
Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases


Abstract
:1. Introduction
2. Regulation of Nrf2
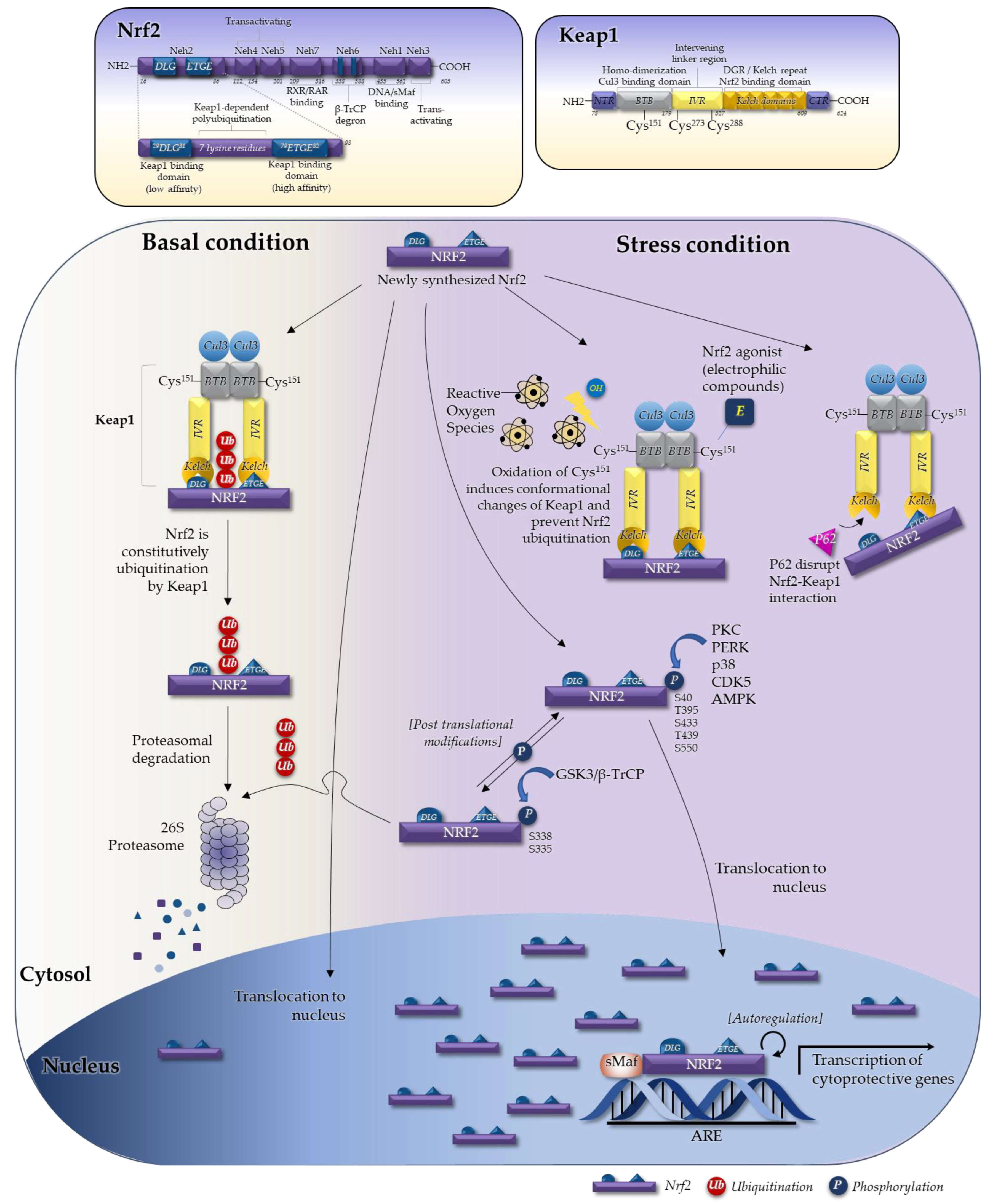
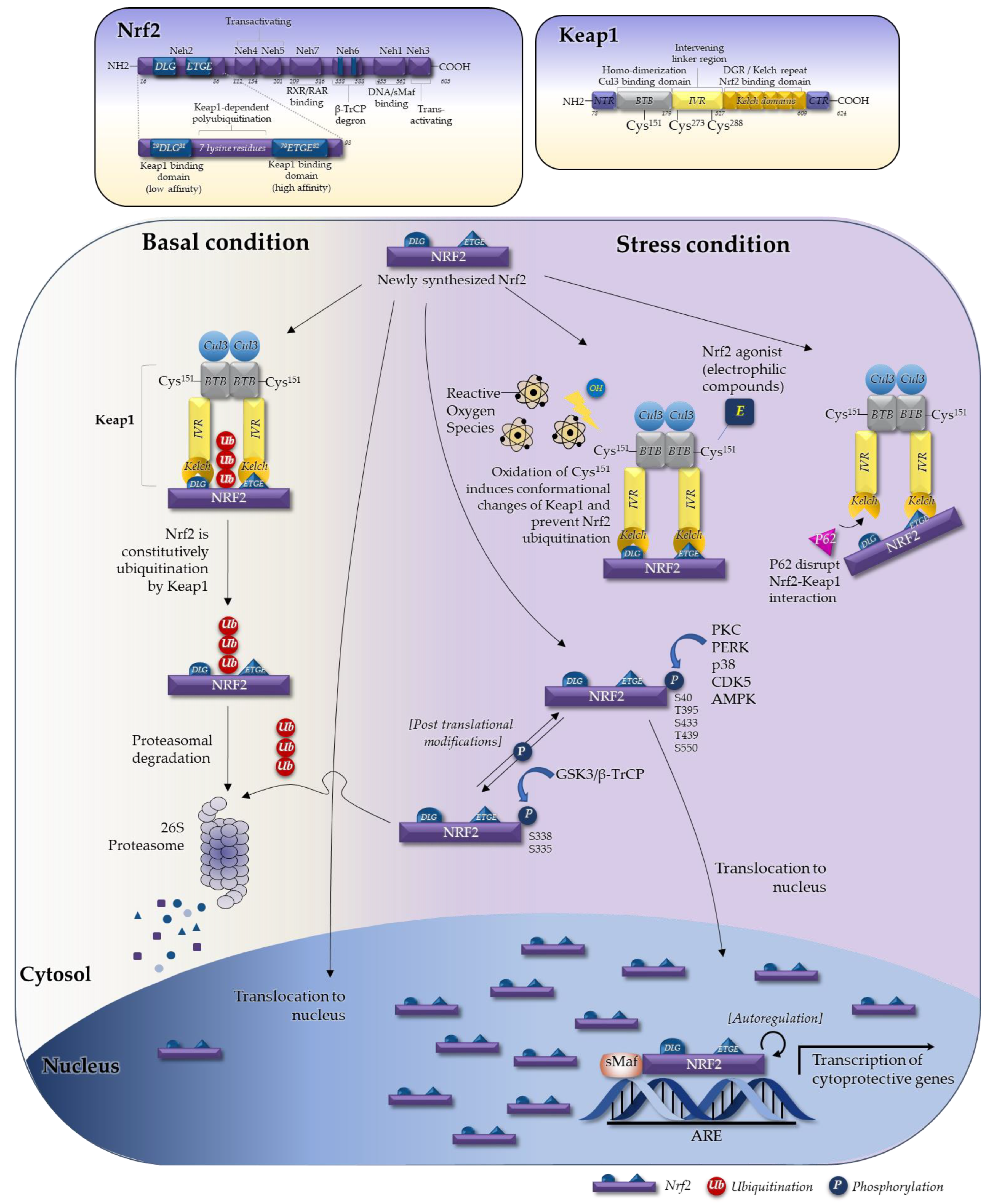
2.1. The Seven Domains of Nrf2 and Their Functions
2.2. The Keap1–Nrf2 System in Regulating Nrf2
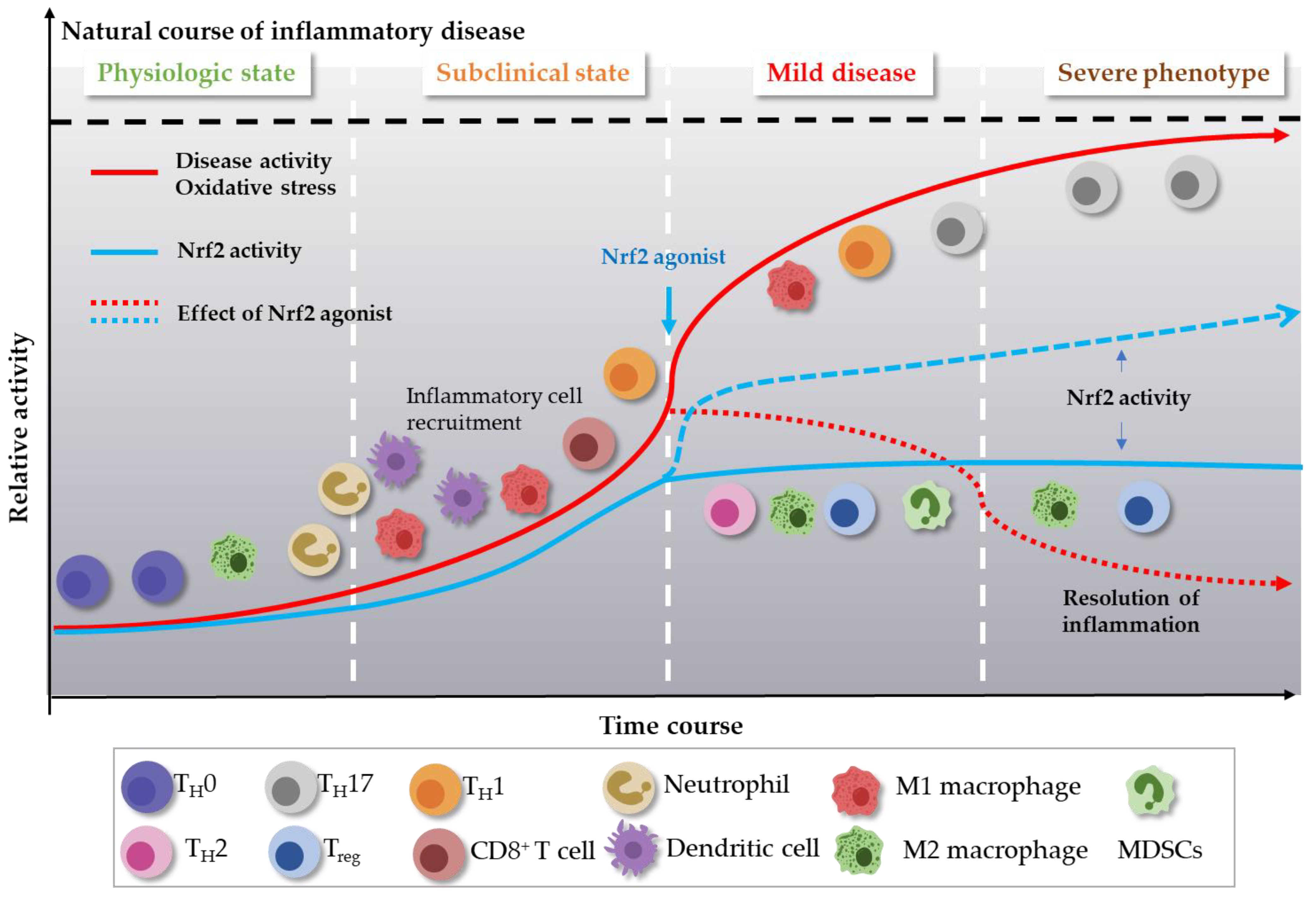
3. Role of Nrf2 Agonism in Chronic Inflammatory Diseases
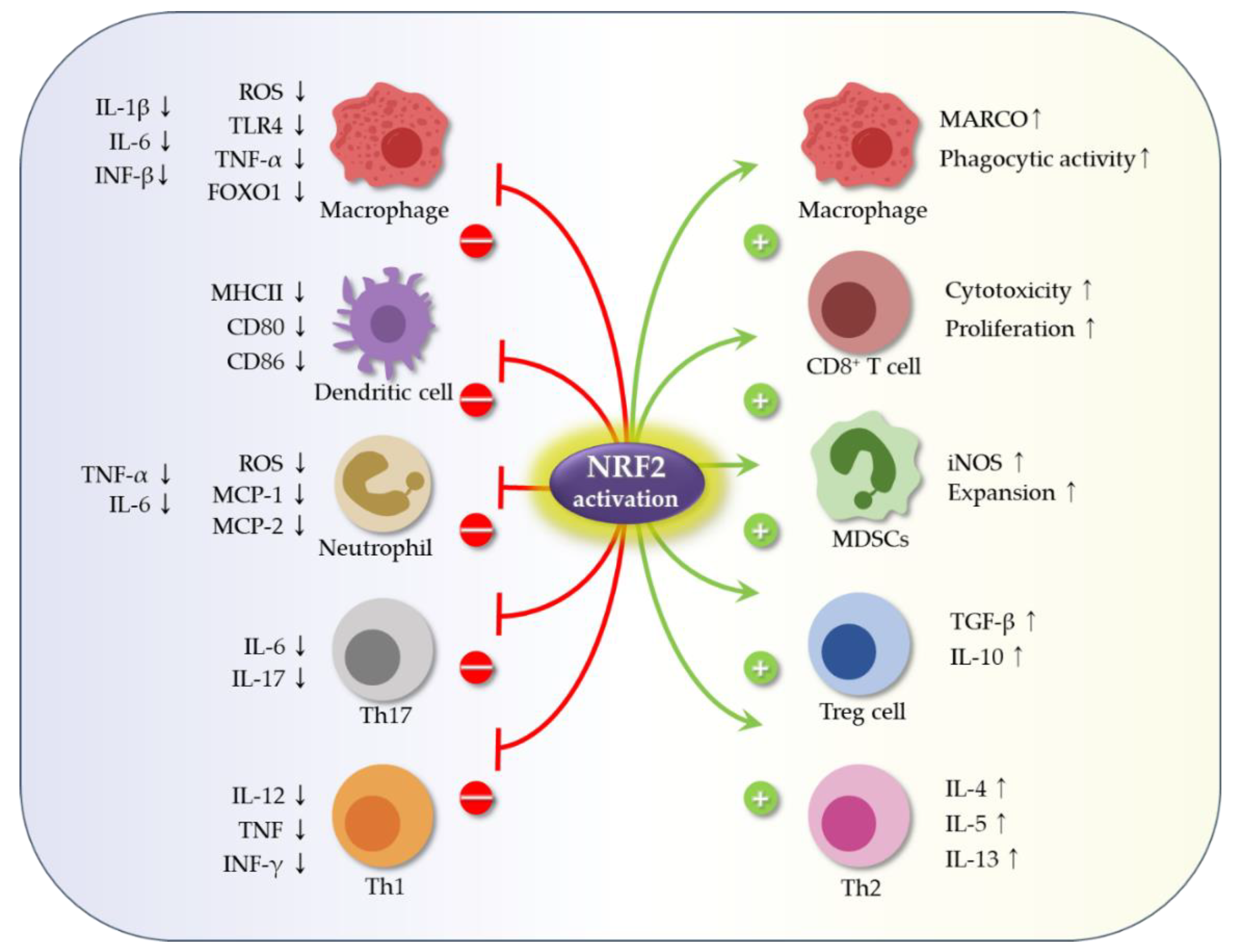
3.1. Role Nrf2 in Immune Cells and in Production of Inflammatory Mediators
3.1.1. The Role of Nrf2 in Immune Cell Function
3.1.2. The Inhibitory Role of Nrf2 in Production of Inflammatory Mediators (Cytokines and Proteases)
3.2. Obesity and Metabolic Syndrome
3.3. Inflammatory and Autoimmune Diseases
3.3.1. Inflammatory Bowel Disease
3.3.2. Systemic Lupus Erythematous
3.3.3. Rheumatoid Arthritis (RA)
4. Nrf2 Agonism in Diabetes and Its Complications
4.1. Diabetes
4.2. Diabetic Complications
5. Recent Development of Nrf2-Related Drugs and Performance in Clinical Trials
5.1. Nrf2 Pharmacological Activators under Clinical Trial
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abed, D.A.; Goldstein, M.; Albanyan, H.; Jin, H.; Hu, L. Discovery of direct inhibitors of Keap1–Nrf2 protein–protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating nrf2–keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayalan Naidu, S.; Dinkova-Kostova, A.T. KEAP1, a cysteine-based sensor and a drug target for the prevention and treatment of chronic disease. Open Biol. 2020, 10, 200105. [Google Scholar] [CrossRef]
- Uddin, M.J.; Kim, E.H.; Hannan, M.A.; Ha, H. Pharmacotherapy against Oxidative Stress in Chronic Kidney Disease: Promising Small Molecule Natural Products Targeting Nrf2-HO-1 Signaling. Antioxidants 2021, 10, 258. [Google Scholar] [CrossRef]
- Ucar, B.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- Galicia-Moreno, M.; Lucano-Landeros, S.; Monroy-Ramirez, H.C.; Silva-Gomez, J.; Gutierrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Roles of Nrf2 in Liver Diseases: Molecular, Pharmacological, and Epigenetic Aspects. Antioxidants 2020, 9, 980. [Google Scholar] [CrossRef]
- Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. Pharmacological Applications of Nrf2 Inhibitors as Potential Antineoplastic Drugs. Int. J. Mol. Sci. 2019, 20, 2025. [Google Scholar] [CrossRef] [Green Version]
- Sykiotis, G.P.; Bohmann, D. Stress-Activated Cap’n’collar Transcription Factors in Aging and Human Disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar]
- Hohmann, M.S.; Zaninelli, T.H.; Staurengo-Ferrari, L.; Manchope, M.F.; Badaro-Garcia, S.; De Freitas, A.; Casagrande, R.; Verri, W.A. Nrf2 in Immune Responses During Inflammation. In The Role of Toll-Like Receptor 4 in Infectious and Non Infectious Inflammation; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2020; pp. 23–49. [Google Scholar]
- Canning, P.; Sorrell, F.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, H.; Katsuoka, F.; Engel, J.D.; Yamamoto, M. Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1-Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 6379–6384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, M.B.; Solovieva, V.; Blank, V. The small MAF transcription factors MAFF, MAFG and MAFK: Current knowledge and perspectives. Biochim. Biophys. Acta 2012, 1823, 1841–1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohashi, H.; Yamamoto, M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Zhang, D.D.; Lo, S.-C.; Cross, J.V.; Templeton, D.J.; Hannink, M. Keap1 Is a Redox-Regulated Substrate Adaptor Protein for a Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol. 2004, 24, 10941–10953. [Google Scholar] [CrossRef] [Green Version]
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The Carboxy-Terminal Neh3 Domain of Nrf2 Is Required for Transcriptional Activation. Mol. Cell. Biol. 2005, 25, 10895–10906. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated Turnover of Nrf2 Is Determined by at Least Two Separate Protein Domains, the Redox-sensitive Neh2 Degron and the Redox-insensitive Neh6 Degron. J. Biol. Chem. 2004, 279, 31556–31567. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108. [Google Scholar] [CrossRef] [Green Version]
- Katoh, Y.; Iida, K.; Kang, M.-I.; Kobayashi, A.; Mizukami, M.; Tong, K.I.; McMahon, M.; Hayes, J.; Itoh, K.; Yamamoto, M. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch. Biochem. Biophys. 2005, 433, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Lv, Y.-F.; Zhao, J.-L.; You, Q.-D.; Jiang, Z.-Y. Regulation of Nrf2 by phosphorylation: Consequences for biological function and therapeutic implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Keum, Y.S. Regulation of the Keap1/Nrf2 system by chemopreventive sulforaphane: Implications of posttranslational modifications. Ann. N. Y. Acad Sci. 2011, 1229, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Chin, Y.E.; Zhang, D.D. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol. Cell Biol. 2009, 29, 2658–2672. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Park, H.; Park, H.J.; Choi, K.-H.; Sadikot, R.T.; Cha, J.; Joo, M. Glycosylation enables aesculin to activate Nrf2. Sci. Rep. 2016, 6, 29956. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Yu, S.; Zhang, C.; Kong, A.-N.T. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic. Biol. Med. 2015, 88, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Wu, R.; Guo, Y.; Kong, A.-N.T. Regulation of Keap1–Nrf2 signaling: The role of epigenetics. Curr. Opin. Toxicol. 2016, 1, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Ashrafizadeh, M.; Ahmadi, Z.; Samarghandian, S.; Mohammadinejad, R.; Yaribeygi, H.; Sathyapalan, T.; Sahebkar, A. MicroRNA-mediated regulation of Nrf2 signaling pathway: Implications in disease therapy and protection against oxidative stress. Life Sci. 2020, 244, 117329. [Google Scholar] [CrossRef]
- Quiles, J.M.; Pepin, M.E.; Sunny, S.; Shelar, S.B.; Challa, A.K.; Dalley, B.; Hoidal, J.R.; Pogwizd, S.M.; Wende, A.R.; Rajasekaran, N.S. Identification of Nrf2-responsive microRNA networks as putative mediators of myocardial reductive stress. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Papp, D.; Lenti, K.; Modos, D.; Fazekas, D.; Dúl, Z.; Turei, D.; Földvári-Nagy, L.; Nussinov, R.; Csermely, P.; Korcsmáros, T. The NRF2-related interactome and regulome contain multifunctional proteins and fine-tuned autoregulatory loops. FEBS Lett. 2012, 586, 1795–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, J.W.; Jaiswal, A.K. An Autoregulatory Loop between Nrf2 and Cul3-Rbx1 Controls Their Cellular Abundance. J. Biol. Chem. 2010, 285, 21349–21358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Katoh, Y.; Kusunoki, H.; Itoh, K.; Tanaka, T.; Yamamoto, M. Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol. 2006, 26, 2887–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.I.; Padmanabhan, B.; Kobayashi, A.; Shang, C.; Hirotsu, Y.; Yokoyama, S.; Yamamoto, M. Different Electrostatic Potentials Define ETGE and DLG Motifs as Hinge and Latch in Oxidative Stress Response. Mol. Cell. Biol. 2007, 27, 7511–7521. [Google Scholar] [CrossRef] [Green Version]
- Madden, S.; Itzhaki, L.S. Structural and mechanistic insights into the Keap1-Nrf2 system as a route to drug discovery. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2020, 1868, 140405. [Google Scholar] [CrossRef]
- Fukutomi, T.; Takagi, K.; Mizushima, T.; Ohuchi, N.; Yamamoto, M. Kinetic, Thermodynamic, and Structural Characterizations of the Association between Nrf2-DLGex Degron and Keap1. Mol. Cell. Biol. 2014, 34, 832–846. [Google Scholar] [CrossRef] [Green Version]
- Horie, Y.; Suzuki, T.; Inoue, J.; Iso, T.; Wells, G.; Moore, T.W.; Mizushima, T.; Dinkova-Kostova, A.T.; Kasai, T.; Kamei, T.; et al. Molecular basis for the disruption of Keap1–Nrf2 interaction via Hinge & Latch mechanism. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Kobayashi, M.; Itoh, K.; Suzuki, T.; Osanai, H.; Nishikawa, K.; Katoh, Y.; Takagi, Y.; Yamamoto, M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells 2002, 7, 807–820. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Kopacz, A.; Kloska, D.; Forman, H.J.; Jozkowicz, A.; Grochot-Przeczek, A. Beyond repression of Nrf2: An update on Keap1. Free Radic. Biol. Med. 2020, 157, 63–74. [Google Scholar] [CrossRef]
- Katsuoka, F.; Motohashi, H.; Engel, J.D.; Yamamoto, M. Nrf2 Transcriptionally Activates the mafG Gene through an Antioxidant Response Element. J. Biol. Chem. 2005, 280, 4483–4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuki, A.; Yamamoto, M. Cis-element architecture of Nrf2–sMaf heterodimer binding sites and its relation to diseases. Arch. Pharmacal Res. 2019, 43, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Yamamoto, M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Curt, M.J.C.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Vasileva, L.V.; Savova, M.S.; Amirova, K.M.; Dinkova-Kostova, A.T.; Georgiev, M.I. Obesity and NRF2-mediated cytoprotection: Where is the missing link? Pharmacol. Res. 2020, 156, 104760. [Google Scholar] [CrossRef]
- Wang, Z.; Zuo, Z.; Li, L.; Ren, S.; Gao, T.; Fu, J.; Hou, Y.; Chen, Y.; Pi, J. Nrf2 in adipocytes. Arch. Pharmacal Res. 2020, 43, 350–360. [Google Scholar] [CrossRef]
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107. [Google Scholar] [CrossRef]
- Yuan, H.; Xu, Y.; Luo, Y.; Wang, N.-X.; Xiao, J.-H. Role of Nrf2 in cell senescence regulation. Mol. Cell. Biochem. 2021, 476, 247–259. [Google Scholar] [CrossRef]
- Panieri, E.; Telkoparan-Akillilar, P.; Suzen, S.; Saso, L. The NRF2/KEAP1 Axis in the Regulation of Tumor Metabolism: Mechanisms and Therapeutic Perspectives. Biomolecules 2020, 10, 791. [Google Scholar] [CrossRef]
- Li, S.; Eguchi, N.; Lau, H.; Ichii, H. The Role of the Nrf2 Signaling in Obesity and Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6973. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; de Galarreta, C.M.R.; Cuadrado, A. Regulation of Heme Oxygenase-1 Expression through the Phosphatidylinositol 3-Kinase/Akt Pathway and the Nrf2 Transcription Factor in Response to the Antioxidant Phytochemical Carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Wang, P.; Qiao, Y.; Jiang, C.; Ge, Y.; Flickinger, B.; Malhotra, D.K.; Dworkin, L.D.; Liu, Z.; Gong, R. GSK3beta-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species 2019, 8, 312–322. [Google Scholar] [CrossRef]
- Kurinna, S.; Werner, S. NRF2 and microRNAs: New but awaited relations. Biochem. Soc. Trans. 2015, 43, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Chambel, S.S.; Santos-Gonçalves, A.; Duarte, T.L. The Dual Role of Nrf2 in Nonalcoholic Fatty Liver Disease: Regulation of Antioxidant Defenses and Hepatic Lipid Metabolism. BioMed Res. Int. 2015, 2015, 597134. [Google Scholar] [CrossRef] [Green Version]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxid. Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing Nrf2 Pathway by Disruption of Keap1 in Myeloid Leukocytes Protects against Sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 Skewing by Activation of Nrf2 in CD4+T Cells. J. Immunol. 2012, 188, 1630–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.; Reddy, S.P.; Rabb, H. T Lymphocyte–Specific Activation of Nrf2 Protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Chen, H.; Ding, Q.; Xu, X.; Yu, B.; Huang, Z. Nuclear Factor Erythroid 2-related Factor 2 Deficiency Exacerbates Lupus Nephritis in B6/lpr mice by Regulating Th17 Cell Function. Sci. Rep. 2016, 6, 38619. [Google Scholar] [CrossRef] [Green Version]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef] [Green Version]
- Harvey, C.J.; Thimmulappa, R.K.; Sethi, S.; Kong, X.; Yarmus, L.; Brown, R.H.; Feller-Kopman, D.; Wise, R.; Biswal, S. Targeting Nrf2 Signaling Improves Bacterial Clearance by Alveolar Macrophages in Patients with COPD and in a Mouse Model. Sci. Transl. Med. 2011, 3, 78ra32. [Google Scholar] [CrossRef] [Green Version]
- Sha, L.K.; Sha, W.; Kuchler, L.; Daiber, A.; Giegerich, A.K.; Weigert, A.; Knape, T.; Snodgrass, R.; Schröder, K.; Brandes, R.P.; et al. Loss of Nrf2 in bone marrow-derived macrophages impairs antigen-driven CD8+ T cell function by limiting GSH and Cys availability. Free Radic. Biol. Med. 2015, 83, 77–88. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Klemm, P.; Rajendiran, A.; Fragoulis, A.; Wruck, C.; Schippers, A.; Wagner, N.; Bopp, T.; Tenbrock, K.; Ohl, K. Nrf2 expression driven by Foxp3 specific deletion of Keap1 results in loss of immune tolerance in mice. Eur. J. Immunol. 2020, 50, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Satoh, H.; Moriguchi, T.; Taguchi, K.; Takai, J.; Maher, J.M.; Suzuki, T.; Winnard, P.T., Jr.; Raman, V.; Ebina, M.; Nukiwa, T.; et al. Nrf2-deficiency creates a responsive microenvironment for metastasis to the lung. Carcinogenesis 2010, 31, 1833–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Wenzel, P.; Kossmann, S.; Münzel, T.; Daiber, A. Redox regulation of cardiovascular inflammation—Immunomodulatory function of mitochondrial and Nox-derived reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2017, 109, 48–60. [Google Scholar] [CrossRef]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.F.; Kuo, H.P.; Liu, M.; Chou, C.K.; Xia, W.; Du, Y.; Shen, J.; Chen, C.T.; Huo, L.; Hsu, M.C.; et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 2009, 36, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Li, H.; Liu, Q.; Liu, F.; Tang, L.; Li, C.; Yuan, Y.; Zhan, Y.; Xu, W.; Li, W.; et al. Nuclear factor p65 interacts with Keap1 to repress the Nrf2-ARE pathway. Cell. Signal. 2011, 23, 883–892. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martin-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-kappaB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.; Qu, Q.; Zhang, W.; Wang, X.; Fang, J.; Xue, J.; Liu, Z.; He, S. NRF2 Exerts Anti-Inflammatory Effects in LPS-Induced gEECs by Inhibiting the Activation of the NF-kappaB. Mediat. Inflamm. 2021, 2021, 9960721. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.J.; Kim, H. Lutein as a Modulator of Oxidative Stress-Mediated Inflammatory Diseases. Antioxidants 2021, 10, 1448. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.D.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO-1 Pathway by Nardochinoid C Inhibits Inflammation and Oxidative Stress in Lipopolysaccharide-Stimulated Macrophages. Front. Pharm. 2018, 9, 911. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, L.; Lin, Z.; Hu, S.; Shi, Y.; Jiang, Z.; Zhao, J.; Zhou, Y.; Wu, Y.; Tian, N.; Sun, L.; et al. Itaconate attenuates osteoarthritis by inhibiting STING/NF-kappaB axis in chondrocytes and promoting M2 polarization in macrophages. Biochem. Pharm. 2022, 198, 114935. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Tang, C.; Wang, X.; Xie, Y.; Cai, X.; Yu, N.; Hu, Y.; Zheng, Z. 4-Octyl Itaconate Activates Nrf2 Signaling to Inhibit Pro-Inflammatory Cytokine Production in Peripheral Blood Mononuclear Cells of Systemic Lupus Erythematosus Patients. Cell. Physiol. Biochem. 2018, 51, 979–990. [Google Scholar] [CrossRef]
- Zhang, S.; Jiao, Y.; Li, C.; Liang, X.; Jia, H.; Nie, Z.; Zhang, Y. Dimethyl Itaconate Alleviates the Inflammatory Responses of Macrophages in Sepsis. Inflammation 2021, 44, 549–557. [Google Scholar] [CrossRef]
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Seibert, K.; Zhang, Y.; Leahy, K.; Hauser, S.; Masferrer, J.; Perkins, W.; Lee, L.; Isakson, P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. USA 1994, 91, 12013–12017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Yan, Y.; Zhang, R.; Xiong, H. Regulation of iNOS on Immune Cells and Its Role in Diseases. Int. J. Mol. Sci. 2018, 19, 3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.; Dinkova-Kostova, A.; Walsh, S.V.; Tsujita, T.; Dillon, J.; Ashford, M.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martin-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.-O.; Wang, H.; Kong, A.-N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Okuda, Y.; Sakoda, S.; Bernard, C.C.; Fujimura, H.; Saeki, Y.; Kishimoto, T.; Yanagihara, T. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int. Immunol. 1998, 10, 703–708. [Google Scholar] [CrossRef]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17–producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1β, TNF-α and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflam. 2010, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Jin, J.; Hu, W.; Chen, Q.; Yang, J.; Wu, Y.; Zhou, Y.; Sun, L.; Gao, W.; Zhang, X.; et al. Betulin Alleviates the Inflammatory Response in Mouse Chondrocytes and Ameliorates Osteoarthritis via AKT/Nrf2/HO-1/NF-kappaB Axis. Front. Pharm. 2021, 12, 754038. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chen, J.H.; Tsai, C.F.; Wu, C.T.; Wu, M.H.; Chang, P.C.; Yeh, W.L. Nicardipine Inhibits Breast Cancer Migration via Nrf2/HO-1 Axis and Matrix Metalloproteinase-9 Regulation. Front. Pharm. 2021, 12, 710978. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-C.; Jeon, W.-K.; Hong, H.-Y.; Jeon, K.-B.; Hahn, J.-H.; Kim, Y.-M.; Numazawa, S.; Yosida, T.; Park, E.-H.; Lim, C.-J. The anti-inflammatory activity of Phellinus linteus (Berk. & M.A. Curt.) is mediated through the PKCδ/Nrf2/ARE signaling to up-regulation of heme oxygenase-1. J. Ethnopharmacol. 2007, 113, 240–247. [Google Scholar] [CrossRef]
- Lee, S.H.; Sohn, D.H.; Jin, X.Y.; Kim, S.W.; Choi, S.C.; Seo, G.S. 2′,4′,6′-Tris(methoxymethoxy) chalcone protects against trinitrobenzene sulfonic acid-induced colitis and blocks tumor necrosis factor-α-induced intestinal epithelial inflammation via heme oxygenase 1-dependent and independent pathways. Biochem. Pharmacol. 2007, 74, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, S.; Zhang, Q.; Fang, Y.; Yu, Y.; Zhu, L.; Liu, Y.; Gong, W.; Zhao, L.; Qin, L.; et al. Curculigoside attenuates oxidative stress and osteoclastogenesis via modulating Nrf2/NF-kappaB signaling pathway in RAW264.7 cells. J. Ethnopharmacol. 2021, 275, 114129. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.-A.; Lee, I.-K. The Role of Nrf2: Adipocyte Differentiation, Obesity, and Insulin Resistance. Oxidative Med. Cell. Longev. 2013, 2013, 184598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, S.; Jiang, X.; Wang, Y.H.; Li, F.; Wang, Y.G.; Zheng, Y.; Cai, L. The role of the Nrf2/Keap1 pathway in obesity and metabolic syndrome. Rev. Endocr. Metab. Disord. 2015, 16, 35–45. [Google Scholar] [CrossRef]
- Shin, S.; Wakabayashi, N.; Misra, V.; Biswal, S.; Lee, G.H.; Agoston, E.S.; Yamamoto, M.; Kensler, T.W. NRF2 Modulates Aryl Hydrocarbon Receptor Signaling: Influence on Adipogenesis. Mol. Cell. Biol. 2007, 27, 7188–7197. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.; Xue, P.; Bai, Y.; Liu, D.; Woods, C.G.; Yarborough, K.; Fu, J.; Zhang, Q.; Sun, G.; Collins, S.; et al. Nuclear factor erythroid-derived factor 2-related factor 2 regulates transcription of CCAAT/enhancer-binding protein beta during adipogenesis. Free. Radic. Biol. Med. 2012, 52, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Leung, L.; Xue, P.; Wang, W.; Hou, Y.; Liu, D.; Yehuda-Shnaidman, E.; Lee, C.; Lau, J.; Kurtz, T.W.; et al. Deficiency in the Nuclear Factor E2-related Factor-2 Transcription Factor Results in Impaired Adipogenesis and Protects against Diet-induced Obesity. J. Biol. Chem. 2010, 285, 9292–9300. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Wakabayashi, J.; Yates, M.S.; Wakabayashi, N.; Dolan, P.M.; Aja, S.; Liby, K.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W. Role of Nrf2 in prevention of high-fat diet-induced obesity by synthetic triterpenoid CDDO-Imidazolide. Eur. J. Pharmacol. 2009, 620, 138–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slocum, S.L.; Skoko, J.J.; Wakabayashi, N.; Aja, S.; Yamamoto, M.; Kensler, T.W.; Chartoumpekis, D.V. Keap1/Nrf2 pathway activation leads to a repressed hepatic gluconeogenic and lipogenic program in mice on a high-fat diet. Arch. Biochem. Biophys 2016, 591, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Shao, W.; Chiang, Y.; Foltz, W.; Zhang, Z.; Ling, W.; Fantus, I.G.; Jin, T. Oltipraz upregulates the nuclear factor (erythroid-derived 2)-like 2 [corrected](NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 2011, 54, 922–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, P.; Hou, Y.; Chen, Y.; Yang, B.; Fu, J.; Zheng, H.; Yarborough, K.; Woods, C.G.; Liu, D.; Yamamoto, M.; et al. Adipose Deficiency of Nrf2 in ob/ob Mice Results in Severe Metabolic Syndrome. Diabetes 2013, 62, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Palliyaguru, D.L.; Wakabayashi, N.; Fazzari, M.; Khoo, N.K.H.; Schopfer, F.J.; Sipula, I.; Yagishita, Y.; Michalopoulos, G.K.; O’Doherty, R.M.; et al. Nrf2 deletion from adipocytes, but not hepatocytes, potentiates systemic metabolic dysfunction after long-term high-fat diet-induced obesity in mice. Am. J. Physiol. Metab. 2018, 315, E180–E195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Huang, J.; Shen, C.; Liu, Y.; He, S.; Sun, J.; Yu, B. NRF2 deficiency increases obesity susceptibility in a mouse menopausal model. PLoS ONE 2020, 15, e0228559. [Google Scholar] [CrossRef] [Green Version]
- Ferraz-Bannitz, R.; Welendorf, C.R.; Coelho, P.O.; Salgado, W.; Nonino, C.B.; Beraldo, R.A.; Foss-Freitas, M.C. Bariatric surgery can acutely modulate ER-stress and inflammation on subcutaneous adipose tissue in non-diabetic patients with obesity. Diabetol. Metab. Syndr. 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Chartoumpekis, D.V.; Ziros, P.G.; Psyrogiannis, A.I.; Papavassiliou, A.G.; Kyriazopoulou, V.E.; Sykiotis, G.P.; Habeos, I.G. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes 2011, 60, 2465–2473. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-K.J.; Wu, K.C.; Liu, J.; Klaassen, C.D. Nrf2 deficiency improves glucose tolerance in mice fed a high-fat diet. Toxicol. Appl. Pharmacol. 2012, 264, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kulkarni, S.R.; Donepudi, A.C.; More, V.R.; Slitt, A.L. Enhanced Nrf2 Activity Worsens Insulin Resistance, Impairs Lipid Accumulation in Adipose Tissue, and Increases Hepatic Steatosis in Leptin-Deficient Mice. Diabetes 2012, 61, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.-H.; Jang, J.Y.; Oh, S.; Yoon, J.-H.; Jo, D.-G.; Yun, U.J.; Park, K.W. Nrf2 induces Ucp1 expression in adipocytes in response to β3-AR stimulation and enhances oxygen consumption in high-fat diet-fed obese mice. BMB Rep. 2021, 54, 419. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Chang, S.-H.; Yang, D.K.; Song, N.-J.; Yun, U.J.; Park, K.W.; Bell, A.; Chiappe, L.M. Sesamol Increases Ucp1 Expression in White Adipose Tissues and Stimulates Energy Expenditure in High-Fat Diet-Fed Obese Mice. Nutrients 2020, 12, 1459. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68 (Suppl. S3), s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Proctor, D.; Scott, F.I.; Falck-Ytter, Y.; Feuerstein, J.D. AGA Technical Review on the Medical Management of Moderate to Severe Luminal and Perianal Fistulizing Crohn’s Disease. Gastroenterology 2021, 160, 2512–2556.e9. [Google Scholar] [CrossRef]
- Feuerstein, J.D.; Ho, E.Y.; Shmidt, E.; Singh, H.; Falck-Ytter, Y.; Sultan, S.; Terdiman, J.P.; Cohen, B.L.; Chachu, K.; Day, L.; et al. AGA Clinical Practice Guidelines on the Medical Management of Moderate to Severe Luminal and Perianal Fistulizing Crohn’s Disease. Gastroenterology 2021, 160, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, J.D.; Isaacs, K.L.; Schneider, Y.; Siddique, S.M.; Falck-Ytter, Y.; Singh, S.; Chachu, K.; Day, L.; Lebwohl, B.; Muniraj, T.; et al. AGA Clinical Practice Guidelines on the Management of Moderate to Severe Ulcerative Colitis. Gastroenterology 2020, 158, 1450–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Allegretti, J.R.; Siddique, S.M.; Terdiman, J.P. AGA Technical Review on the Management of Moderate to Severe Ulcerative Colitis. Gastroenterology 2020, 158, 1465–1496.e17. [Google Scholar] [CrossRef] [Green Version]
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Olivares-Villagómez, D.; Van Kaer, L. Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol. 2018, 39, 264–275. [Google Scholar] [CrossRef]
- Alzoghaibi, M.A. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J. Gastroenterol. 2013, 19, 6540–6547. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: Updated experimental and clinical evidence. Exp. Biol. Med. 2012, 237, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Yanaka, A. Role of NRF2 in protection of the gastrointestinal tract against oxidative stress. J. Clin. Biochem. Nutr. 2018, 63, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iborra, M.; Moret, I.; Rausell, F.; Bastida, G.; Aguas, M.; Cerrillo, E.; Nos, P.; Beltran, B. Role of oxidative stress and antioxidant enzymes in Crohn’s disease. Biochem. Soc. Trans. 2011, 39, 1102–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanaka, A. Contribution of NRF2 in Gastrointestinal Protection from Oxidative Injury. Curr. Pharm. Des. 2018, 24, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, M.; Swierczynski, M.; Fichna, J.; Piechota-Polanczyk, A. The Nrf2 in the pathophysiology of the intestine: Molecular mechanisms and therapeutic implications for inflammatory bowel diseases. Pharmacol. Res. 2021, 163, 105243. [Google Scholar] [CrossRef]
- Wen, Z.; Liu, W.; Li, X.; Chen, W.; Liu, Z.; Wen, J.; Liu, Z. A Protective Role of the NRF2-Keap1 Pathway in Maintaining Intestinal Barrier Function. Oxidative Med. Cell. Longev. 2019, 2019, 1759149. [Google Scholar] [CrossRef] [Green Version]
- Lau, W.L.; Liu, S.-M.; Pahlevan, S.; Yuan, J.; Khazaeli, M.; Ni, Z.; Chan, J.Y.; Vaziri, N.D. Role of Nrf2 Dysfunction in Uremia-Associated Intestinal Inflammation and Epithelial Barrier Disruption. Dig. Dis. Sci. 2015, 60, 1215–1222. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bao, Z.; Xu, X.; Chao, H.; Lin, C.; Li, Z.; Liu, Y.; Wang, X.; You, Y.; Liu, N.; et al. Extracellular Signal-Regulated Kinase/Nuclear Factor-Erythroid2-like2/Heme Oxygenase-1 Pathway-Mediated Mitophagy Alleviates Traumatic Brain Injury-Induced Intestinal Mucosa Damage and Epithelial Barrier Dysfunction. J. Neurotrauma. 2017, 34, 2119–2131. [Google Scholar] [CrossRef]
- Rodríguez-Ramiro, I.; Ramos, S.; Bravo, L.; Goya, L.; Martín, M. Ángeles Procyanidin B2 induces Nrf2 translocation and glutathione S-transferase P1 expression via ERKs and p38-MAPK pathways and protect human colonic cells against oxidative stress. Z. Ernährungswissenschaft 2012, 51, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Cheng, Y.; Li, X.; Wang, F.; Lu, Z.; Xiao, X.; Wang, Y. Biogenic Nanoselenium Particles Effectively Attenuate Oxidative Stress-Induced Intestinal Epithelial Barrier Injury by Activating the Nrf2 Antioxidant Pathway. ACS Appl. Mater. Interfaces 2017, 9, 14724–14740. [Google Scholar] [CrossRef]
- Fan, X.; Staitieh, B.S.; Jensen, J.S.; Mould, K.J.; Greenberg, J.A.; Joshi, P.C.; Koval, M.; Guidot, D.M. Activating the Nrf2-mediated antioxidant response element restores barrier function in the alveolar epithelium of HIV-1 transgenic rats. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L267–L277. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hu, Y.; Fang, Y.; Djukic, Z.; Yamamoto, M.; Shaheen, N.J.; Orlando, R.C.; Chen, X. Nrf2 deficiency impairs the barrier function of mouse oesophageal epithelium. Gut 2014, 63, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Theiss, A.L.; Vijay–Kumar, M.; Obertone, T.S.; Jones, D.P.; Hansen, J.M.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. Prohibitin Is a Novel Regulator of Antioxidant Response That Attenuates Colonic Inflammation in Mice. Gastroenterology 2009, 137, 199–208.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.-Y.; Li, Y.-D.; Cui, Y.-K.; Wu, C.; Hong, Y.-X.; Li, G.; Wu, Y.; Jie, L.-J.; Wang, Y.; Li, G.-R. The Natural Flavone Acacetin Confers Cardiomyocyte Protection Against Hypoxia/Reoxygenation Injury via AMPK-Mediated Activation of Nrf2 Signaling Pathway. Front. Pharmacol. 2018, 9, 497. [Google Scholar] [CrossRef] [PubMed]
- Osburn, W.O.; Karim, B.; Dolan, P.M.; Liu, G.; Yamamoto, M.; Huso, D.L.; Kensler, T.W. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int. J. Cancer 2007, 121, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Khor, T.O.; Huang, M.-T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.-N. Nrf2-Deficient Mice Have an Increased Susceptibility to Dextran Sulfate Sodium–Induced Colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabzevary-Ghahfarokhi, M.; Shohan, M.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Bagheri, N.; Shafigh, M.; Tahmasbi, K. The regulatory role of Nrf2 in antioxidants phase2 enzymes and IL-17A expression in patients with ulcerative colitis. Pathol. Res. Pr. 2018, 214, 1149–1155. [Google Scholar] [CrossRef]
- Cao, J.; Lu, M.; Yan, W.; Li, L.; Ma, H. Dehydroepiandrosterone alleviates intestinal inflammatory damage via GPR30-mediated Nrf2 activation and NLRP3 inflammasome inhibition in colitis mice. Free Radic. Biol. Med. 2021, 172, 386–402. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear Factor E2-Related Factor-2 Negatively Regulates NLRP3 Inflammasome Activity by Inhibiting Reactive Oxygen Species-Induced NLRP3 Priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [Green Version]
- Gwak, S.-Y.; Kim, S.-J.; Park, J.; Kim, S.H.; Joe, Y.; Lee, H.-N.; Kim, W.; Muna, I.A.; Na, H.-K.; Chung, H.T.; et al. Potential Role of Heme Oxygenase-1 in the Resolution of Experimentally Induced Colitis through Regulation of Macrophage Polarization. Gut Liver 2021. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, L.R.; Stonesifer, E.; Small, J.S.; Liby, K.T. The synthetic triterpenoid (CDDO-Im) inhibits STAT3, as well as IL-17, and improves DSS-induced colitis in mice. Inflammopharmacology 2014, 22, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhou, W.; Zhang, X.; Lu, P.; Du, Q.; Tao, L.; Ding, Y.; Wang, Y.; Hu, R. Dimethyl fumarate ameliorates dextran sulfate sodium-induced murine experimental colitis by activating Nrf2 and suppressing NLRP3 inflammasome activation. Biochem. Pharmacol. 2016, 112, 37–49. [Google Scholar] [CrossRef]
- Casili, G.; Cordaro, M.; Impellizzeri, D.; Bruschetta, G.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Reduces Inflammatory Responses in Experimental Colitis. J. Crohn’s Colitis 2016, 10, 472–483. [Google Scholar] [CrossRef]
- Yu, Y.; Zheng, C.; Lu, X.; Deng, C.; Xu, Q.; Guo, W.; Wu, Q.; Wang, Q.; Liu, C.; Huang, X.; et al. GB1a Ameliorates Ulcerative Colitis via Regulation of the NF-kappaB and Nrf2 Signaling Pathways in an Experimental Model. Front. Med. 2021, 8, 654867. [Google Scholar]
- Qiu, S.; Li, P.; Zhao, H.; Li, X. Maresin 1 alleviates dextran sulfate sodium-induced ulcerative colitis by regulating NRF2 and TLR4/NF-kB signaling pathway. Int. Immunopharmacol. 2020, 78, 106018. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Wang, X.W.; Ma, Y.; Hao, J.H.; Wang, J.S.; Lu, Y.Q.; Liu, Y.L.; Wang, X.F.; Zhang, L. Nrf2-induced miR-23a-27a-24-2 cluster modulates damage repair of intestinal mucosa by targeting the Bach1/HO-1 axis in inflammatory bowel diseases. Free Radic. Biol. Med. 2021, 163, 1–9. [Google Scholar]
- Jia, H.; Zhang, Y.; Si, X.; Jin, Y.; Jiang, D.; Dai, Z.; Wu, Z. Quercetin Alleviates Oxidative Damage by Activating Nuclear Factor Erythroid 2-Related Factor 2 Signaling in Porcine Enterocytes. Nutrients 2021, 13, 375. [Google Scholar] [CrossRef]
- Luo, M.; Luo, Y. Imperatorin Relieved Ulcerative Colitis by Regulating the Nrf-2/ARE/HO-1 Pathway in Rats. Inflammation 2021, 44, 558–569. [Google Scholar] [CrossRef]
- Saber, S.; Khalil, R.M.; Abdo, W.S.; Nassif, D.; El-Ahwany, E. Olmesartan ameliorates chemically-induced ulcerative colitis in rats via modulating NFκB and Nrf-2/HO-1 signaling crosstalk. Toxicol. Appl. Pharmacol. 2019, 364, 120–132. [Google Scholar] [CrossRef]
- Ramaswamy, M.; Tummala, R.; Streicher, K.; da Costa, A.N.; Brohawn, P.Z. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 11286. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.B.; Lima, C.A.D.; Vajgel, G.; Sandrin-Garcia, P. The Role of NLRP3 Inflammasome in Lupus Nephritis. Int. J. Mol. Sci. 2021, 22, 12476. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Tsokos, G.C. The role of CD8+ T-cell systemic lupus erythematosus pathogenesis: An update. Curr. Opin. Rheumatol. 2021, 33, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Atisha-Fregoso, Y.; Toz, B.; Diamond, B. Meant to B: B cells as a therapeutic target in systemic lupus erythematosus. J. Clin. Investig. 2021, 131, 12. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.; Anupam, K.; Kaushal, J.; Gautam, P.; Sharma, A.; Bhatnagar, A. Altered oxidative stress markers in relation to T cells, NK cells & killer immunoglobulin receptors that are associated with disease activity in SLE patients. Lupus 2020, 29, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan Autoimmune Inflammation, Enhanced Lymphoproliferation, and Impaired Homeostasis of Reactive Oxygen Species in Mice Lacking the Antioxidant-Activated Transcription Factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morito, N.; Yoh, K.; Hirayama, A.; Itoh, K.; Nose, M.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 deficiency improves autoimmune nephritis caused by the fas mutation lpr. Kidney Int. 2004, 65, 1703–1713. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Zhuang, H.; Lee, P.Y.; Li, M.; Yang, L.; Nigrovic, P.A.; Reeves, W.H. NF-E2–Related Factor 2 Regulates Interferon Receptor Expression and Alters Macrophage Polarization in Lupus. Arthritis Rheumatol. 2020, 72, 1707–1720. [Google Scholar] [CrossRef]
- Wu, Y.; He, S.; Bai, B.; Zhang, L.; Xue, L.; Lin, Z.; Yang, X.; Zhu, F.; He, P.; Tang, W.; et al. Therapeutic effects of the artemisinin analog SM934 on lupus-prone MRL/lpr mice via inhibition of TLR-triggered B-cell activation and plasma cell formation. Cell. Mol. Immunol. 2016, 13, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Goess, C.; Terrillon, S.; Mayo, M.; Bousquet, P.; Wallace, C.; Hart, M.; Mathieu, S.; Twomey, R.; Donnelly-Roberts, D.; Namovic, M.; et al. NRF2 activator A-1396076 ameliorates inflammation in autoimmune disease models by inhibiting antigen dependent T cell activation. J. Transl. Autoimmun. 2021, 4, 100079. [Google Scholar] [CrossRef]
- Wu, T.; Ye, Y.; Min, S.-Y.; Zhu, J.; Khobahy, E.; Zhou, J.; Yan, M.; Hemachandran, S.; Pathak, S.; Zhou, X.J.; et al. Prevention of Murine Lupus Nephritis by Targeting Multiple Signaling Axes and Oxidative Stress Using a Synthetic Triterpenoid. Arthritis Rheumatol. 2014, 66, 3129–3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebihara, S.; Tajima, H.; Ono, M. Nuclear factor erythroid 2-related factor 2 is a critical target for the treatment of glucocorticoid-resistant lupus nephritis. Arthritis Res. Ther. 2016, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D.J.K.I. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-κB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Sharma, A.; Bhatnagar, A. Role of oxidative stress in pathophysiology of rheumatoid arthritis: Insights into NRF2-KEAP1 signalling. Autoimmunity 2021, 54, 385–397. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, G.; Wang, T.; Cao, W.; Zhang, L.; Chen, X. Nrf2–Keap1 pathway–mediated effects of resveratrol on oxidative stress and apoptosis in hydrogen peroxide–treated rheumatoid arthritis fibroblast-like synoviocytes. Ann. N. Y. Acad. Sci. 2019, 1457, 166–178. [Google Scholar] [CrossRef]
- Lal, R.; Dhaliwal, J.; Dhaliwal, N.; Dharavath, R.N.; Chopra, K. Activation of the Nrf2/HO-1 signaling pathway by dimethyl fumarate ameliorates complete Freund’s adjuvant-induced arthritis in rats. Eur. J. Pharm. 2021, 899, 174044. [Google Scholar] [CrossRef]
- Chabaud, M.; Fossiez, F.; Taupin, J.L.; Miossec, P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J. Immunol. 1998, 161, 409–414. [Google Scholar]
- Cornelissen, F.; van Hamburg, J.P.; Lubberts, E. The IL-12/IL-23 axis and its role in Th17 cell development, pathology and plasticity in arthritis. Curr. Opin. Investig. Drugs 2009, 10, 452–462. [Google Scholar]
- Miossec, P.; Korn, T.; Kuchroo, V.K. Interleukin-17 and Type 17 Helper T Cells. N. Engl. J. Med. 2009, 361, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.T.; Lin, C.C.; Lin, S.C.; Wang, J.H.; Tsai, S.W. Kurarinone Attenuates Collagen-Induced Arthritis in Mice by Inhibiting Th1/Th17 Cell Responses and Oxidative Stress. Int. J. Mol. Sci. 2021, 22, 4002. [Google Scholar] [CrossRef] [PubMed]
- Castejon, M.L.; Alarcon-de-la-Lastra, C.; Rosillo, M.A.; Montoya, T.; Fernandez-Bolanos, J.G.; Gonzalez-Benjumea, A.; Sanchez-Hidalgo, M. A New Peracetylated Oleuropein Derivative Ameliorates Joint Inflammation and Destruction in a Murine Collagen-Induced Arthritis Model via Activation of the Nrf-2/Ho-1 Antioxidant Pathway and Suppression of MAPKs and NF-kappaB Activation. Nutrients 2021, 13, 311. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhu, Y.; Gao, Y.; Qi, D.; Zhao, L.; Zhao, L.; Liu, C.; Tao, T.; Zhou, C.; Sun, X.; et al. NR1D1 modulates synovial inflammation and bone destruction in rheumatoid arthritis. Cell Death Dis. 2020, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Li, Y.; Yao, C.; Liu, X.; Liu, J.; Yu, B. DC32, a Dihydroartemisinin Derivative, Ameliorates Collagen-Induced Arthritis Through an Nrf2-p62-Keap1 Feedback Loop. Front. Immunol. 2018, 9, 2762. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Z.; He, X.; Li, Z.; Shi, B.; Cai, F. beta-Sitosterol-loaded solid lipid nanoparticles ameliorate complete Freund’s adjuvant-induced arthritis in rats: Involvement of NF-small ka, CyrillicB and HO-1/Nrf-2 pathway. Drug Deliv. 2020, 27, 1329–1341. [Google Scholar] [CrossRef]
- Puppala, E.R.; Jain, S.; Saha, P.; Rachamalla, M.; Np, S.; Yalamarthi, S.S.; Abubakar, M.; Chaudhary, A.; Chamundeswari, D.; Usn, M.; et al. Perillyl alcohol attenuates rheumatoid arthritis via regulating TLR4/NF-kappaB and Keap1/Nrf2 signaling pathways: A comprehensive study onin-vitro and in-vivo experimental models. Phytomedicine 2022, 97, 153926. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44 (Suppl. S1), S15–S33. [Google Scholar] [CrossRef]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2016, 66, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Pullen, T.J.; Rutter, G.A. When less is more: The forbidden fruits of gene repression in the adult beta-cell. Diabetes Obes. Metab. 2013, 15, 503–512. [Google Scholar] [CrossRef]
- Lenzen, S.; Drinkgern, J.; Tiedge, M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic. Biol. Med. 1996, 20, 463–466. [Google Scholar] [CrossRef]
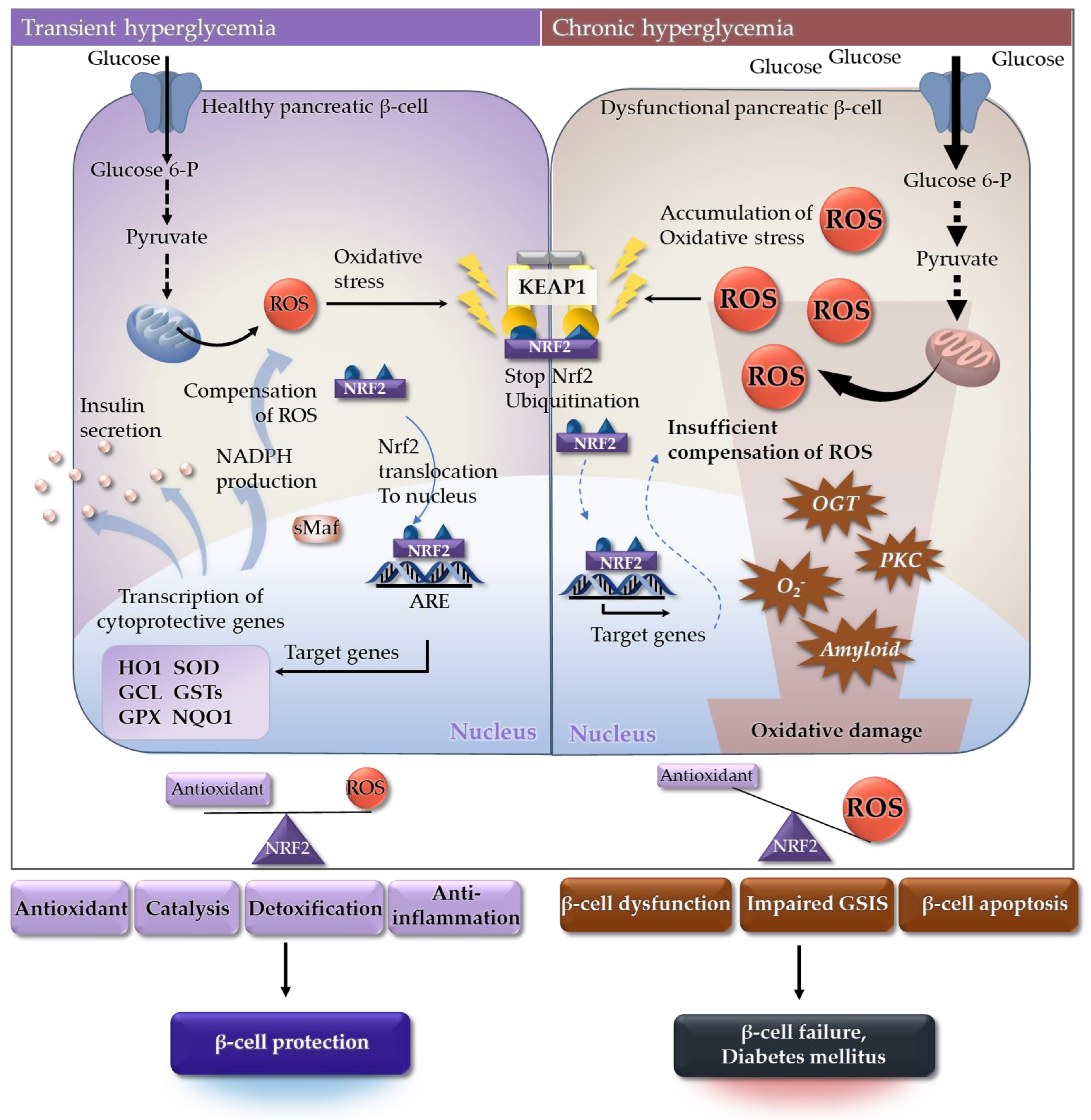
- Gerber, P.A.; Rutter, G.A. The Role of Oxidative Stress and Hypoxia in Pancreatic Beta-Cell Dysfunction in Diabetes Mellitus. Antioxid. Redox Signal. 2017, 26, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Ihara, Y.; Toyokuni, S.; Uchida, K.; Odaka, H.; Tanaka, T.; Ikeda, H.; Hiai, H.; Seino, Y.; Yamada, Y. Hyperglycemia causes oxidative stress in pancreatic beta-cells of GK rats, a model of type 2 diabetes. Diabetes 1999, 48, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C. Glucolipotoxicity, β-Cells, and Diabetes: The Emperor Has No Clothes. Diabetes 2020, 69, 273–278. [Google Scholar] [CrossRef]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic beta-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1–Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumel-Alterzon, S.; Katz, L.S.; Brill, G.; Garcia-Ocaña, A.; Scott, D.K. Nrf2: The Master and Captain of Beta Cell Fate. Trends Endocrinol. Metab. 2020, 32, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Zhang, Q.; Wong, V.; Floering, L.M.; Daniel, K.; Reece, J.M.; Deeney, J.T.; Andersen, M.E.; Corkey, B.E.; et al. Reactive Oxygen Species as a Signal in Glucose-Stimulated Insulin Secretion. Diabetes 2007, 56, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Pi, J.; Zhang, Q.; Fu, J.; Woods, C.G.; Hou, Y.; Corkey, B.; Collins, S.; Andersen, M. ROS signaling, oxidative stress and Nrf2 in pancreatic beta-cell function. Toxicol. Appl. Pharmacol. 2010, 244, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Bhakkiyalakshmi, E.; Sireesh, D.; Rajaguru, P.; Paulmurugan, R.; Ramkumar, K.M. The emerging role of redox-sensitive Nrf2–Keap1 pathway in diabetes. Pharmacol. Res. 2015, 91, 104–114. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Sharma, E.; Kumar, A.; Grover, M.; Bungau, S. Unfolding Nrf2 in diabetes mellitus. Mol. Biol. Rep. 2021, 48, 927–939. [Google Scholar] [CrossRef]
- Morgan, D.; Oliveira-Emilio, H.R.; Keane, D.; Hirata, A.E.; Da Rocha, M.S.; Bordin, S.; Curi, R.; Newsholme, P.; Carpinelli, A.R. Glucose, palmitate and pro-inflammatory cytokines modulate production and activity of a phagocyte-like NADPH oxidase in rat pancreatic islets and a clonal beta cell line. Diabetologia 2007, 50, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zraika, S.; Hull, R.L.; Udayasankar, J.; Aston-Mourney, K.; Subramanian, S.L.; Kisilevsky, R.; Szarek, W.A.; Kahn, S.E. Oxidative stress is induced by islet amyloid formation and time-dependently mediates amyloid-induced beta cell apoptosis. Diabetologia 2009, 52, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagida, K.; Maejima, Y.; Santoso, P.; Otgon-Uul, Z.; Yang, Y.; Sakuma, K.; Shimomura, K.; Yada, T. Hexosamine pathway but not interstitial changes mediates glucotoxicity in pancreatic beta-cells as assessed by cytosolic Ca2+ response to glucose. Aging 2014, 6, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, S.K.; Bhatt, R.; Kumar, A. Reactive oxygen species: Sources, consequences and targeted therapy in type 2 diabetes. J. Drug Target. 2017, 25, 93–101. [Google Scholar] [CrossRef]
- Wang, X.; Hai, C.X. ROS acts as a double-edged sword in the pathogenesis of type 2 diabetes mellitus: Is Nrf2 a potential target for the treatment? Mini Rev. Med. Chem. 2011, 11, 1082–1092. [Google Scholar] [CrossRef]
- Kjorholt, C.; Akerfeldt, M.C.; Biden, T.J.; Laybutt, D.R. Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of beta-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes 2005, 54, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Uruno, A.; Furusawa, Y.; Yagishita, Y.; Fukutomi, T.; Muramatsu, H.; Negishi, T.; Sugawara, A.; Kensler, T.W.; Yamamoto, M. The Keap1-Nrf2 System Prevents Onset of Diabetes Mellitus. Mol. Cell. Biol. 2013, 33, 2996–3010. [Google Scholar] [CrossRef] [Green Version]
- David, J.A.; Rifkin, W.J.; Rabbani, P.S.; Ceradini, D.J. The Nrf2/Keap1/ARE Pathway and Oxidative Stress as a Therapeutic Target in Type II Diabetes Mellitus. J. Diabetes Res. 2017, 2017, 4826724. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Sathyapalan, T.; Atkin, S.L.; Sahebkar, A. Molecular Mechanisms Linking Oxidative Stress and Diabetes Mellitus. Oxidative Med. Cell. Longev. 2020, 2020, 8609213. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Nichols, G.A.; Qian, L.; Munis, M.A.; Harrison, T.N.; Li, Z.; Wei, R.; Weiss, T.; Rajpathak, S.; Reynolds, K. Prevalence and incidence of microvascular and macrovascular complications over 15 years among patients with incident type 2 diabetes. BMJ Open Diabetes Res. Care 2021, 9, e001847. [Google Scholar] [CrossRef]
- Gregg, E.W.; Cheng, Y.J.; Saydah, S.; Cowie, C.; Garfield, S.; Geiss, L.; Barker, L. Trends in death rates among U.S. adults with and without diabetes between 1997 and 2006: Findings from the National Health Interview Survey. Diabetes Care 2012, 35, 1252–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, J.A.; Creager, M.A. Vascular Complications of Diabetes. Circ. Res. 2016, 118, 1771–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacobini, C.; Vitale, M.; Pesce, C.; Pugliese, G.; Menini, S. Diabetic Complications and Oxidative Stress: A 20-Year Voyage Back in Time and Back to the Future. Antioxidants 2021, 10, 727. [Google Scholar] [CrossRef]
- Gupte, A.A.; Lyon, C.J.; Hsueh, W.A. Nuclear Factor (Erythroid-Derived 2)-Like-2 Factor (Nrf2), a Key Regulator of the Antioxidant Response to Protect Against Atherosclerosis and Nonalcoholic Steatohepatitis. Curr. Diabetes Rep. 2013, 13, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, K.; Kolodgie, F.D.; Lutter, C.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arter. Thromb. Vasc. Biol. 2017, 37, 191–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, J.; Itoh, K. Role of Nrf2 in the pathogenesis of atherosclerosis. Free Radic. Biol. Med. 2015, 88, 221–232. [Google Scholar] [CrossRef]
- Negi, C.K.; Jena, G. Nrf2, a novel molecular target to reduce type 1 diabetes associated secondary complications: The basic considerations. Eur. J. Pharmacol. 2019, 843, 12–26. [Google Scholar] [CrossRef]
- Wu, J.; Sun, X.; Jiang, Z.; Jiang, J.; Xu, L.; Tian, A.; Sun, X.; Meng, H.; Li, Y.; Huang, W.; et al. Protective role of NRF2 in macrovascular complications of diabetes. J. Cell. Mol. Med. 2020, 24, 8903–8917. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done? Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Nakamura, N.; Ojima, A.; Nishino, Y.; Yamagishi, S.-I. Sulforaphane reduces advanced glycation end products (AGEs)-induced inflammation in endothelial cells and rat aorta. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 797–807. [Google Scholar] [CrossRef]
- Pereira, A.; Fernandes, R.; Crisóstomo, J.; Seiça, R.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14357. [Google Scholar] [CrossRef] [Green Version]
- Shawky, N.M.; Pichavaram, P.; Shehatou, G.S.; Suddek, G.M.; Gameil, N.M.; Jun, J.Y.; Segar, L. Sulforaphane improves dysregulated metabolic profile and inhibits leptin-induced VSMC proliferation: Implications toward suppression of neointima formation after arterial injury in western diet-fed obese mice. J. Nutr. Biochem. 2016, 32, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Z.; Sun, W.; Tan, Y.; Liu, Y.; Zheng, Y.; Liu, Q.; Cai, L.; Sun, J. Sulforaphane Attenuation of Type 2 Diabetes-Induced Aortic Damage Was Associated with the Upregulation of Nrf2 Expression and Function. Oxidative Med. Cell. Longev. 2014, 2014, 123963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourakis, S.; Timpani, C.A.; De Haan, J.B.; Gueven, N.; Fischer, D.; Rybalka, E. Dimethyl Fumarate and Its Esters: A Drug with Broad Clinical Utility? Pharmaceuticals 2020, 13, 306. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Abdelaziz, R.R.; Hamed, M.F.; Nader, M.A.; Shehatou, G.S. Dimethyl fumarate ameliorates diabetes-associated vascular complications through ROS-TXNIP-NLRP3 inflammasome pathway. Life Sci. 2020, 256, 117887. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Behl, T.; Sehgal, A.; Bhatia, S.; Jaglan, D.; Bungau, S. Therapeutic potential of Nrf-2 pathway in the treatment of diabetic neuropathy and nephropathy. Mol. Biol. Rep. 2021, 48, 2761–2774. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Du, L.; Hao, M.; Zhou, X.; Xuan, Q.; Apu, C.; Sun, Y.; Lu, Q.; Yin, X. Keap1/Nrf2/ARE signaling unfolds therapeutic targets for redox imbalanced-mediated diseases and diabetic nephropathy. Biomed Pharm. 2020, 123, 109732. [Google Scholar] [CrossRef]
- Li, B.; Liu, S.; Miao, L.; Cai, L. Prevention of Diabetic Complications by Activation of Nrf2: Diabetic Cardiomyopathy and Nephropathy. Exp. Diabetes Res. 2012, 2012, 216512. [Google Scholar] [CrossRef]
- Landis, R.C.; Quimby, K.R.; Greenidge, A.R. M1/M2 Macrophages in Diabetic Nephropathy: Nrf2/HO-1 as Therapeutic Targets. Curr. Pharm. Des. 2018, 24, 2241–2249. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic Potential of Nrf2 Activators in Streptozotocin-Induced Diabetic Nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef] [Green Version]
- Negi, G.; Kumar, A.; Sharma, S.S. Nrf2 and NF-kappaB modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Curr. Neurovasc. Res. 2011, 8, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.V.; Sundaresan, N.R.; Gupta, M.P.; White, C. Defective Nrf2-dependent redox signalling contributes to microvascular dysfunction in type 2 diabetes. Cardiovasc. Res. 2013, 100, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batliwala, S.; Xavier, C.; Liu, Y.; Wu, H.; Pang, I.-H. Involvement of Nrf2 in Ocular Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 1703810. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2021. Diabetes Care 2021, 44 (Suppl. S1), S151–S167. [Google Scholar] [CrossRef]
- Song, Y.; Ding, W.; Bei, Y.; Xiao, Y.; Tong, H.-D.; Wang, L.-B.; Ai, L.-Y. Insulin is a potential antioxidant for diabetes-associated cognitive decline via regulating Nrf2 dependent antioxidant enzymes. Biomed. Pharmacother. 2018, 104, 474–484. [Google Scholar] [CrossRef]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription Factor Nrf2-Mediated Antioxidant Defense System in the Development of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.M.; Barber, A.; Spagnuolo, C.; Russo, G.L.; Daglia, M.; Sobarzo-Sanchez, E. Nrf2 as molecular target for polyphenols: A novel therapeutic strategy in diabetic retinopathy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 293–312. [Google Scholar] [CrossRef]
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through activating NRF2 and inhibiting ER stress. Aging 2018, 10, 2954–2972. [Google Scholar] [CrossRef]
- Kumar, A.; Mittal, R. Nrf2: A potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef]
- Vavougios, G.; Zarogiannis, S.G.; Doskas, T. The putative interplay between DJ-1/NRF2 and Dimethyl Fumarate: A potentially important pharmacological target. Mult. Scler. Relat. Disord. 2018, 21, 88–91. [Google Scholar] [CrossRef] [PubMed]
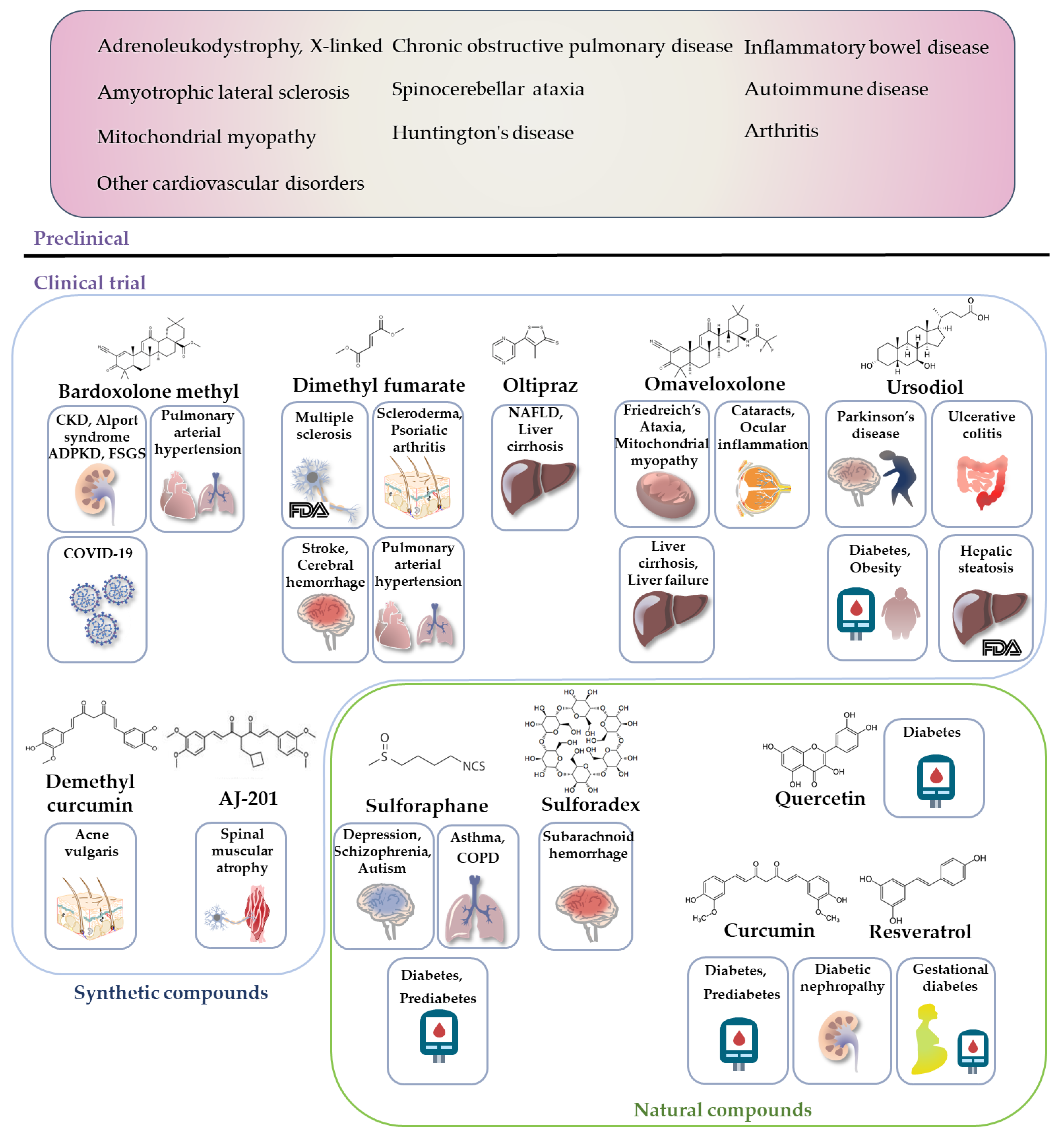
- Yagishita, Y.; Gatbonton-Schwager, T.N.; McCallum, M.L.; Kensler, T.W. Current Landscape of NRF2 Biomarkers in Clinical Trials. Antioxidants 2020, 9, 716. [Google Scholar] [CrossRef] [PubMed]
- Hishikawa, A.; Hayashi, K.; Itoh, H. Transcription Factors as Therapeutic Targets in Chronic Kidney Disease. Molecules 2018, 23, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenvinkel, P.; Warady, B.; Pergola, P.; Inker, L.; Nozu, K.; Appel, G.; Meyer, C.; Block, G.; Knebelmann, B.; Silva, A.; et al. Study Design and Baseline Characteristics of the CARDINAL Trial: A Phase 3 Study of Bardoxolone Methyl in Patients with Alport Syndrome. Am. J. Nephrol. 2021, 52, 180–189. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Toto, R.D. Bardoxolone-the Phoenix? J. Am. Soc. Nephrol. 2018, 29, 360–361. [Google Scholar] [CrossRef]
- Gai, L.; Zhu, Y.; Zhang, C.; Meng, X. Targeting Canonical and Non-Canonical STAT Signaling Pathways in Renal Diseases. Cells 2021, 10, 1610. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Chertow, G.M.; Devarajan, P.; Levin, A.; Andreoli, S.P.; Bangalore, S.; Warady, B.A. Chronic Inflammation in Chronic Kidney Disease Progression: Role of Nrf2. Kidney Int. Rep. 2021, 6, 1775–1787. [Google Scholar] [CrossRef]
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2021, 32, 35–47. [Google Scholar] [CrossRef]
- Lynch, D.R.; Chin, M.P.; Delatycki, M.B.; Subramony, S.H.; Corti, M.; Hoyle, J.C.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Mathews, K.D.; et al. Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann. Neurol. 2020, 89, 212–225. [Google Scholar] [CrossRef]
- Madsen, K.L.; Buch, A.E.; Cohen, B.H.; Falk, M.J.; Goldsberry, A.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Muraresku, C.C.; Meyer, C.; et al. Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 2020, 94, e687–e698. [Google Scholar] [CrossRef] [PubMed]
- Yamawaki, K.; Kanda, H.; Shimazaki, R. Nrf2 activator for the treatment of kidney diseases. Toxicol. Appl. Pharmacol. 2018, 360, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.M.; Ram, C.; Murty, U.S.; Sahu, B.D. A review on herbal Nrf2 activators with preclinical evidence in cardiovascular diseases. Phytother. Res. 2021, 35, 5068–5102. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margoni, M.; Rinaldi, F.; Perini, P.; Gallo, P. Therapy of Pediatric-Onset Multiple Sclerosis: State of the Art, Challenges, and Opportunities. Front. Neurol. 2021, 12, 676095. [Google Scholar] [CrossRef] [PubMed]
- Zolty, R. Novel Experimental Therapies for Treatment of Pulmonary Arterial Hypertension. J. Exp. Pharmacol. 2021, 13, 817–857. [Google Scholar] [CrossRef]
- Ishizaki, K.; Imada, T.; Tsurufuji, M. Hepatoprotective bile acid ‘ursodeoxycholic acid (UDCA)’: Property and difference as bile acids. Hepatol. Res. 2005, 33, 174–177. [Google Scholar] [CrossRef]
- Okada, K.; Shoda, J.; Taguchi, K.; Maher, J.M.; Ishizaki, K.; Inoue, Y.; Ohtsuki, M.; Goto, N.; Takeda, K.; Utsunomiya, H.; et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am. J. Physiol. Liver Physiol. 2008, 295, G735–G747. [Google Scholar] [CrossRef]
- Arisawa, S.; Ishida, K.; Kameyama, N.; Ueyama, J.; Hattori, A.; Tatsumi, Y.; Hayashi, H.; Yano, M.; Hayashi, K.; Katano, Y.; et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem. Pharmacol. 2009, 77, 858–866. [Google Scholar] [CrossRef]
- Kawata, K.; Kobayashi, Y.; Souda, K.; Kawamura, K.; Sumiyoshi, S.; Takahashi, Y.; Noritake, H.; Watanabe, S.; Suehiro, T.; Nakamura, H. Enhanced Hepatic Nrf2 Activation After Ursodeoxycholic Acid Treatment in Patients with Primary Biliary Cirrhosis. Antioxid. Redox Signal. 2010, 13, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Matencio, A.; Navarro-Orcajada, S.; González-Ramón, A.; García-Carmona, F.; López-Nicolás, J.M. Recent advances in the treatment of Niemann pick disease type C: A mini-review. Int. J. Pharm. 2020, 584, 119440. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Begum, N.; Mutha, S.; Bakshi, V. Beneficial effect of Curcumin in Letrozole induced polycystic ovary syndrome. Asian Pac. J. Reprod. 2016, 5, 116–122. [Google Scholar] [CrossRef]
- Kim, W.; Kim, B.G.; Lee, J.S.; Lee, C.K.; Yeon, J.E.; Chang, M.S.; Kim, J.H.; Kim, H.; Yi, S.; Cho, J.-Y.; et al. Randomised clinical trial: The efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 45, 1073–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Kim, Y.M.; Choi, J.Y.; Han, J.-Y.; Jang, J.W.; Cho, S.-H.; Um, S.H.; Chon, C.Y.; Lee, D.H.; Jang, J.-J.; et al. Oltipraz therapy in patients with liver fibrosis or cirrhosis: A randomized, double-blind, placebo-controlled phase II trial. J. Pharm. Pharmacol. 2011, 63, 627–635. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Zolnourian, A.H.; Franklin, S.; Galea, I.; Bulters, D.O. Study protocol for SFX-01 after subarachnoid haemorrhage (SAS): A multicentre randomised double-blinded, placebo controlled trial. BMJ Open 2020, 10, e028514. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Ci, X.; Man, X.; Li, J.; Wei, Z.; Zhang, S. Food-Derived Pharmacological Modulators of the Nrf2/ARE Pathway: Their Role in the Treatment of Diseases. Molecules 2021, 26, 1016. [Google Scholar] [CrossRef]
- Zhang, Y. The molecular basis that unifies the metabolism, cellular uptake and chemopreventive activities of dietary isothiocyanates. Carcinogenesis 2012, 33, 2–9. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Animal Model | Nrf2 Gain-of Function (Including Nrf2 Agonist) or Loss-of-Function | Phenotype | Refs. |
|---|---|---|---|---|
| Obesity | C57BL/6J mice WT and Nrf2-disrupted; high fat diet (HFD) | Nrf2 loss-of-function, Nrf2 agonist (CDDO-Im) | Improvement of obesity and suppression of lipogenesis by CDDO-Im. This beneficial is lost in Nrf2-deficient mice | [112] |
| C57BL/6J mice WT and Keap1-hypo; HFD | Nrf2 gain-of function (Hypomorphic Keap1 allele) | Hypomorphic Keap1 allele mice (model of Nrf2 activation) gain less weight, show ameliorated glucose tolerance, and develop less hepatic steatosis. Keap1-hypo livers exhibit activated AMPK signaling | [113] | |
| C57BL/6J mice; HFD | Nrf2 agonist (Otipraz) | Administration of Nrf2 activator reverses the detrimental effects of HFD-induced obesity. | [114] | |
| C57BL/6J ob/ob mice | Nrf2 loss-of-function (Global and adipocyte-specific) | Global, as well as adipose-specific, ablation of Nrf2 in ob/ob mice results in severe metabolic syndrome | [115] | |
| C57BL/6J HFD | Nrf2 loss-of-function (Adipocyte-specific Nrf2−/−) | Adipocyte-specific Nrf2−/− impaires glucose tolerance, higher fasting glucose levels, and higher levels of cholesterol and non-esterified fatty acids. | [116] | |
| ob/ob mice | Nrf2 gain-of function (Keap1−/−) | Reduces epididymal fat mass and body weight | [121] | |
| Inflammatory Bowel Disease | salmonella typhimurium challenge model, dextran sodium sulfate (DSS)-induced colitis model | Nrf2 gain-of function by prohibitin B overexpression (transgenic mice) | Prohibitin B transenic mice exhibit decreases oxidative stress and improved colitis | [144] |
| DSS-induced colitis | Nrf2 loss-of-function (Mitogen-activated protein kinase phosphatase 1 KO; Mkp-1−/−) | Mkp-1−/− mice are more susceptible to DSS-induced colitis | [145] | |
| DSS-induced colitis | Nrf2-deficient mice | Increased inflammation and mucosal damage | [146] | |
| DSS-induced colitis | Dehydroepiandrosterone (DHEA); acivates Nrf2 via G protein-coupled receptor 30 (GPR30)-dependent pathway | DHEA inhibits intestinal inflammation and improves barrier function in DSS-induced colitis model | [149] | |
| DSS-induced colitis | Nrf2 inhibition by treating HO-1 inhibitor zinc protoporphyrin IX (ZnPP) | Administration of ZnPP blunts the resolution of DSS-induced intestinal inflammation and expression of the proresolving M2 macrophage marker CD206 | [151] | |
| DSS-induced colitis | Nrf2 activation by CDDO-Im | Administration of CDDO-Im improves the altered colonic histology, and cytokine | [153] | |
| DSS-induced colitis | Nrf2 activation by GB1a | GB1a administration reverses loss of body weight and disease activity index scores in experimental colitis | [154] | |
| DSS-induced colitis | Nrf2 activation by dimethyl fumarate (DMF) | DMF attenuates the shortening of colons and alleviated colonic inflammation | [51,154,155] | |
| DSS-induced colitis | Nrf2 activation by Maresin 1 and Nrf2 inhibition by ML385 | Maresin 1 attenuates experimental colitis by reducing activation of TLR4/NF-κB. ML385 reverses the protective effects of maresin 1 markedly | [157] | |
| 4,6-trinitro-benzenesulfonic acid (TNBS) induced colitis | Nrf2 activation by Imperatorin | Imperatorin administration alleviates the symptoms of ulcerative colitis and inhibited the secretion of TNF-α and IL-6 | [160] | |
| Acetic acid (AA)-induced colitis in rats | Nrf2 activation by Olmerartan | Olmerartan ameliorates colon injury and inflammatory signs | [161] | |
| Systemic Lupus Erythematosus | Female Nrf2−/− mice | Nrf2-deficient mice | Multiorgan inflammatory lesions Apearance of anti-double-stranded DNA antibodies in young adulthoodintravascular Pemature death due to rapidly progressing membranoproliferative glomerular nephritis | [167] |
| B6/lpr mouse (sponatenous lupus nephritis model) | Nrf2-deficient mice | Nrf2 deficiency increases lupus nephritis and Th17 cell numners in B6/lpr mice | [66] | |
| MRL/lpr mouse | Nrf2-deficient mice | Nrf2 deficiency increases life span, improves nephritis. Immunologic abnormalities as well as hypergammaglobulinemia is correctetd. | [168] | |
| NZB/W mouse (spontaneous lupus nephritis model) | Nrf2 activation by A-1396076 | A-1396076 dampens inflammation in an IFN-α-accelerated NZB/W mouse lupus nephritis model | [171] | |
| B6.Sle1.Sle3 mouse and MRL/lpr mouse | Nrf2 activation by CDDO-Me | CDDO-Me reduces severity of lupus disease by attenuating MEK-1/2, ERK, and STAT-3 signaling in CD4+ T cells, as well as oxidative stress in B6.Sle1.Sle3 mice or MRL/lpr mice | [172] | |
| Pristane-induced lupus nephritis mice | Nrf2 activation by DMF | DMF ameliorates pristane-induced lupus nephritis mice, and showes stronger anti-inflammatory and organ-protective effects than glucocorticoids | [173] | |
| Pristane-induced lupus nephritis mice | Nrf2 activation by sulphoraphane | Sulphoraphane suppresses pritane-induced lupus nephritis | [174] | |
| Rheumatoid Arthritis | Complete Freund’s adjuvant-induced arthritis in rats | Nrf2 activation by DMF | DMF ameliorates complete Freund’s adjuvant-induced arthritis by suppressing oxidative stress and inflammatory mediators, and by increasing local Nrf2 and HO-1 concentration in the involved joints | [178] |
| Collagen-induced arthritis in DBA/1 mice | Nrf2 activation by kurarinone | Kurarinone reduces arthritis severity of CIA mice, as well as their levels of proinflammatory cytokines in the serum and paw tissues | [182] | |
| Collagen-induced arthritis in DBA/1 mice | Nrf2 activation by oleuropein | Oleuropein containing diet prevents histological damage and arthritic score development | [183] | |
| Collagen-induced arthritis in DBA/1 mice | NR1D1 activation by SR9009 increases Nrf2-associated enzymes. | SR9009 significantly suppresses synovial hyperplasia, infiltration of inflammatory cells, and destruction of cartilage and bone in mice with CIA | [184]. | |
| Collagen-induced arthritis in DBA/1 mice | Nrf2 activation by DC32, a dihydroartemisinin derivative | DC32 significantly alleviates footpad inflammation, reduce cartilage degradation | [185] |
| Compound Name [Mechanism] | Disease Target | ClinicalTrials.gov Identifier | Status | Phase |
|---|---|---|---|---|
| Synthetic compounds | ||||
| Bardoxolone methyl (CDDO-Me, BARD, RTA-402) [Electrophilic compunds] | Obesity | NCT04018339 | Completed | I |
| Hereditary nephritis (Alport syndrome) | NCT03019185 | Completed | II/III | |
| Autosomal dominant polycystic kidney disease (ADPKD) | NCT03918447 | Recruiting | III | |
| Pulmonary hypertension | NCT03068130 | Terminated | III | |
| Connective tissue disease-associated pulmonary arterial hypertension | NCT02657356 | Terminated | III | |
| Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection; Coronavirus disease 2019 (COVID-19) | NCT04494646 | Completed | II/III | |
| Focal segmental glomerulosclerosis (FSGS) | NCT03366337 | Completed | II | |
| Diabetic kidney disease | NCT00811889 | Completed | II | |
| NCT00550849 | Terminated | I/II | ||
| NCT00664027 | Completed | II | ||
| NCT03550443 | Active, not recruiting | III | ||
| Type 2 diabetes | NCT02316821 | Completed | II | |
| NCT01053936 | Completed | II | ||
| NCT01053936 | Completed | II | ||
| CKD associated with type 1 diabetes | NCT03366337 | Completed | II | |
| CKD associated with type 2 diabetes | NCT01351675 | Terminated | III | |
| Chronic kidney disease | NCT04702997 | Active, not recruiting | II | |
| Dimethyl fumarate (Brand name Tecifidera®) [Electrophilic compunds] | Pediatric multiple sclerosis, relapsing-remitting | NCT03870763 | Recruiting | III |
| Multiple sclerosis | NCT02097849 | Completed | II | |
| Ischemic stroke | NCT04891497 | Not yet recruiting | II | |
| Obstructive sleep apnea | NCT02438137 | Completed | II | |
| Pulmonary hypertension Scleroderma | NCT02981082 | Terminated | I | |
| Age-related macular degeneration (AMD) | NCT04292080 | Not yet recruiting | II | |
| Psoriatic arthritis | NCT02475304 | Withdrawn | II | |
| Oltipraz (CB-1400) [Electrophilic compunds] | Non-alcoholic fatty liver disease (NAFLD) | NCT04142749 | Recruiting | II/III |
| Omaveloxolone [Electrophilic compunds] | Friedreich’s ataxia | NCT02255435 | Active, not recruiting | II/III |
| Mitochondrial myopathy | NCT02255422 | Completed | II | |
| Cataracts | NCT02128113 | Completed | II | |
| Ocular inflammation | NCT02065375 | Completed | II | |
| Liver cirrhosis, liver failure | NCT03902002 | Completed | I | |
| Ursodiol (Ursodeoxycholic acid, brand names Actigall® or Urso®) [Electrophilic compunds] | Parkinson’s disease | NCT03840005 | Completed | II |
| Ulcerative colitis | NCT03724175 | Recruiting | II/III | |
| Type 2 diabetes | NCT02033876 | Completed | II | |
| Hepatic steatosis | NCT03664596 | Completed | II | |
| Retinopathy | NCT02841306 | Completed | I | |
| Dimethyl curcumin (AJ-101, ASC-J9) [Electrophilic compunds] | Acne vulgaris | NCT00525499 | Completed | II |
| Inflammatory acne | NCT01289574 | Completed | II | |
| AJ-201 (ALZ-002, ASC-JM-17) [Electrophilic compunds] | Spinal and bulbar muscular atrophy | NCT04392830 | Completed | I |
| Natural compounds | ||||
| Sulforaphane (SFN) [Electrophilic compunds] | Type 2 diabetes | NCT02801448 | Completed | II |
| Cognitive disorders | NCT04252261 | Not yet recruiting | II | |
| Chronic obstructive pulmonary disease (COPD) | NCT01318603 | Completed | II | |
| Asthma | NCT00994604 | Completed | NA | |
| Schizoaffective disorder, Schizophrenia | NCT02810964 | Completed | II | |
| Autism spectrum disorders | NCT02654743 | Completed | II | |
| Sulforadex (SFX-01) [Electrophilic compunds] | Subarachnoid hemorrhage | NCT02614742 | Completed | II |
| Curcumin [Electrophilic compunds] | Prediabetes | NCT03917784 | Unknown | IV |
| Diabetic nephropathy | NCT03262363 | Unknown | II/III | |
| Type 2 diabetes | NCT02529969 | Unknown | II/III | |
| NCT01052597 | Unknown | IV | ||
| NCT01052025 | Unknown | IV | ||
| Resveratrol [Electrophilic compunds] | Diabetic nephropathy | NCT02704494 | Completed | I |
| Gestational diabetes | NCT01997762 | Unknown | IV | |
| Type 2 diabetes | NCT01677611 | Completed | I | |
| NCT01158417 | Unknown | II/III | ||
| NCT02244879 | Completed | III | ||
| NCT02216552 | Completed | II/III | ||
| NCT01354977 | Completed | II | ||
| Quercetin [Electrophilic compunds] | Type 2 diabetes | NCT00065676 | Completed | II |
| NCT01839344 | Completed | II | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-J.; Jeon, J.-H. Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. Int. J. Mol. Sci. 2022, 23, 2846. https://doi.org/10.3390/ijms23052846
Kim M-J, Jeon J-H. Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. International Journal of Molecular Sciences. 2022; 23(5):2846. https://doi.org/10.3390/ijms23052846
Chicago/Turabian StyleKim, Min-Ji, and Jae-Han Jeon. 2022. "Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases" International Journal of Molecular Sciences 23, no. 5: 2846. https://doi.org/10.3390/ijms23052846
APA StyleKim, M.-J., & Jeon, J.-H. (2022). Recent Advances in Understanding Nrf2 Agonism and Its Potential Clinical Application to Metabolic and Inflammatory Diseases. International Journal of Molecular Sciences, 23(5), 2846. https://doi.org/10.3390/ijms23052846