
Transcriptome Profiling and Functional Validation of RING-Type E3 Ligases in Halophyte Sesuvium verrucosum under Salinity Stress

 , and
, and
Abstract
:1. Introduction
2. Results
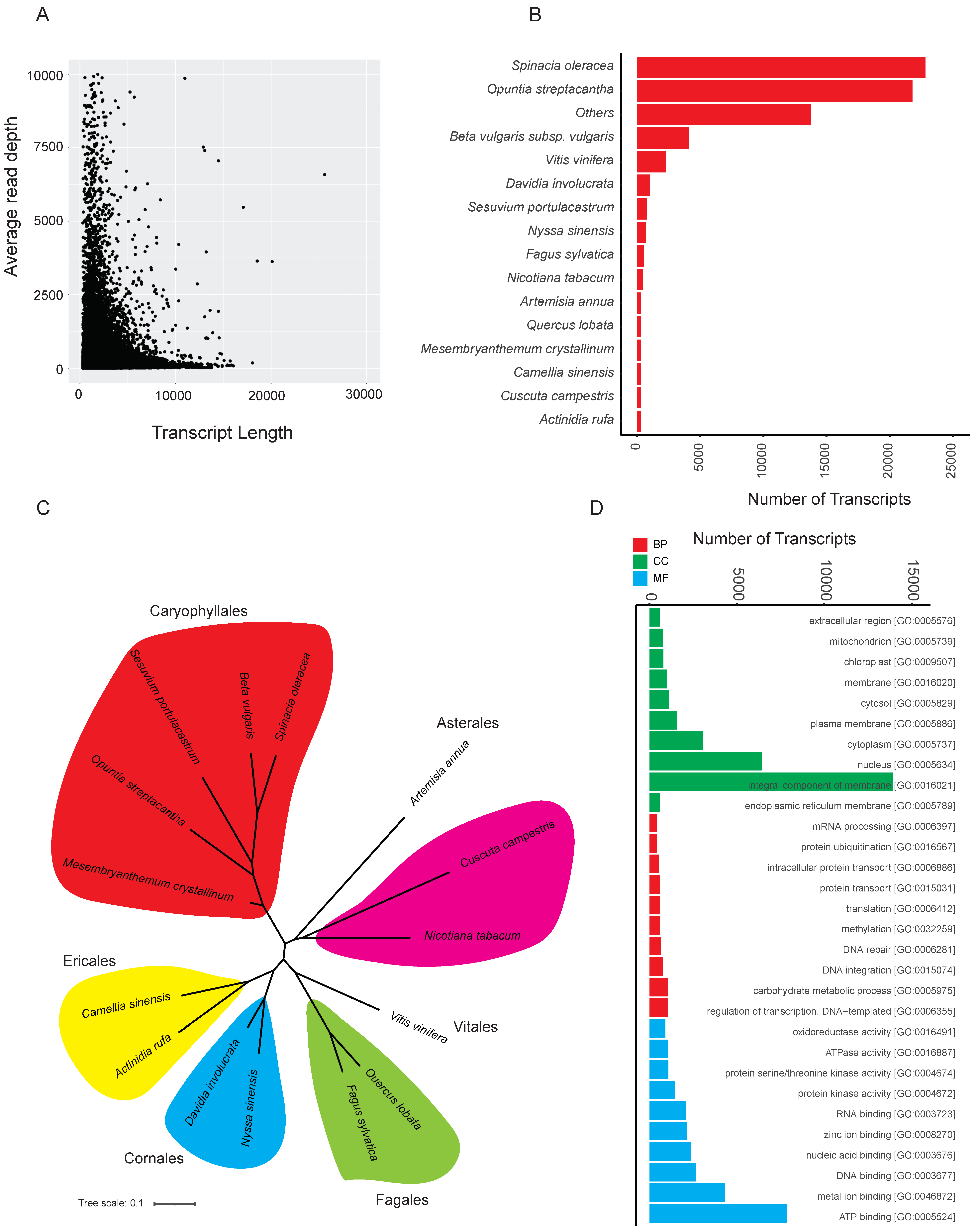
2.1. RNA-Seq Quality Check, De Novo Assembly, and Annotation
2.2. Positive Selection and Ortholog-Based Analysis
2.3. Classification and Comparison of E3 Ligase Abundance
2.4. Differential Gene Expression Analysis
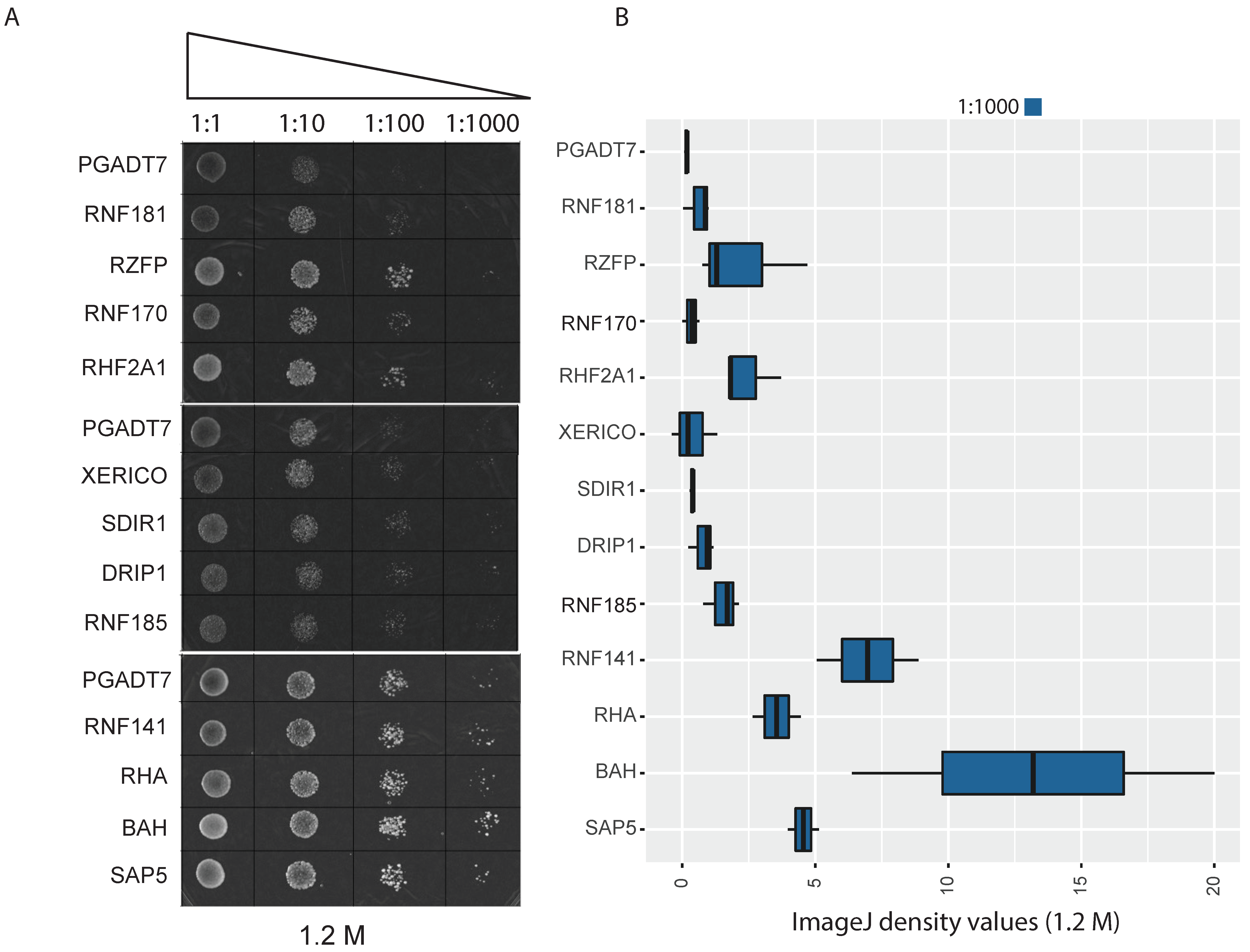
2.5. Yeast Functional Analysis of E3 Single-RING E3 Ligases
3. Discussion
4. Materials and Methods
4.1. Sample Preparation, RNA Isolation, and Sequencing
4.2. Transcriptome Analysis
4.3. Differential Expression, Functional Annotation, and Characterization of E3 Ligases
4.4. Positive Selection and Ortholog-Based Analysis
4.5. RT-qPCR Validation
4.6. Functional Analysis in Yeast
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahmad, P.; Prasad, M.N.V. Environmental Adaptations and Stress Tolerance of Plants in the Era of Climate Change; Springer Science & Business Media: New York, NY, USA, 2011. [Google Scholar]
- Sadanandom, A.; Bailey, M.; Ewan, R.; Lee, J.; Nelis, S. The ubiquitin–proteasome system: Central modifier of plant signalling. New Phytol. 2012, 196, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.V.; Oliveira, M.M.; Saibo, N.J.; Lourenço, T.F. Modulation of Abiotic Stress Responses in Rice by E3-Ubiquitin Ligases: A Promising Way to Develop Stress-Tolerant Crops. Front. Plant Sci. 2021, 12, 368. [Google Scholar] [CrossRef] [PubMed]
- Vierstra, R.D. The ubiquitin–26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 2009, 10, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [Green Version]
- Romero-Barrios, N.; Vert, G. Proteasome-independent functions of lysine-63 polyubiquitination in plants. New Phytol. 2018, 217, 995–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Yang, M.; Wang, P.; Zhao, Y.; Ma, C. Interplay between the ubiquitin proteasome system and ubiquitin-mediated autophagy in plants. Cells 2020, 9, 2219. [Google Scholar] [CrossRef]
- Vierstra, R.D. The expanding universe of ubiquitin and ubiquitin-like modifiers. Plant Physiol. 2012, 160, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Dametto, A.; Buffon, G.; dos Reis Blasi, É.A.; Sperotto, R.A. Ubiquitination pathway as a target to develop abiotic stress tolerance in rice. Plant Signal. Behav. 2015, 10, e1057369. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.Q.; Xue, H.W. The ubiquitin-proteasome system in plant responses to environments. Plant Cell Environ. 2019, 42, 2931–2944. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, C.; Li, Y.; Zheng, N.; Chen, H.; Zhao, Q.; Gao, T.; Guo, H.; Xie, Q. SDIR1 is a RING finger E3 ligase that positively regulates stress-responsive abscisic acid signaling in Arabidopsis. Plant Cell 2007, 19, 1912–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Cui, F.; Wu, Y.; Lou, L.; Liu, L.; Tian, M.; Ning, Y.; Shu, K.; Tang, S.; Xie, Q. The RING finger ubiquitin E3 ligase SDIR1 targets SDIR1-INTERACTING PROTEIN1 for degradation to modulate the salt stress response and ABA signaling in Arabidopsis. Plant Cell 2015, 27, 214–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Wu, Y.; Zhang, Y.; Liu, L.; Ning, Y.; Wang, D.; Tong, H.; Chen, S.; Chu, C.; Xie, Q. OsSDIR1 overexpression greatly improves drought tolerance in transgenic rice. Plant Mol. Biol. 2011, 76, 145–156. [Google Scholar] [CrossRef] [PubMed]
- El-Awady, M.A.; Hassan, M.M.; Al-Sodany, Y.M. Isolation and characterization of salt tolerant endophytic and rhizospheric plant growth-promoting bacteria (PGPB) associated with the halophyte plant (Sesuvium verrucosum) grown in KSA. Int. J. Appl. Sci. Biotechnol. 2015, 3, 552–560. [Google Scholar] [CrossRef]
- Lokhande, V.H.; Gor, B.K.; Desai, N.S.; Nikam, T.D.; Suprasanna, P. Sesuvium portulacastrum, a plant for drought, salt stress, sand fixation, food and phytoremediation. A review. Agron. Sustain. Dev. 2013, 33, 329–348. [Google Scholar] [CrossRef] [Green Version]
- Lastiri-Hernández, M.A.; Álvarez-Bernal, D.; Ochoa-Estrada, S.; Contreras-Ramos, S.M. Potential of Bacopa monnieri (L.) Wettst and Sesuvium verrucosum Raf. as an agronomic management alternative to recover the productivity of saline soils. Int. J. Phytoremediation 2020, 22, 343–352. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Fang, H.; Gough, J. DcGO: Database of domain-centric ontologies on functions, phenotypes, diseases and more. Nucleic Acids Res. 2013, 41, D536–D544. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.-M.; Tan, Y.; Chu, H.-J.; Sun, M.-X.; Xing, J.-C. Transcriptome sequencing revealed molecular mechanisms underlying tolerance of Suaeda salsa to saline stress. PLoS ONE 2019, 14, e0219979. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Cogill, S.; Qiu, Y.; Li, Z.; Zhou, M.; Hu, Q.; Chang, Z.; Noorai, R.E.; Xia, X.; Saski, C. Comparative transcriptome profiling provides insights into plant salt tolerance in seashore paspalum (Paspalum vaginatum). BMC Genom. 2020, 21, 131. [Google Scholar] [CrossRef] [PubMed]
- Shu, K.; Yang, W. E3 ubiquitin ligases: Ubiquitous actors in plant development and abiotic stress responses. Plant Cell Physiol. 2017, 58, 1461–1476. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Li, L.; Su, Z. plantsUPS: A database of plants’ Ubiquitin Proteasome System. BMC Genom. 2009, 10, 227. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.L.; Hauksdóttir, H.; Troy, A.; Herschleb, J.; Kraft, E.; Callis, J. Functional Analysis of the RING-Type Ubiquitin Ligase Family of Arabidopsis. Plant Physiol. 2005, 137, 13–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Mogami, J.; Yamaguchi-Shinozaki, K. ABA-dependent and ABA-independent signaling in response to osmotic stress in plants. Curr. Opin. Plant Biol. 2014, 21, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Qin, F.; Sakuma, Y.; Tran, L.-S.P.; Maruyama, K.; Kidokoro, S.; Fujita, Y.; Fujita, M.; Umezawa, T.; Sawano, Y.; Miyazono, K.-I. Arabidopsis DREB2A-interacting proteins function as RING E3 ligases and negatively regulate plant drought stress–responsive gene expression. Plant Cell 2008, 20, 1693–1707. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Fokar, M.; Abdelmageed, H.; Allen, R.D. Arabidopsis SAP5 functions as a positive regulator of stress responses and exhibits E3 ubiquitin ligase activity. Plant Mol. Biol. 2011, 75, 451–466. [Google Scholar] [CrossRef]
- Zhang, N.; Yin, Y.; Liu, X.; Tong, S.; Xing, J.; Zhang, Y.; Pudake, R.N.; Izquierdo, E.M.; Peng, H.; Xin, M. The E3 ligase TaSAP5 alters drought stress responses by promoting the degradation of DRIP proteins. Plant Physiol. 2017, 175, 1878–1892. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.H.; Yang, S.H.; Han, K.H. Upregulation of an Arabidopsis RING-H2 gene, XERICO, confers drought tolerance through increased abscisic acid biosynthesis. Plant J. 2006, 47, 343–355. [Google Scholar] [CrossRef]
- Zeng, D.-E.; Hou, P.; Xiao, F.; Liu, Y. Overexpression of Arabidopsis XERICO gene confers enhanced drought and salt stress tolerance in rice (Oryza sativa L.). J. Plant Biochem. Biotechnol. 2015, 24, 56–64. [Google Scholar] [CrossRef]
- Brugière, N.; Zhang, W.; Xu, Q.; Scolaro, E.J.; Lu, C.; Kahsay, R.Y.; Kise, R.; Trecker, L.; Williams, R.W.; Hakimi, S. Overexpression of RING domain E3 ligase ZmXerico1 confers drought tolerance through regulation of ABA homeostasis. Plant Physiol. 2017, 175, 1350–1369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Hao, Z.; Ning, Y.; Wang, G.-L. SINA E3 ubiquitin ligases: Versatile moderators of plant growth and stress response. Mol. Plant 2019, 12, 610–612. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Jantasuriyarat, C.; Zhao, Q.; Zhang, H.; Chen, S.; Liu, J.; Liu, L.; Tang, S.; Park, C.H.; Wang, X. The SINA E3 ligase OsDIS1 negatively regulates drought response in rice. Plant Physiol. 2011, 157, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Bao, Y.; Wang, C.; Jiang, C.; Pan, J.; Zhang, G.; Liu, H.; Zhang, H. The tumor necrosis factor receptor-associated factor (TRAF)-like family protein SEVEN IN ABSENTIA 2 (SINA2) promotes drought tolerance in an ABA-dependent manner in A rabidopsis. New Phytol. 2014, 202, 174–187. [Google Scholar] [CrossRef]
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Report. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.researchgate.net/publication/281238574_FastQC_A_Quality_Control_tool_for_High_Throughput_Sequence_Data (accessed on 15 May 2021).
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Springer: Berlin/Heidelberg, Germany, 2019; pp. 227–245. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 121. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.J.; Brewer, M.S. FUSTr: A tool to find gene Families Under Selection in Transcriptomes. PeerJ 2018, 6, e4234. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Miele, V.; Penel, S.; Duret, L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinform. 2011, 12, 116. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tran, K.; Green, E.M. Assessing yeast cell survival following hydrogen peroxide exposure. Bio-Protocol 2019, 9, e3149. [Google Scholar] [CrossRef]
- Petropavlovskiy, A.A.; Tauro, M.G.; Lajoie, P.; Duennwald, M.L. A quantitative imaging-based protocol for yeast growth and survival on agar plates. STAR Protoc. 2020, 1, 100182. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Assembly | After Clustering | * Final Transcriptome | |
|---|---|---|---|
| Total transcripts | 301,627 | 195,255 | 131,454 |
| Total bases | 424,665,332 | 258,421,237 | 207,568,729 |
| Read alignment % | 98 | 95 | 92 |
| BUSCO validation | ~99.5% (96% complete) | ~99.5% (96% complete) | ~99.3% (95.8% complete) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thayale Purayil, F.; Sudalaimuthuasari, N.; Li, L.; Aljneibi, R.; Al Shamsi, A.M.K.; David, N.; Kottackal, M.; AlZaabi, M.; Balan, J.; Kurup, S.S.; et al. Transcriptome Profiling and Functional Validation of RING-Type E3 Ligases in Halophyte Sesuvium verrucosum under Salinity Stress. Int. J. Mol. Sci. 2022, 23, 2821. https://doi.org/10.3390/ijms23052821
Thayale Purayil F, Sudalaimuthuasari N, Li L, Aljneibi R, Al Shamsi AMK, David N, Kottackal M, AlZaabi M, Balan J, Kurup SS, et al. Transcriptome Profiling and Functional Validation of RING-Type E3 Ligases in Halophyte Sesuvium verrucosum under Salinity Stress. International Journal of Molecular Sciences. 2022; 23(5):2821. https://doi.org/10.3390/ijms23052821
Chicago/Turabian StyleThayale Purayil, Fayas, Naganeeswaran Sudalaimuthuasari, Ling Li, Ruwan Aljneibi, Aysha Mohammed Khamis Al Shamsi, Nelson David, Martin Kottackal, Mariam AlZaabi, Jithin Balan, Shyam S. Kurup, and et al. 2022. "Transcriptome Profiling and Functional Validation of RING-Type E3 Ligases in Halophyte Sesuvium verrucosum under Salinity Stress" International Journal of Molecular Sciences 23, no. 5: 2821. https://doi.org/10.3390/ijms23052821