] [Green Version]
- Mantas, D.; Kostakis, I.D.; Machairas, N.; Markopoulos, C. White blood cell and platelet indices as prognostic markers in patients with invasive ductal breast carcinoma. Oncol. Lett. 2016, 12, 1610–1614. [Google Scholar] [CrossRef] [Green Version]
- Matić, I.Z.; Kolundžija, B.; Damjanović, A.; Spasić, J.; Radosavljević, D.; Đorđić Crnogorac, M.; Grozdanić, N.; Juranić, Z.D. Peripheral White Blood Cell Subsets in Metastatic Colorectal Cancer Patients Treated with Cetuximab: The Potential Clinical Relevance. Front. Immunol. 2017, 8, 1886. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil Diversity in Health and Disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Gagliardi, S.; Milani, P.; Sardone, V.; Pansarasa, O.; Cereda, C. From Transcriptome to Noncoding RNAs: Implications in ALS Mechanism. Neurol. Res. Int. 2012, 2012, 278725. [Google Scholar] [CrossRef] [Green Version]
- Butti, Z.; Patten, S.A. RNA dysregulation in amyotrophic lateral sclerosis. Front. Genet. 2019, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, N.J.; Zhang, Y.-J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.-F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.P.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Mougeot, J.L.C.; Li, Z.; Price, A.E.; Wright, F.A.; Brooks, B.R. Microarray analysis of peripheral blood lymphocytes from ALS patients and the SAFE detection of the KEGG ALS pathway. BMC Med. Genom. 2011, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Kapeli, K.; Pratt, G.A.; Vu, A.Q.; Hutt, K.R.; Martinez, F.J.; Sundararaman, B.; Batra, R.; Freese, P.; Lambert, N.J.; Huelga, S.C.; et al. Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses. Nat. Commun. 2016, 7, 12143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Jin, H.; Kasarskis, E.J.; Zhu, H. ALS mutations of FUS suppress protein translation and disrupt the regulation of nonsense-mediated decay. Proc. Natl. Acad. Sci. USA 2018, 115, E11904–E11913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wang, Z.; Zhang, J.; Zhao, X.; Xu, P.; Liu, X.; Li, M.; Lv, C.; Song, X. Crosstalk of mRNA, miRNA, lncRNA, and circRNA and Their Regulatory Pattern in Pulmonary Fibrosis. Mol. Ther. Nucleic Acids 2019, 18, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, Z. Efficient backsplicing produces translatable circular mRNAs. RNA 2015, 21, 172–179. [Google Scholar] [CrossRef] [Green Version]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Errichelli, L.; Dini Modigliani, S.; Laneve, P.; Colantoni, A.; Legnini, I.; Capauto, D.; Rosa, A.; De Santis, R.; Scarfò, R.; Peruzzi, G.; et al. FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat. Commun. 2017, 8, 14741. [Google Scholar] [CrossRef]
- Gagliardi, S.; Pandini, C.; Garofalo, M.; Bordoni, M.; Pansarasa, O.; Cereda, C. Long non coding RNAs and ALS: Still much to do. Non-Coding RNA Res. 2018, 3, 226–231. [Google Scholar] [CrossRef]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008, 454, 126–130. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef]
- Moens, T.G.; Mizielinska, S.; Niccoli, T.; Mitchell, J.S.; Thoeng, A.; Ridler, C.E.; Grönke, S.; Esser, J.; Heslegrave, A.; Zetterberg, H.; et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 2018, 135, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, S.; Cova, E.; Davin, A.; Guareschi, S.; Abel, K.; Alvisi, E.; Laforenza, U.; Ghidoni, R.; Cashman, J.R.; Ceroni, M.; et al. SOD1 mRNA expression in sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2010, 39, 198–203. [Google Scholar] [CrossRef]
- Lu, L.; Zheng, L.; Viera, L.; Suswam, E.; Li, Y.; Li, X.; Estévez, A.G.; King, P.H. Mutant Cu/Zn-superoxide dismutase associated with amyotrophic lateral sclerosis destabilizes vascular endothelial growth factor mRNA and downregulates its expression. J. Neurosci. 2007, 27, 7929–7938. [Google Scholar] [CrossRef] [Green Version]
- Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- Aguirre, T.; Van Den Bosch, L.; Goetschalckx, K.; Tilkin, P.; Mathijs, G.; Cassiman, J.J.; Robberecht, W. Increased sensitivity of fibroblasts from amyotrophic lateral sclerosis patients to oxidative stress. Ann. Neurol. 1998, 43, 452–457. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Curti, D.; Malaspina, A.; Facchetti, G.; Camana, C.; Mazzini, L.; Tosca, P.; Zerbi, F.; Ceroni, M. Amyotrophic lateral sclerosis: Oxidative energy metabolism and calcium homeostasis in peripheral blood lymphocytes. Neurology 1996, 47, 1060–1064. [Google Scholar] [CrossRef]
- Ono, S. The skin in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kuźma-Kozakiewicz, M.; Kaźmierczak, B.; Chudy, A.; Gajewska, B.; Barańczyk-Kuźma, A. Alteration of Motor Protein Expression Involved in Bidirectional Transport in Peripheral Blood Mononuclear Cells of Patients with Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2016, 16, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Prabhakar, S.; Abburi, C.; Sharma, N.K.; Anand, A. Vascular endothelial growth factor-A and chemokine ligand (CCL2) genes are upregulated in peripheral blood mononuclear cells in Indian amyotrophic lateral sclerosis patients. J. Neuroinflamm. 2011, 8, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Nachmany, H.; Wald, S.; Abekasis, M.; Bulvik, S.; Weil, M. Two potential biomarkers identified in mesenchymal stem cells and leukocytes of patients with sporadic amyotrophic lateral sclerosis. Dis. Markers 2012, 32, 211–220. [Google Scholar] [CrossRef]
- Vrabec, K.; Boštjančič, E.; Koritnik, B.; Leonardis, L.; Dolenc Grošelj, L.; Zidar, J.; Rogelj, B.; Glavač, D.; Ravnik-Glavač, M. Differential Expression of Several miRNAs and the Host Genes AATK and DNM2 in Leukocytes of Sporadic ALS Patients. Front. Mol. Neurosci. 2018, 11, 106. [Google Scholar] [CrossRef]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.-J.; van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; van Es, M.A.; Blauw, H.M.; van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLoS ONE 2018, 13, e0198874. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Hooten, K.G.; Sieglaff, D.H.; Zhang, A.; Kalyana-Sundaram, S.; Traini, C.M.; Halsey, W.S.; Hughes, A.M.; Sathe, G.M.; et al. Characterization of Gene Expression Phenotype in Amyotrophic Lateral Sclerosis Monocytes. JAMA Neurol. 2017, 74, 677–685. [Google Scholar] [CrossRef]
- Ravnik-Glavač, M.; Glavač, D. Circulating RNAs as Potential Biomarkers in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 1714. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, U.G.; Milla, V.; Cynthia Stafford, M.Y.; Bjourson, A.J.; Duddy, W.; Duguez, S.M.-R. A Systematic Review of Suggested Molecular Strata, Biomarkers and Their Tissue Sources in ALS. Front. Neurol. 2019, 10, 400. [Google Scholar] [CrossRef] [Green Version]
- De Felice, B.; Manfellotto, F.; Fiorentino, G.; Annunziata, A.; Biffali, E.; Pannone, R.; Federico, A. Wide-Ranging Analysis of MicroRNA Profiles in Sporadic Amyotrophic Lateral Sclerosis Using Next-Generation Sequencing. Front. Genet. 2018, 9, 310. [Google Scholar] [CrossRef]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, Q.; Chen, X.; Li, C.; Cao, B.; Ou, R.; Hadano, S.; Shang, H.-F. Aberration of miRNAs Expression in Leukocytes from Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2016, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Zeng, Y.; Qian, Y.; Dong, J.; Zhang, Z.; Zhang, J. MicroRNA let-7c-5p improves neurological outcomes in a murine model of traumatic brain injury by suppressing neuroinflammation and regulating microglial activation. Brain Res. 2018, 1685, 91–104. [Google Scholar] [CrossRef]
- Parisi, C.; Napoli, G.; Amadio, S.; Spalloni, A.; Apolloni, S.; Longone, P.; Volonté, C. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016, 23, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, H. miR-451 elevation relieves inflammatory pain by suppressing microglial activation-evoked inflammatory response via targeting TLR4. Cell Tissue Res. 2018, 374, 487–495. [Google Scholar] [CrossRef]
- Kos, A.; de Mooij-Malsen, A.J.; van Bokhoven, H.; Kaplan, B.B.; Martens, G.J.; Kolk, S.M.; Aschrafi, A. MicroRNA-338 modulates cortical neuronal placement and polarity. RNA Biol. 2017, 14, 905–913. [Google Scholar] [CrossRef]
- Zolboot, N.; Du, J.X.; Zampa, F.; Lippi, G. MicroRNAs Instruct and Maintain Cell Type Diversity in the Nervous System. Front. Mol. Neurosci. 2021, 14, 69. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Dykes-Hoberg, M.; Rothstein, J.D. Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann. Neurol. 2004, 55, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef] [Green Version]
- Di Pietro, L.; Baranzini, M.; Berardinelli, M.G.; Lattanzi, W.; Monforte, M.; Tasca, G.; Conte, A.; Logroscino, G.; Michetti, F.; Ricci, E.; et al. Potential therapeutic targets for ALS: MIR206, MIR208b and MIR499 are modulated during disease progression in the skeletal muscle of patients. Sci. Rep. 2017, 7, 9538. [Google Scholar] [CrossRef] [PubMed]
- Doose, G.; Haake, A.; Bernhart, S.H.; López, C.; Duggimpudi, S.; Wojciech, F.; Bergmann, A.K.; Borkhardt, A.; Burkhardt, B.; Claviez, A.; et al. MINCR is a MYC-induced lncRNA able to modulate MYC’s transcriptional network in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5261–E5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-H.; Yang, Y.; Wu, X.-C.; Zhang, M.-D.; Weng, M.-Z.; Zhou, D.; Wang, J.-D.; Quan, Z.-W. Long non-coding RNA MINCR promotes gallbladder cancer progression through stimulating EZH2 expression. Cancer Lett. 2016, 380, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Zhang, X.; Lu, Y.; Hao, S.; Zhang, Z.; Yang, Y. Expression and Significance of LncRNA-MINCR and CDK2 mRNA in Primary Hepatocellular Carcinoma. Comb. Chem. High Throughput Screen. 2019, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gu, T.; Lu, Z.; Qiu, L.; Xiao, G.; Zhu, X.; Li, F.; Yu, H.; Li, G.; Liu, H. Roles of MYC-targeting long non-coding RNA MINCR in cell cycle regulation and apoptosis in non-small cell lung Cancer. Respir. Res. 2019, 20, 202. [Google Scholar] [CrossRef]
- Lyu, Q.; Jin, L.; Yang, X.; Zhang, F. LncRNA MINCR activates Wnt/β-catenin signals to promote cell proliferation and migration in oral squamous cell carcinoma. Pathol. Res. Pract. 2019, 215, 924–930. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, Y.; Chen, Z. LncRNA MINCR regulates irradiation resistance in nasopharyngeal carcinoma cells via the microRNA-223/ZEB1 axis. Cell Cycle 2020, 19, 53–66. [Google Scholar] [CrossRef]
- Li, Z.; Xie, X.; Fan, X.; Li, X. Long Non-coding RNA MINCR Regulates miR-876-5p/GSPT1 Axis to Aggravate Glioma Progression. Neurochem. Res. 2020, 45, 1690–1699. [Google Scholar] [CrossRef]
- Pandini, C.; Garofalo, M.; Rey, F.; Garau, J.; Zucca, S.; Sproviero, D.; Bordoni, M.; Berzero, G.; Davin, A.; Poloni, T.E.; et al. MINCR: A long non-coding RNA shared between cancer and neurodegeneration. Genomics 2021, 113, 4039–4051. [Google Scholar] [CrossRef]
- Dolinar, A.; Koritnik, B.; Glavač, D.; Ravnik-Glavač, M. Circular RNAs as Potential Blood Biomarkers in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 8052–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.S.; Dokholyan, N. V SOD1 oligomers in amyotrophic lateral sclerosis. Curr. Opin. Struct. Biol. 2021, 66, 225–230. [Google Scholar] [CrossRef]
- Park, H.R.; Yang, E.J. Oxidative Stress as a Therapeutic Target in Amyotrophic Lateral Sclerosis: Opportunities and Limitations. Diagnostics 2021, 11, 1546. [Google Scholar] [CrossRef] [PubMed]
- Cova, E.; Cereda, C.; Galli, A.; Curti, D.; Finotti, C.; Di Poto, C.; Corato, M.; Mazzini, G.; Ceroni, M. Modified expression of Bcl-2 and SOD1 proteins in lymphocytes from sporadic ALS patients. Neurosci. Lett. 2006, 399, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Pandini, C.; Bordoni, M.; Jacchetti, E.; Diamanti, L.; Carelli, S.; Raimondi, M.T.; Sproviero, D.; Crippa, V.; Carra, S.; et al. RNA Molecular Signature Profiling in PBMCs of Sporadic ALS Patients: HSP70 Overexpression Is Associated with Nuclear SOD1. Cells 2022, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- De Marco, G.; Lupino, E.; Calvo, A.; Moglia, C.; Buccinnà, B.; Grifoni, S.; Ramondetti, C.; Lomartire, A.; Rinaudo, M.T.; Piccinini, M.; et al. Cytoplasmic accumulation of TDP-43 in circulating lymphomonocytes of ALS patients with and without TARDBP mutations. Acta Neuropathol. 2011, 121, 611–622. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Liu, W.; Li, X.; Chen, Q.; Liu, T.; Gao, S.; Deng, M. The fused in sarcoma protein forms cytoplasmic aggregates in motor neurons derived from integration-free induced pluripotent stem cells generated from a patient with familial amyotrophic lateral sclerosis carrying the FUS-P525L mutation. Neurogenetics 2015, 16, 223–231. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Drufuca, L.; Diamanti, L.; Sproviero, D.; Trotti, R.; Bernuzzi, S.; La Salvia, S.; Gagliardi, S.; Ceroni, M.; et al. Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis. Model. Mech. 2018, 11, dmm031625. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Ranum, L.P.W. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum. Mol. Genet. 2013, 22, R45–R51. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Dimachkie, M.M.; Agbas, A. Blood-Based Biomarkers for Amyotrophic Lateral Sclerosis; Araki, T., Ed.; Exon Publications: Brisbane, Australia, 2021; ISBN 978-0-6450017-7-8. [Google Scholar]
- Nonaka, T.; Masuda-Suzukake, M.; Hosokawa, M.; Shimozawa, A.; Hirai, S.; Okado, H.; Hasegawa, M. C9ORF72 dipeptide repeat poly-GA inclusions promote intracellular aggregation of phosphorylated TDP-43. Hum. Mol. Genet. 2018, 27, 2658–2670. [Google Scholar] [CrossRef] [Green Version]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.-J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kodavati, M.; Britz, G.W.; Hegde, M.L. DNA Damage and Repair Deficiency in ALS/FTD-Associated Neurodegeneration: From Molecular Mechanisms to Therapeutic Implication. Front. Mol. Neurosci. 2021, 14, 784361. [Google Scholar] [CrossRef]
- Vats, A.; Gourie-Devi, M.; Ahuja, K.; Sharma, A.; Wajid, S.; Ganguly, N.K.; Taneja, V. Expression analysis of protein homeostasis pathways in the peripheral blood mononuclear cells of sporadic amyotrophic lateral sclerosis patients. J. Neurol. Sci. 2018, 387, 85–91. [Google Scholar] [CrossRef]
- Brettschneider, J.; Lehmensiek, V.; Mogel, H.; Pfeifle, M.; Dorst, J.; Hendrich, C.; Ludolph, A.C.; Tumani, H. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS). Neurosci. Lett. 2010, 468, 23–27. [Google Scholar] [CrossRef]
- Ryberg, H.; An, J.; Darko, S.; Lustgarten, J.L.; Jaffa, M.; Gopalakrishnan, V.; Lacomis, D.; Cudkowicz, M.; Bowser, R. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve 2010, 42, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Conraux, L.; Pech, C.; Guerraoui, H.; Loyaux, D.; Ferrara, P.; Guillemot, J.-C.; Meininger, V.; Pradat, P.-F.; Salachas, F.; Bruneteau, G.; et al. Plasma peptide biomarker discovery for amyotrophic lateral sclerosis by MALDI-TOF mass spectrometry profiling. PLoS ONE 2013, 8, e79733. [Google Scholar] [CrossRef]
- Wormser, U.; Mandrioli, J.; Vinceti, M.; Fini, N.; Sintov, A.; Brodsky, B.; Proskura, E.; Finkelstein, Y. Reduced levels of alpha-1-antitrypsin in cerebrospinal fluid of amyotrophic lateral sclerosis patients: A novel approach for a potential treatment. J. Neuroinflamm. 2016, 13, 131. [Google Scholar] [CrossRef] [Green Version]
- Končarević, S.; Lößner, C.; Kuhn, K.; Prinz, T.; Pike, I.; Zucht, H.-D. In-depth profiling of the peripheral blood mononuclear cells proteome for clinical blood proteomics. Int. J. Proteom. 2014, 2014, 129259. [Google Scholar] [CrossRef] [PubMed]
- Nardo, G.; Pozzi, S.; Mantovani, S.; Garbelli, S.; Marinou, K.; Basso, M.; Mora, G.; Bendotti, C.; Bonetto, V. Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxid. Redox Signal. 2009, 11, 1559–1567. [Google Scholar] [CrossRef]
- Filareti, M.; Luotti, S.; Pasetto, L.; Pignataro, M.; Paolella, K.; Messina, P.; Pupillo, E.; Filosto, M.; Lunetta, C.; Mandrioli, J.; et al. Decreased Levels of Foldase and Chaperone Proteins Are Associated with an Early-Onset Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, A.; Puentes, F.; Amor, S. Disease origin and progression in amyotrophic lateral sclerosis: An immunology perspective. Int. Immunol. 2015, 27, 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roser, A.-E.; Tönges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef] [PubMed]
- Giraudon, P.; Vincent, P.; Vuaillat, C. T-cells in neuronal injury and repair: Semaphorins and related T-cell signals. Neuromol. Med. 2005, 7, 207–216. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Thonhoff, J.R.; Thome, A.D.; Faridar, A.; Wang, J.; Wen, S.; Ornelas, L.; Sareen, D.; Goodridge, H.S.; et al. Immunosuppressive Functions of M2 Macrophages Derived from iPSCs of Patients with ALS and Healthy Controls. iScience 2020, 23, 101192. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, W.; Thonhoff, J.R.; Wang, J.; Wen, S.; Appel, S.H. Increased activation ability of monocytes from ALS patients. Exp. Neurol. 2020, 328, 113259. [Google Scholar] [CrossRef]
- Swindell, W.R.; Kruse, C.P.S.; List, E.O.; Berryman, D.E.; Kopchick, J.J. ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J. Transl. Med. 2019, 17, 170. [Google Scholar] [CrossRef]
- Arosio, B.; D’Addario, C.; Gussago, C.; Casati, M.; Tedone, E.; Ferri, E.; Nicolini, P.; Rossi, P.D.; Maccarrone, M.; Mari, D. Peripheral blood mononuclear cells as a laboratory to study dementia in the elderly. Biomed Res. Int. 2014, 2014, 169203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pansarasa, O.; Garofalo, M.; Scarian, E.; Dragoni, F.; Garau, J.; Di Gerlando, R.; Diamanti, L.; Bordoni, M.; Gagliardi, S. Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 23, 2580. https://doi.org/10.3390/ijms23052580
Pansarasa O, Garofalo M, Scarian E, Dragoni F, Garau J, Di Gerlando R, Diamanti L, Bordoni M, Gagliardi S. Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2022; 23(5):2580. https://doi.org/10.3390/ijms23052580
Chicago/Turabian StylePansarasa, Orietta, Maria Garofalo, Eveljn Scarian, Francesca Dragoni, Jessica Garau, Rosalinda Di Gerlando, Luca Diamanti, Matteo Bordoni, and Stella Gagliardi. 2022. "Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 23, no. 5: 2580. https://doi.org/10.3390/ijms23052580
APA StylePansarasa, O., Garofalo, M., Scarian, E., Dragoni, F., Garau, J., Di Gerlando, R., Diamanti, L., Bordoni, M., & Gagliardi, S. (2022). Biomarkers in Human Peripheral Blood Mononuclear Cells: The State of the Art in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 23(5), 2580. https://doi.org/10.3390/ijms23052580