
Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review


Abstract
: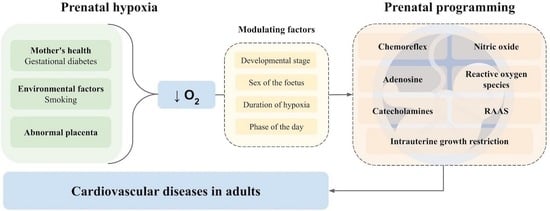
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Oxygen | Duration; Time | Animal Model | Birth Body Weight | Ref. |
|---|---|---|---|---|
| 6.5% | 1–20 ED; 8 h per day: 80 s hypoxia and 120 s normoxia; 18 cycles per hour | Sprague Dawley rats | ↓ | [18] |
| 7% | 13–14 ED; 3 h | Wistar rats | ↓ | [19] |
| 7% | 18 ED; 3 h | Wistar rats | = | [20] |
| 9% | 15–21 ED; 6 h per day | Sprague Dawley rats | = | [21] |
| 9.5–10% | 12, 24, 48, 120 h immediately prior to delivery at term | Sprague Dawley rats | ↓ | [22] |
| 10% | 5–19 ED | Sprague Dawley rats | ↓ | [23] |
| 10% | 5–20 ED | Sprague Dawley rats | ↓ | [13,24,25,26,27] |
| 10% | 15–20 ED | Wistar rats | ↓ | [28,29] |
| 10% | from 121 ED–NA | sheep | = | [30] |
| 10 ± 0.5% | 5–20 ED | Sprague Dawley rats | ↓ | [31] |
| 10.5% | 15–20 ED; 4 h per day | Sprague Dawley rats | = | [32] |
| 10.5% | 4–21 ED | Sprague Dawley rats | ↓ | [33] |
| 10.5% | 15–21 ED | Sprague Dawley rats | ↓ | [34,35] |
| 10.5% | 11–17.5 ED | BALB/c mice | ↓ | [36] |
| 10.5% | last 15 days of gravidity | guinea pigs | ↓ | [37] |
| 10 ± 1% | 7–21 ED; 3 h per day | Sprague Dawley rats | ↓ | [38] |
| 11% | 15–21 ED | rats | ↓ | [39] |
| 11.5% | 13–20 ED | Sprague Dawley rats | ↓ | [40] |
| 12% | 15–19 ED | Sprague Dawley rats | = | [41] |
| 12% | 14.5–21 ED | CD-1 mice | ↓ | [11] |
| 13% | 6–20 ED | Wistar rats | = | [2,12,42] |
| 13–14% | 6–20 ED | Wistar rats | = | [43] |
| 14% | 6–18 ED | C57BL/J6 mice | = | [44] |
| 15% | 19 ED–delivery; 10 min; 6 times per day | Sprague Dawley rats | = | [45] |
| NA | NA | Jackson Black C-57 mice | = | [46] |
| 280–300 mmHg; 8000 m above sea level | 14 ED–delivery; 2 h per day | C57BL/6 mice | = | [47] |
| PaO2 13 mmHg | 14 days | sheep | = | [48] |
| 3820 m above sea level | 30–120 ED | sheep | = | [49] |
| 4000 m above sea level on first day, 5000 m above sea level on the second to fifth day | 14–18 ED, 8 h per day | rats | ↓ | [50] |
| chronic anaemia | NA | sheep | = | [51] |
| 9000 m above sea level; PaO2 42 mmHg | 14–19 ED, once 4 h | albino rats | ↓ | [52] |
| NA | 105–138 ED | sheep | ↓ | [53] |
2. Methodology
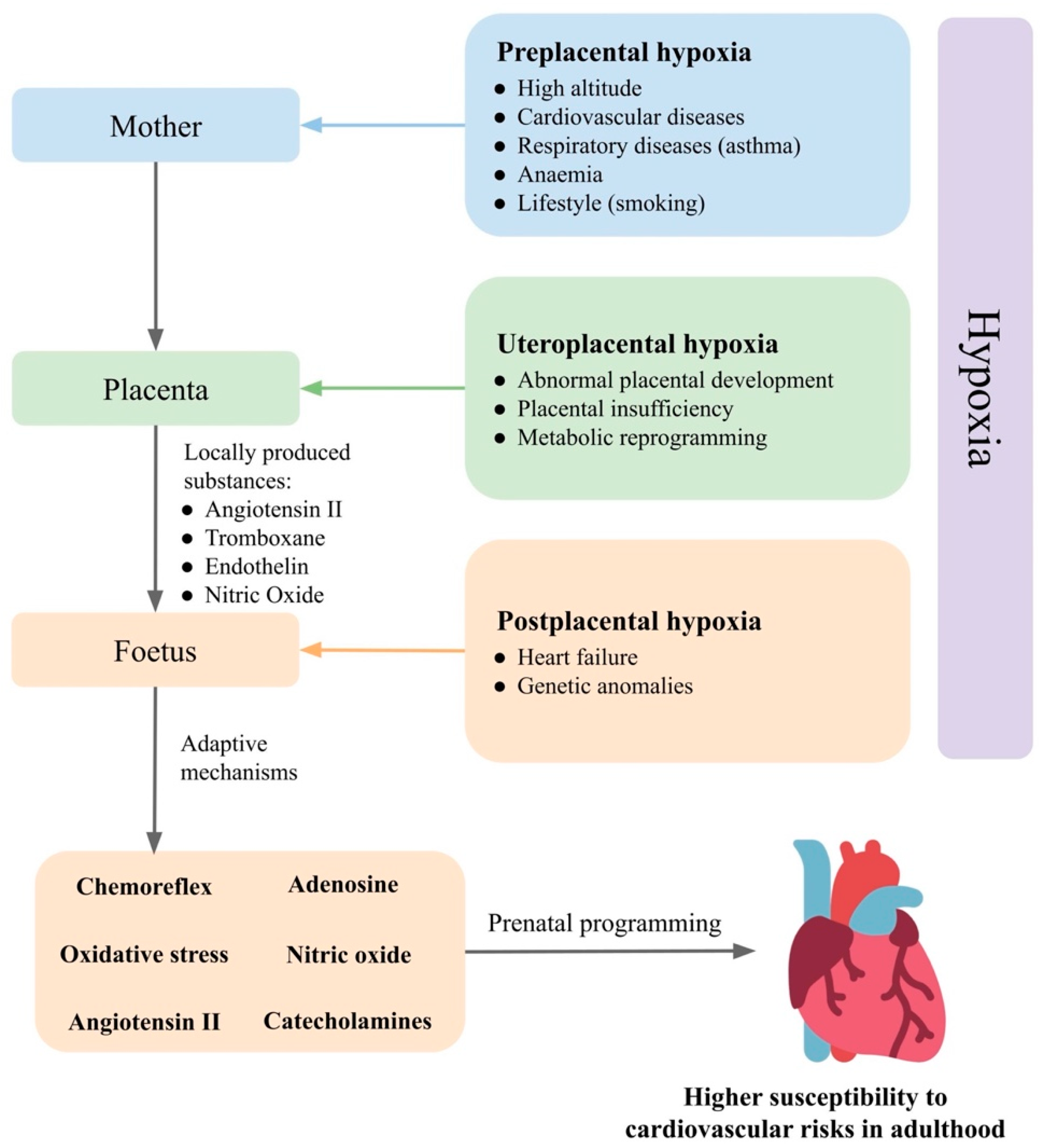
3. Prenatal Hypoxia
3.1. Foetus
- (1)
- Reflex response mediated by the chemoreflex and α1-adrenergic signalling in peripheral vessels [70,71,72]. Hypoxia also directly affects chromaffin cells in the adrenal medulla, thereby stimulating the release of catecholamines. Chromaffin cells of the adrenal medulla have a chemosensitive function until a sympathetic innervation develops between the adrenal glands and the central nervous system [73].
- (2)
- (3)
- (4)
- (5)
- Maternal factors that pass from the mother to the foetus. Environmental hypoxia, during which both the mother and the foetus are hypoxic, causes adaptive changes not only at the level of the foetus but also in the mother. Some of the mother’s adaptation mechanisms may also affect foetal development [81].
| Prenatal Hypoxia Type | Animal Model | Hypoxia Outcomes | Ref. | |
|---|---|---|---|---|
| Adenosine | Arterial PaO2 15 mmHg; 1 h | Sheep | Foetal acidosis, mean arterial pressure increase, a transient heart rate decrease | [77] |
| Hypoxia/anoxia; 20 or 60 min | A1R+/+, A1R+/− and A1R−/− C57BL mice, hippocampal slices, isolated brainstem spinal cord | Reduction in field excitatory postsynaptic potential | [82] | |
| 10% O2; 7.5–10.5 ED | A1AR+/+ and A1AR-deficient C57BL/6 mice | Growth retardation, less stabilized HIF-1α protein and cardiac gene expression in A1AR−/− embryos | [83] | |
| 10–12% O2; 30 min; 122–128 ED | Western sheep | Cortical blood flow increase, attenuated by a non-selective adenosine receptor antagonist | [84] | |
| NO, ROS | 13% O2; 6–20 ED | Wistar rats | Foetus: aortic thickening, enhanced nitrotyrosine staining and increased cardiac HSP70 expression. Adult offspring: impaired NO-dependent relaxation, increased myocardial contractility | [2] |
| 12% O2; for 4, 7, or 10 days; 58–62 ED | Hartley-Duncan guinea pigs | Increased eNOS mRNA in foetal ventricles, not altered K+-channel activation in response to acetylcholine-stimulated coronary dilation | [59] | |
| 40–50% uteroplacental artery ligation; 25 ED | New Zealand white rabbits | Normal left and right ventricular thickness, increased vessel dilatation; HIF-1α, eNOS, p-eNOS, and iNOS induction suggesting increased NO and oxidative stress in the hearts | [85] | |
| 13% O2; most of gestation (prior to day 5) | Wistar rats | Maternal and placental oxidative stress—prevented by maternal treatment with vitamin C | [42] | |
| 13% O2; 6–20 ED | Wistar rats | Increased LF/HF HRV ratio and baroreflex gain—prevented by vitamin C | [86] | |
| Acute: 10% O2; 0.5 h, 127 ± 1 ED; chronic: 10% O2; 105–138 ED | Welsh Mountain sheep | Mitochondria-derived oxidative stress, endothelial dysfunction and hypertension in adult offspring | [53] | |
| 6% O2; 0.5 h | Welsh Mountain sheep | Increased redistribution of blood flow and the glycemic and plasma catecholamine responses | [87] | |
| 14 ± 0.5% O2; 1–19 ED (embryos underwent euthanasia) | Bovans Brown eggs | Cardiac systolic dysfunction, impaired cardiac contractility and relaxability, increased cardiac sympathetic dominance, endothelial dysfunction in peripheral circulations | [88] | |
| Conceived, gestated, born and studied at Putre Research Station (3600 m above sea level) | Sheep (neonates) | Worsened carotid blood flow, vascular responses to potassium, serotonin, methacholine, and melatonin; diminished endothelial response via NO-independent mechanisms in isolated arteries | [89] | |
| 10.5% O2; 15–21 ED | Sprague Dawley rats | Revealed reprogramming of the mitochondrion | [90] | |
| 11% O2; 15–21 ED | Sprague Dawley rats | Male and female foetuses: increased oxidative stress in placentas; 7-month-old male and female offspring: cardiac diastolic dysfunction; 13-month-old female offspring: reduced vascular sensitivity to methacholine, 13-month-old male offspring: decreased vascular sensitivity to phenylephrine | [91] | |
| 13–14% O2; 6–20 ED | Wistar rats | Increased α1-adrenergic reactivity of the cardiovascular system, enhanced reactive hyperemia, sympathetic dominance, hypercontractility and diastolic dysfunction in the heart | [92] | |
| 7% O2; 2 h; 50–55 ED; foetal hearts were harvested at the end of hypoxia | Guinea pigs | Decreased heart ATP, lipid peroxides, 4-hydroxynonenal and malondialdehyde; increased apoptotic index, unremarkable foetal heart morphology, normal postpartum neonatal cardiac function and cerebral histology | [93] | |
| Acute: 220–240 mmHg; 10,000 m above sea level; 4–5 min; 18 ED–delivery; chronic: 280–300 mmHg; 8000 m above sea level); 2 h; 14 ED–delivery | C57BL/6 mice | Acute hypoxia: decreased basal O2 consumption rate and intensity of oxidative phosphorylation by the brain mitochondria of newborn, the activation of the respiratory complex II; chronic hypoxia: increased basal O2 consumption rate and oxidative phosphorylation intensity | [47] | |
| RAAS | 10.5% O2; 6–21 ED | Sprague Dawley rats | Foetal growth restriction, impaired trophoblast invasion and uteroplacental vascular remodeling, increased plasma ET-1 levels, prepro-ET-1 mRNA, ET-1 type A receptor and AT1 receptor in the kidney and placenta | [78] |
| 12% O2; from 14.5 ED | CD1 mice | Weaning: both sexes: increased susceptibility to salt-induced cardiac fibrosis; male: renal fibrosis by high salt, increased renal renin mRNA; 12 months: both sexes: increased renal renin mRNA expression and concentrations, male: increased AT1a mRNA expression | [94] | |
| 10.5% O2; 4–21 ED | Sprague Dawley rats | Increased superoxide production and decreased SOD expression, enhanced NADPH4, but not NADPH1 or NADPH2 in foetal aortas; increased Ang II-mediated vessel contractions in foetal thoracic aortas blocked by losartan | [33] | |
| Acute isocapnic hypoxaemia (foetal PaO2 12–14 mmHg); 1 h; 110/114–124/128 ED | Sheep foetuses | No effects in foetal heart rate, mean arterial pressure, baro- or chemoreflexes, femoral blood flow, femoral vascular resistance or foetal growth | [48] | |
| Reflex | Aortic PaO2 12–15 mmHg without alterations in foetal PaCO2; 1 h; 124 ED | Welsh Mountain sheep foetuses | Transient bradycardia, femoral vasoconstriction and increases in plasma noradrenaline and adrenaline; the NO clamp: persisted bradycardia, greater peripheral vasoconstrictor and catecholaminergic responses—enhanced the chemoreflex sensitivity | [70] |
| PaO2 15 mmHg; 137–144 ED | Border Leicester Merino cross sheep | Reduced and delayed the IA-type current | [73] | |
| Aortic PaO2 10–11 mmHg without alterations in foetal PaCO2; 1 h; 117–118 ED | Sheep foetuses | Bradycardia, increased arterial blood pressure, femoral vasoconstriction, blood glucose, lactate concentrations, plasma epinephrine and norepinephrine | [95] | |
| Foetal arterial oxygen saturation by 47.3% (uterine blood flow restriction); 118–126 ED | Sheep foetuses | Bradycardia, not in denervated foetuses, followed by a tachycardia; increased foetal heart rate in denervated foetuses; transiently increased foetal blood pressure in intact foetuses and decrease in denervated foetuses; increased cerebral blood flow in both intact and denervated foetuses; decreased carotid vascular resistance in denervated foetuses | [96] | |
| 10% O2; 5–20 ED | Sprague Dawley rats | Decreased dopamine content in the carotid bodies; until 3 weeks after birth: hyperventilation and disturbed metabolism | [31] | |
| 10% O2; 5–20 ED | Sprague Dawley rats | Evaluated resting ventilation and ventilatory response; periphery: reduced tyrosine hydroxylase activity within the first postnatal week and enhanced later; central areas: upregulated tyrosine hydroxylase activity within the first postnatal week and downregulated later | [27] |
3.1.1. Reflex Response
3.1.2. Adenosine
3.1.3. Nitric Oxide, A Reactive Oxygen Species
3.1.4. Renin-Angiotensin-Aldosterone System
3.1.5. Morphological-Functional Changes in the Heart and Blood Vessels
3.2. Placenta
3.3. Mother
4. Factors Affecting the Consequences of Hypoxia
4.1. Sex
4.2. Circadian Variability
4.2.1. Hypoxia-Inducible Factors
4.2.2. Consequences
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhatnagar, A. Environmental determinants of cardiovascular disease. Circ. Res. 2017, 121, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 2012, 7, e31017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, E.J.; Kim, Y.J. What is fetal programming? A lifetime health is under the control of in utero health. Obstet. Gynecol. Sci. 2017, 60, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Brameld, J.M.; Buttery, P.J.; Dawson, J.M.; Harper, J.M.M. Nutritional and hormonal control of skeletal-muscle cell growth and differentiation. Proc. Nutr. Soc. 1998, 57, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert-Barness, E. Teratogenic causes of malformations. Ann. Clin. Lab. Sci. 2010, 40, 99–114. [Google Scholar] [PubMed]
- Armengaud, J.B.; Yzydorczyk, C.; Siddeek, B.; Peyter, A.C.; Simeoni, U. Intrauterine growth restriction: Clinical consequences on health and disease at adulthood. Reprod. Toxicol. 2021, 99, 168–176. [Google Scholar] [CrossRef]
- Barker, D.J.P. The fetal and infant origins of adult disease. Br. Med. J. 1990, 301, 1111. [Google Scholar] [CrossRef] [Green Version]
- Brain, K.L.; Allison, B.J.; Niu, Y.; Cross, C.M.; Itani, N.; Kane, A.D.; Herrera, E.A.; Skeffington, K.L.; Botting, K.J.; Giussani, D.A. Intervention against hypertension in the next generation programmed by developmental hypoxia. PLoS Biol. 2019, 17, e2006552. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Qi, L.; Fan, X.; Tao, H.; Zhang, M.; Gao, Q.; Liu, Y.; Xu, T.; Zhang, P.; Su, H.; et al. Prenatal hypoxia affected endothelium-dependent vasodilation in mesenteric arteries of aged offspring via increased oxidative stress. Hypertens. Res. 2019, 42, 863–875. [Google Scholar] [CrossRef]
- Patterson, A.J.; Chen, M.; Xue, Q.; Xiao, D.; Zhang, L. Chronic Prenatal Hypoxia Induces Epigenetic Programming of PKCε Gene Repression in Rat Hearts. Circ. Res. 2010, 107, 365–373. [Google Scholar] [CrossRef] [Green Version]
- Walton, S.L.; Singh, R.R.; Tan, T.; Paravicini, T.M.; Moritz, K.M. Late gestational hypoxia and a postnatal high salt diet programs endothelial dysfunction and arterial stiffness in adult mouse offspring. J. Physiol. 2016, 594, 1451–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, E.A.; Camm, E.J.; Cross, C.M.; Mullender, J.L.; Wooding, F.B.P.; Giussani, D.A. Morphological and Functional Alterations in the Aorta of the Chronically Hypoxic Fetal Rat. J. Vasc. Res. 2011, 49, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Peyronnet, J.; Dalmaz, Y.; Ehrström, M.; Mamet, J.; Roux, J.C.; Pequignot, J.M.; Thorén, P.H.; Lagercrantz, H. Long-lasting adverse effects of prenatal hypoxia on developing autonomic nervous system and cardiovascular parameters in rats. Pflugers Archiv. Eur. J. Physiol. 2002, 443, 858–865. [Google Scholar] [CrossRef]
- Svitok, P.; Molcan, L.; Stebelova, K.; Vesela, A.; Sedlackova, N.; Ujhazy, E.; Mach, M.; Zeman, M. Prenatal hypoxia in rats increased blood pressure and sympathetic drive of the adult offspring. Hypertens. Res. 2016, 39, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Rook, W.; Johnson, C.D.; Coney, A.M.; Marshall, J.M. Prenatal hypoxia leads to increased muscle sympathetic nerve activity, sympathetic hyperinnervation, premature blunting of neuropeptide y signaling, and hypertension in adult life. Hypertension 2014, 64, 1321–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostadal, B.; Ostadalova, I.; Szarszoi, O.; Netuka, I.; Olejnickova, V.; Hlavackova, M. Sex-dependent effect of perinatal hypoxia on cardiac tolerance to oxygen deprivation in adults. Can. J. Physiol. Pharmacol. 2021, 99, 1–8. [Google Scholar] [CrossRef]
- Giussani, D.A.; Davidge, S.T. Developmental programming of cardiovascular disease by prenatal hypoxia. J. Dev. Origins Health Dis. 2013, 4, 328–337. [Google Scholar] [CrossRef]
- Iqbal, W.; Ciriello, J. Effect of maternal chronic intermittent hypoxia during gestation on offspring growth in the rat. Am. J. Obstet. Gynecol. 2013, 209, 564.e1–564.e9. [Google Scholar] [CrossRef]
- Zhuravin, I.A.; Dubrovskaya, N.M.; Tumanova, N.L. Postnatal physiological development of rats after acute prenatal hypoxia. Neurosci. Behav. Physiol. 2004, 34, 809–816. [Google Scholar] [CrossRef]
- Dubrovskaya, N.M.; Zhuravin, I.A. Ontogenetic characteristics of behavior in rats subjected to hypoxia on day 14 or day 18 of embryogenesis. Neurosci. Behav. Physiol. 2010, 40, 231–238. [Google Scholar] [CrossRef]
- Hermans, R.H.M.; Longo, L.D.; Mcgivern, R.F. Decreased postnatal testosterone and corticosterone concentrations in rats following acute intermittent prenatal hypoxia without alterations in adult male sex behavior. Neurotoxicol. Teratol. 1994, 16, 201–206. [Google Scholar] [CrossRef]
- Minior, V.K.; Levine, B.; Ferber, A.; Guller, S.; Divon, M.Y. Nucleated red blood cells as a marker of acute and chronic fetal hypoxia in a rat model. Rambam Maimonides Med. J. 2017, 8, e0025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, O.; Daire, J.L.; Dalmaz, Y.; Fontaine, R.H.; Krueger, R.C.; Sebag, G.; Evrard, P.; Gressens, P.; Verney, C. Gestational hypoxia induces white matter damage in neonatal rats: A new model of periventricular leukomalacia. Brain Pathol. 2004, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joseph, V.; Mamet, J.; Lee, F.; Dalmaz, Y.; van Reeth, O. Prenatal hypoxia impairs circadian synchronisation and response of the biological clock to light in adult rats. J. Physiol. 2002, 543, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Mamet, J.; Peyronnet, J.; Roux, J.C.; Perrin, D.; Cottet-Emard, J.M.; Pequignot, J.M.; Lagercrantz, H.; Dalmaz, Y. Long-term prenatal hypoxia alters maturation of adrenal medulla in rat. Pediatr. Res. 2002, 51, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Perrin, D.; Mamet, J.; Scarna, H.; Roux, J.C.; Bérod, A.; Dalmaz, Y. Long-term prenatal hypoxia alters maturation of brain catecholaminergic systems and motor behavior in rats. Synapse 2004, 54, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Peyronnet, J.; Roux, J.C.; Mamet, J.; Perrin, D.; Lachuer, J.; Pequignot, J.M.; Dalmaz, Y. Developmental plasticity of the carotid chemoafferent pathway in rats that are hypoxic during the prenatal period. Eur. J. Neurosci. 2007, 26, 2865–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camm, E.J.; Hansell, J.A.; Kane, A.D.; Herrera, E.A.; Lewis, C.; Wong, S.; Morrell, N.W.; Giussani, D.A. Partial contributions of developmental hypoxia and undernutrition to prenatal alterations in somatic growth and cardiovascular structure and function. Am. J. Obstet. Gynecol. 2010, 203, 495.e24–495.e34. [Google Scholar] [CrossRef]
- Camm, E.J.; Martin-Gronert, M.S.; Wright, N.L.; Hansell, J.A.; Ozanne, S.E.; Giussani, D.A. Prenatal hypoxia independent of undernutrition promotes molecular markers of insulin resistance in adult offspring. FASEB J. 2011, 25, 420–427. [Google Scholar] [CrossRef]
- Allison, B.J.; Brain, K.L.; Niu, Y.; Kane, A.D.; Herrera, E.A.; Thakor, A.S.; Botting, K.J.; Cross, C.M.; Itani, N.; Shaw, C.J.; et al. Altered cardiovascular defense to hypotensive stress in the chronically hypoxic fetus. Hypertension 2020, 76, 1195–1207. [Google Scholar] [CrossRef]
- Peyronnet, J.; Roux, J.C.; Géloën, A.; Tang, L.Q.; Pequignot, J.M.; Lagercrantz, H.; Dalmaz, Y. Prenatal hypoxia, impairs the postnatal development of neural and functional chemoafferent pathway in rat. J. Physiol. 2000, 524, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Hermans, R.H.M.; Hunter, D.E.; Mcgivern, R.F.; Cain, C.D.; Longo, L.D. Behavioral sequelae in young rats of acute intermittent antenatal hypoxia. Neurotoxicol. Teratol. 1992, 14, 119–129. [Google Scholar] [CrossRef]
- Zhu, X.; Gao, Q.; Tu, Q.; Zhong, Y.; Zhu, D.; Mao, C.; Xu, Z. Prenatal hypoxia enhanced angiotensin II-mediated vasoconstriction via increased oxidative signaling in fetal rats. Reprod. Toxicol. 2016, 60, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Bae, S.; Zhang, L. Effect of prenatal hypoxia on heat stress-mediated cardioprotection in adult rat heart. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1712–H1719. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Chen, W.; Ostrowski, R.P.; Ma, Q.; Souvenir, R.; Zhang, L.; Zhang, J.H.; Tang, J. Maternal hypoxia increases the activity of MMPs and decreases the expression of TIMPs in the brain of neonatal rats. Dev. Neurobiol. 2010, 70, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.W.; Larcombe, A.N.; Berry, L.J.; Morton, J.S.; Davidge, S.T.; James, A.L.; Noble, P.B. Foetal growth restriction in mice modifies postnatal airway responsiveness in an age and sex-dependent manner. Clin. Sci. 2018, 132, 273–284. [Google Scholar] [CrossRef]
- Thompson, L.P.; Chen, L.; Polster, B.M.; Pinkas, G.; Song, H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1232–R1241. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, Z.; Lu, G.; Lin, L.; Ferrari, M. Hypoxia during pregnancy in rats leads to early morphological changes of atherosclerosis in adult offspring. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1321–H1328. [Google Scholar] [CrossRef] [Green Version]
- Bourque, S.L.; Gragasin, F.S.; Quon, A.L.; Mansour, Y.; Morton, J.S.; Davidge, S.T. Prenatal hypoxia causes long-term alterations in vascular endothelin-1 function in aged male, but not female, offspring. Hypertension 2013, 62, 753–758. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.W.; Morton, J.S.; Davidge, S.T.; Larcombe, A.N.; James, A.L.; Donovan, G.M.; Noble, P.B. Increased heterogeneity of airway Calibre in adult rats after hypoxia-induced intrauterine growth restriction. Respirology 2017, 22, 1329–1335. [Google Scholar] [CrossRef]
- Vargas, V.E.; Gurung, S.; Grant, B.; Hyatt, K.; Singleton, K.; Myers, S.M.; Saunders, D.; Njoku, C.; Towner, R.; Myers, D.A. Gestational hypoxia disrupts the neonatal leptin surge and programs hyperphagia and obesity in male offspring in the sprague-dawley rat. PLoS ONE 2017, 12, e0185272. [Google Scholar] [CrossRef] [PubMed]
- Richter, H.G.; Camm, E.J.; Modi, B.N.; Naeem, F.; Cross, C.M.; Cindrova-Davies, T.; Spasic-Boskovic, O.; Dunster, C.; Mudway, I.S.; Kelly, F.J.; et al. Ascorbate prevents placental oxidative stress and enhances birth weight in hypoxic pregnancy in rats. J. Physiol. 2012, 590, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, A.M.; Camm, E.J.; Sferruzzi-Perri, A.N.; Ashmore, T.J.; Yung, H.W.; Cindrova-Davies, T.; Spiroski, A.M.; Sutherland, M.R.; Logan, A.; Austin-Williams, S.; et al. Placental adaptation to early-onset hypoxic pregnancy and mitochondria-targeted antioxidant therapy in a rodent model. Am. J. Pathol. 2018, 188, 2704–2716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellgren, K.T.; Premanandhan, H.; Quinn, C.J.; Trafford, A.W.; Galli, G.L.J. Sex-dependent effects of developmental hypoxia on cardiac mitochondria from adult murine offspring. Free Rad. Biol. Med. 2021, 162, 490–499. [Google Scholar] [CrossRef]
- Wang, R.; Xu, F.; Liu, J. prenatal hypoxia preconditioning improves hypoxic ventilatory response and reduces mortality in neonatal rats. J. Perinat. Med. 2008, 36, 161–167. [Google Scholar] [CrossRef]
- Golan, H.; Kashtutsky, I.; Hallak, M.; Sorokin, Y.; Huleihel, M. Maternal hypoxia during pregnancy delays the development of motor reflexes in newborn mice. Dev. Neurosci. 2004, 26, 24–29. [Google Scholar] [CrossRef]
- Urazov, D.; Astrakhanova, T.A.; Usenko, V.; Mishchenko, T.A.; Schelchkova, N.; Kravchenko, G.A.; Vedunova, V.; Mitroshina, E.V. New aspects of central nervous system adaptation to prenatal hypoxia. Sovremennye Tehnol. Med. 2018, 10, 60–68. [Google Scholar] [CrossRef]
- Steyn, C.; Hanson, M.A. The effect of repeated acute hypoxaemia on fetal cardiovascular development in the sheep. J. Physiol. 1998, 512, 295–306. [Google Scholar] [CrossRef]
- Kamitomo, M.; Longo, L.D.; Gilbert, R.D. Right and left ventricular function in fetal sheep exposed to long-term high-altitude hypoxemia. Am. J. Physiol. Heart Circ. Physiol. 1992, 262, H399–H405. [Google Scholar] [CrossRef]
- Ostádalová, I.; Ostádal, B.; Kolár, F. Effect of prenatal hypoxia on contractile performance and responsiveness to Ca2+ in the isolated perinatal rat heart. Physiol. Res. Acad. Sci. Bohemoslov. 1995, 44, 135–137. [Google Scholar]
- Davis, L.E.; Hohimer, A.R. Hemodynamics and organ blood flow in fetal sheep subjected to chronic anemia. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 1991, 261, R1542–R1548. [Google Scholar] [CrossRef] [PubMed]
- Zubenko, S.I.; Yan, L.; Zhul’kov, M.O.; Lebed’ko, O.A.; Sazonova, E.N. Effects of antenatal hypoxia on tissue homeostasis in the myocardium of albino rats: Early and delayed consequences. Bull. Exp. Biol. Med. 2014, 157, 320–323. [Google Scholar] [CrossRef] [PubMed]
- Botting, K.J.; Skeffington, K.L.; Niu, Y.; Allison, B.J.; Brain, K.L.; Itani, N.; Beck, C.; Logan, A.; Murray, A.J.; Murphy, M.P.; et al. Translatable mitochondria-targeted protection against programmed cardiovascular dysfunction. Sci. Adv. 2020, 6, eabb1929. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Prabhakar, N.R. neural regulation of hypoxia-inducible factors and redox state drives the pathogenesis of hypertension in a rodent model of sleep apnea. J. Appl. Physiol. 2015, 119, 1152–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; di Lorenzo, A.; Jiang, W.; Cantalupo, A.; Sessa, W.C.; Giordano, F.J. Hypoxia-inducible factor-1α in vascular smooth muscle regulates blood pressure homeostasis through a peroxisome proliferator-activated receptor-γ-angiotensin II receptor type 1 axis. Hypertension 2013, 62, 634–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Wang, Z.; Xia, M.; Li, P.L.; van Tassell, B.W.; Abbate, A.; Dhaduk, R.; Li, N. Silencing of hypoxia-inducible factor-1α gene attenuated angiotensin II-induced renal injury in sprague-dawley rats. Hypertension 2011, 58, 657–664. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.F.; Wang, Z.H.; Chen, Z.K.; Lv, G.R.; Ferrari, M. Intermittent maternal hypoxia has an influence on regional expression of endothelial nitric oxide synthase in fetal arteries of rabbits. Pediatr. Res. 2013, 73, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Rood, K.; Lopez, V.; la Frano, M.R.; Fiehn, O.; Zhang, L.; Blood, A.B.; Wilson, S.M. Gestational hypoxia and programing of lung metabolism. Front. Physiol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Thompson, L.P.; Aguan, K.; Pinkas, G.; Weiner, C.P. Chronic hypoxia increases the NO contribution of acetylcholine vasodilation of the fetal guinea pig heart. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2000, 279, R1813–R1820. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, Y.; Jin, F.; Sun, X.; Li, Z.; Liu, Y.; Fang, P.; Shi, H.; Jiang, X. Endothelin-1 induces hypoxia inducible factor 1α expression in pulmonary artery smooth muscle cells. FEBS Lett. 2012, 586. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.L.; Luo, C.W.; Dai, Z.K.; Shaw, K.P.; Chai, C.Y.; Wu, C.C. Hypoxia-inducible factor-1α, vascular endothelial growth factor, inducible nitric oxide synthase, and endothelin-1 expression correlates with angiogenesis in congenital heart disease. Kaohsiung J. Med. Sci. 2016, 32, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Tang, D.; Liu, N.; Xiong, W.; Huang, H.; Li, Y.; Ma, Z.; Zhao, H.; Chen, P.; Qi, X.; et al. Reciprocal regulation between the circadian clock and hypoxia signaling at the genome level in mammals. Cell Metab. 2017, 25, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Zlacká, J.; Zeman, M. Glycolysis under circadian control. Int. J. Molec. Sci. 2021, 22, 13666. [Google Scholar] [CrossRef] [PubMed]
- Hutter, D.; Kingdom, J.; Jaeggi, E. Causes and mechanisms of intrauterine hypoxia and its impact on the fetal cardiovascular system: A review. Int. J. Pediatr. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nye, G.A.; Ingram, E.; Johnstone, E.D.; Jensen, O.E.; Schneider, H.; Lewis, R.M.; Chernyavsky, I.L.; Brownbill, P. Human placental oxygenation in late gestation: Experimental and theoretical approaches. J. Physiol. 2018, 596, 5523–5534. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hashmi, M. Partial pressure of oxygen measurement. Antioxidants 2020, 9, 414. [Google Scholar] [CrossRef]
- Vali, P.; Lakshminrusimha, S. The fetus can teach us: Oxygen and the pulmonary vasculature. Children 2017, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.M.; Mitchell, M.D.; Kumar, S.S. The physiology of intrapartum fetal compromise at term. Am. J. Obstet. Gynecol. 2020, 222, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Parer, J.T. The effect of acute maternal hypoxia on fetal oxygenation and the umbilical circulation in the sheep. Eur. J. Obstet. Gynecol. Reprod. Biol. 1980, 10, 125–136. [Google Scholar] [CrossRef]
- Morrison, S.; Gardner, D.S.; Fletcher, A.J.W.; Bloomfield, M.R.; Giussani, D.A. Enhanced nitric oxide activity offsets peripheral vasoconstriction during acute hypoxaemia via chemoreflex and adrenomedullary actions in the sheep fetus. J. Physiol. 2003, 547, 283–291. [Google Scholar] [CrossRef]
- Giussani, D.A. The fetal brain sparing response to hypoxia: Physiological mechanisms. J. Physiol. 2016, 594, 1215–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, A.D.; Herrera, E.A.; Hansell, J.A.; Giussani, D.A. Statin treatment depresses the fetal defence to acute hypoxia via increasing nitric oxide bioavailability. J. Physiol. 2012, 590, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Rychkov, G.Y.; Adams, M.B.; McMillen, I.C.; Roberts, M.L. Oxygen sensing mechanisms are present in the chromaffin cells of the sheep adrenal medulla before birth. J. Physiol. 1998, 509, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Hou, J.; Ge, J.; Hu, Y.; Ding, Y.; Zhou, Y.; Zhang, H.; Xu, Z.; Zhang, L. Changes of renal AT1/AT2 receptors and structures in ovine fetuses following exposure to long-term hypoxia. Am. J. Nephrol. 2010, 31, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Li, N.; Chen, X.; Gao, Q.; Zhou, X.; Zhang, Y.; Liu, B.; Sun, M.; Xu, Z. Prenatal hypoxia induced dysfunction in cerebral arteries of offspring rats. J. Am. Heart Assoc. 2017, 6, e006630. [Google Scholar] [CrossRef] [Green Version]
- Lane, S.L.; Doyle, A.S.; Bales, E.S.; Lorca, R.A.; Julian, C.G.; Moore, L.G. Increased uterine artery blood flow in hypoxic murine pregnancy is not sufficient to prevent fetal growth restriction. Biol. Reprod. 2020, 102, 660–670. [Google Scholar] [CrossRef]
- Koos, B.J.; Chau, A.; Ogunyemi, D. Adenosine mediates metabolic and cardiovascular responses to hypoxia in fetal sheep. J. Physiol. 1995, 488, 761–766. [Google Scholar] [CrossRef]
- Zhou, J.; Xiao, D.; Hu, Y.; Wang, Z.; Paradis, A.; Mata-Greenwood, E.; Zhang, L. Gestational hypoxia induces preeclampsia-like symptoms via heightened endothelin-1 signaling in pregnant rats. Hypertension 2013, 62, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Kurlak, L.O.; Williams, P.J.; Bulmer, J.N.; Broughton Pipkin, F.; Mistry, H.D. Placental expression of adenosine A2A receptor and hypoxia inducible factor-1 alpha in early pregnancy, term and pre-eclamptic pregnancies: Interactions with placental renin-angiotensin system. Placenta 2015, 36, 611–613. [Google Scholar] [CrossRef]
- Benoit, C.; Zavecz, J.; Wang, Y. Vasoreactivity of chorionic plate arteries in response to vasoconstrictors produced by preeclamptic placentas. Placenta 2007, 28, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Cuffe, J.S.M.; Walton, S.L.; Singh, R.R.; Spiers, J.G.; Bielefeldt-Ohmann, H.; Wilkinson, L.; Little, M.H.; Moritz, K.M. Mid- to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex-specific manner. J. Physiol. 2014, 592, 3127–3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, B.; Halldner, L.; Dunwiddie, T.V.; Masino, S.A.; Poelchen, W.; Giménez-Llort, L.; Escorihuela, R.M.; Fernández-Teruel, A.; Wiesenfeld-Hallin, Z.; Xu, X.J.; et al. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 9407–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendler, C.C.; Amatya, S.; McClaskey, C.; Ghatpande, S.; Fredholm, B.B.; Rivkees, S.A. A1 adenosine receptors play an essential role in protecting the embryo against hypoxia. Proc. Natl. Acad. Sci. USA 2007, 104, 9697–9702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blood, A.B.; Hunter, C.J.; Power, G.G. Adenosine mediates decreased cerebral metabolic rate and increased cerebral flow during acute moderate hypoxia in the near-term fetal sheep. J. Physiol. 2003, 553, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, H.; Alvarado, C.; Cifuentes, J.; Lozano, M.; Rocco, J.; Cabezas, C.; Illanes, S.E.; Eixarch, E.; Hernández-Andrade, E.; Gratacós, E.; et al. Oxidative damage and nitric oxide synthase induction by surgical uteroplacental circulation restriction in the rabbit fetal heart. Prenat. Diagn. 2017, 37, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.D.; Herrera, E.A.; Camm, E.J.; Giussani, D.A. Vitamin C prevents intrauterine programming of in vivo cardiovascular dysfunction in the rat. Circ. J. 2013, 77, 2604–2611. [Google Scholar] [CrossRef] [Green Version]
- Thakor, A.S.; Allison, B.J.; Niu, Y.; Botting, K.J.; Serõn-Ferré, M.; Herrera, E.A.; Giussani, D.A. Melatonin modulates the fetal cardiovascular defense response to acute hypoxia. J. Pineal Res. 2015, 59, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Itani, N.; Skeffington, K.L.; Beck, C.; Niu, Y.; Giussani, D.A. Melatonin rescues cardiovascular dysfunction during hypoxic development in the chick embryo. J. Pineal Res. 2016, 60, 16–26. [Google Scholar] [CrossRef]
- Herrera, E.A.; Macchiavello, R.; Montt, C.; Ebensperger, G.; Díaz, M.; Ramírez, S.; Parer, J.T.; Serõn-Ferré, M.; Reyes, R.V.; Llanos, A.J. Melatonin improves cerebrovascular function and decreases oxidative stress in chronically hypoxic lambs. J. Pineal Res. 2014, 57, 33–42. [Google Scholar] [CrossRef]
- Gao, Y.; Dasgupta, C.; Huang, L.; Song, R.; Zhang, Z.; Zhang, L. Multi-omics integration reveals short and long-term effects of gestational hypoxia on the heart development. Cells 2019, 8, 1608. [Google Scholar] [CrossRef] [Green Version]
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Maternal treatment with a placental-targeted antioxidant (MitoQ) impacts offspring cardiovascular function in a rat model of prenatal hypoxia. Pharmacol. Res. 2018, 134, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Spiroski, A.; Niu, Y.; Nicholas, L.M.; Austin-Williams, S.; Camm, E.J.; Sutherland, M.R.; Ashmore, T.J.; Skeffington, K.L.; Logan, A.; Ozanne, S.E.; et al. Mitochondria antioxidant protection against cardiovascular dysfunction programmed by early-onset gestational hypoxia. FASEB J. 2021, 35, e21446. [Google Scholar] [CrossRef] [PubMed]
- Powell, S.R.; Mantell, L.L.; Garramone, V.; Drexler, S.; Teichberg, S. Acute effects of nonlethal in utero hypoxia on fetal guinea pig heart and lack of persistent cardiac or cerebral effects in the neonate. Biol. Neonate 2004, 86, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Walton, S.L.; Bielefeldt-Ohmann, H.; Singh, R.R.; Li, J.; Paravicini, T.M.; Little, M.H.; Moritz, K.M. Prenatal hypoxia leads to hypertension, renal renin-angiotensin system activation and exacerbates salt-induced pathology in a sex-specific manner. Sci. Rep. 2017, 7, 8241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giussani, D.A.; Gardner, D.S.; Cox, D.T.; Fletcher, A.J.W. Purinergic contribution to circulatory, metabolic, and adrenergic responses to acute hypoxemia in fetal sheep. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2001, 280, R678–R685. [Google Scholar] [CrossRef] [Green Version]
- Stein, P.; White, S.E.; Homan, J.; Hanson, M.A.; Bocking, A.D. Altered fetal cardiovascular responses to prolonged hypoxia after sinoaortic denervation. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 1999, 276, R340–R346. [Google Scholar] [CrossRef]
- DeSesso, J.M. Comparative gestational milestones in vertebrate development. In Developmental and Reproductive Toxicology; CRC Press: Boca Raton, FL, USA, 2016; pp. 107–152. [Google Scholar]
- Zeman, M.; Herichová, I. circadian melatonin production develops faster in birds than in mammals. Gen. Comp. Endocrinol. 2011, 172, 23–30. [Google Scholar] [CrossRef]
- Sekulić, S.; Božić, K.; Bozić, A.; Borota, J.; Ćulić, M. Precocial rodents as new experimental model to study the effects of altered gravitational conditions on fetal development. Microgravity Sci. Technol. 2006, 18, 223–225. [Google Scholar] [CrossRef]
- Carter, A.M. Animal models of human pregnancy and placentation: Alternatives to the mouse. Reproduction 2020, 160, R129–R143. [Google Scholar] [CrossRef]
- Kline, D.D.; Peng, Y.J.; Manalo, D.J.; Semenza, G.L.; Prabhakar, N.R. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. Proc. Natl. Acad. Sci. USA 2002, 99, 821–826. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.J.; Nanduri, J.; Khan, S.A.; Yuan, G.; Wang, N.; Kinsman, B.; Vaddi, D.R.; Kumar, G.K.; Garcia, J.A.; Semenza, G.L.; et al. Hypoxia-inducible factor 2α (HIF-2α) heterozygous-null mice exhibit exaggerated carotid body sensitivity to hypoxia, breathing instability, and hypertension. Proc. Natl. Acad. Sci. USA 2011, 108, 3065–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scortegagna, M.; Ding, K.; Oktay, Y.; Gaur, A.; Thurmond, F.; Yan, L.-J.; Marck, B.T.; Matsumoto, A.M.; Shelton, J.M.; Richardson, J.A.; et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1−/− mice. Nat. Genet. 2003, 35, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.V.; Kotch, L.E.; Agani, F.; Leung, S.W.; Laughner, E.; Wenger, R.H.; Gassmann, M.; Gearhart, J.D.; Lawler, A.M.; Yu, A.Y.; et al. Cellular and developmental control of o 2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998, 12, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournaud, R.; Hidalgo, J.; Yu, H.; Girard, E.; Shimahara, T. Catecholamine secretion from rat foetal adrenal chromaffin cells and hypoxia sensitivity. Pflugers Archiv. Eur. J. Physiol. 2007, 454, 83–92. [Google Scholar] [CrossRef]
- Levitsky, K.L.; López-Barneo, J. Developmental change of T-type Ca2+ channel expression and its role in rat chromaffin cell responsiveness to acute hypoxia. J. Physiol. 2009, 587, 1917–1929. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Zhao, Z.; Porter, G.; Turner, C. Influences of adenosine on the fetus and newborn. Molec. Genet. Metab. 2001, 74, 160–171. [Google Scholar] [CrossRef]
- Rivkees, S.A.; Wendler, C.C. Regulation of cardiovascular development by adenosine and adenosine-mediated embryo protection. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Koos, B.J. Adenosine A 2a receptors and O 2 sensing in development. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2011, 301, R601–R622. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, K.L.; Reller, M.D. Coronary flow regulation in the fetal sheep. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 1999, 277, R1249–R1260. [Google Scholar] [CrossRef]
- Hilaire, C.S.T.; Carroll, S.H.; Chen, H.; Ravid, K. Mechanisms of induction of adenosine receptor genes and its functional significance. J. Cell. Physiol. 2009, 218, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.D.; Kothmann, E.; Giussani, D.A. Detection and response to acute systemic hypoxia. BJA Educ. 2020, 20, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Góngora, M.C.; Harrison, D.G.; Lambeth, J.D.; Dikalov, S.; Griendling, K.K. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via ENOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H673–H679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, E.E.; Almeida, V.R.; Amaral, F.G.; Simon, K.A.; Futuro-Neto, H.A.; Pontes, R.B.; Cespedes, J.G.; Campos, R.R.; Bergamaschi, C.T. Melatonin attenuates renal sympathetic overactivity and reactive oxygen species in the brain in neurogenic hypertension. Hypertens. Res. 2019, 42, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Shi, L.; Xu, F.; Zhang, L.; Xu, Z. Development of fetal brain renin-angiotensin system and hypertension programmed in fetal origins. Prog. Neurobiol. 2009, 87, 252–263. [Google Scholar] [CrossRef] [Green Version]
- Carey, R.M.; Wang, Z.-Q.; Siragy, H.M. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 2000, 35, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.; Zhang, L. Fetal hypoxia and programming of matrix metalloproteinases. Drug Discov. Today 2012, 17. [Google Scholar] [CrossRef] [Green Version]
- Irani, R.A.; Zhang, Y.; Blackwell, S.C.; Zhou, C.C.; Ramin, S.M.; Kellems, R.E.; Xia, Y. The detrimental role of angiotensin receptor agonistic autoantibodies in intrauterine growth restriction seen in preeclampsia. J. Exp. Med. 2009, 206, 2809–2822. [Google Scholar] [CrossRef]
- Moritz, K.M. The effect of the in utero environment on nephrogenesis and renal function. In Kidney Development, Disease, Repair and Regeneration; Elsevier: Amsterdam, The Netherlands, 2015; pp. 177–190. ISBN 9780128004388. [Google Scholar]
- Clark, A.T.; Young, R.J.; Bertram, J.F. In vitro studies on the roles of transforming growth factor-Γ1 in rat metanephric development. Kidney Int. 2001, 59, 1641–1653. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, H.; Nigam, S.K. In vitro branching Tubulogenesis: Implications for developmental and cystic disorders, nephron number, renal repair, and nephron engineering. Kidney Int. 1998, 54, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.R.; Moritz, K.M.; Bertram, J.F.; Cullen-McEwen, L.A. Effects of dexamethasone exposure on rat metanephric development: In vitro and in vivo studies. Am. J. Physiol. Renal Physiol. 2007, 293, F548–F554. [Google Scholar] [CrossRef] [Green Version]
- Hinchliffe, S.A.; Howard, C.V.; Lynch, M.R.J.; Sargent, P.H.; Judd, B.A.; van Velzen, D. Renal developmental arrest in sudden infant death syndrome. Fetal Pediatr. Pathol. 1993, 13, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, T.A.; Saleh, J.L.; McCook, E.C.; Seidler, F.J. Impaired cardiac function during postnatal hypoxia in rats exposed to nicotine prenatally: Implications for perinatal morbidity and mortality, and for sudden infant death syndrome. Teratology 1997, 55, 177–184. [Google Scholar] [CrossRef]
- Ton, A.T.; Biet, M.; Delabre, J.F.; Morin, N.; Dumaine, R. In-utero exposure to nicotine alters the development of the rabbit cardiac conduction system and provides a potential mechanism for sudden infant death syndrome. Arch. Toxicol. 2017, 91, 3947–3960. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Skilton, M.R.; Crispi, F. Human fetal growth restriction: A cardiovascular journey through to adolescence. J. Dev. Origins Health Dis. 2016, 7, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.L.; Botting, K.J.; Dyer, J.L.; Williams, S.J.; Thornburg, K.L.; McMillen, I.C. Restriction of placental function alters heart development in the sheep fetus. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2007, 293, R306–R313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.W.; Zhang, L.; McMillen, I.C.; Botting, K.J.; Duffield, J.A.; Zhang, S.; Suter, C.M.; Brooks, D.A.; Morrison, J.L. Fetal growth restriction and the programming of heart growth and cardiac insulin-like growth factor 2 expression in the lamb. J. Physiol. 2011, 589, 4709–4722. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.M.; Hennessy-Strahs, S.; McGovern, P.E.; Mejaddam, A.Y.; Rossidis, A.C.; Baumgarten, H.D.; Bansal, E.; Villeda, M.; Han, J.; Gou, Z.; et al. Fetal hypoxemia causes abnormal myocardial development in a preterm ex utero fetal ovine model. JCI Insight 2018, 3, e124338. [Google Scholar] [CrossRef] [Green Version]
- Ream, M.; Ray, A.M.; Chandra, R.; Chikaraishi, D.M. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2008, 295, R583–R595. [Google Scholar] [CrossRef] [Green Version]
- Brain, K.L.; Allison, B.J.; Niu, Y.; Cross, C.M.; Itani, N.; Kane, A.D.; Herrera, E.A.; Giussani, D.A. Induction of controlled hypoxic pregnancy in large mammalian species. Physiol. Rep. 2015, 3, e12614. [Google Scholar] [CrossRef] [Green Version]
- Sutovska, H.; Molcan, L.; Koprdova, R.; Piesova, M.; Mach, M.; Zeman, M. Prenatal hypoxia increases blood pressure in male rat offspring and affects their response to artificial light at night. J. Dev. Origins Health Dis. 2021, 12, 587–594. [Google Scholar] [CrossRef]
- Giussani, D.; Lakshman, R.; Spiroski, A.; McIver, L.; Murphy, M. Non-invasive biomarkers for cardiovascular dysfunction programmed in male offspring of adverse pregnancy. Hypertension 2021, 78, 1818–1828. [Google Scholar] [CrossRef]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental origins of chronic disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, S.; Torricos, T.; Fik, E.; Oyala, M.; Echalar, L.; Pullockaran, J.; Tutino, E.; Martin, B.; Belliappa, S.; Balanza, E.; et al. Hypoglycemia and the origin of hypoxia-induced reduction in human fetal growth. PLoS ONE 2010, 5, e8551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, L.C.; Debant, M.; Walker, J.J.; Beech, D.J.; Simpson, N.A.B. Placental blood flow sensing and regulation in fetal growth restriction. Placenta 2021, 113, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Labarrere, C.A.; DiCarlo, H.L.; Bammerlin, E.; Hardin, J.W.; Kim, Y.M.; Chaemsaithong, P.; Haas, D.M.; Kassab, G.S.; Romero, R. Failure of physiologic transformation of spiral arteries, endothelial and trophoblast cell activation, and acute atherosis in the basal plate of the placenta. Am. J. Obstet. Gynecol. 2017, 216, 287.e1–287.e16. [Google Scholar] [CrossRef] [Green Version]
- Cahill, L.S.; Rennie, M.Y.; Hoggarth, J.; Yu, L.X.; Rahman, A.; Kingdom, J.C.; Seed, M.; Macgowan, C.K.; Sled, J.G. Feto- and utero-placental vascular adaptations to chronic maternal hypoxia in the mouse. J. Physiol. 2018, 596, 3285–3297. [Google Scholar] [CrossRef]
- Hung, T.-H.; Burton, G.J. Hypoxia and reoxygenation: A possible mechanism for placental oxidative stress in preeclampsia. Taiwan. J. Obstet. Gynecol. 2006, 45, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy, V.; Prasad, P.D. Role of transporters in placental transfer of drugs. Toxicol. Appl. Pharmacol. 2005, 207, 381–387. [Google Scholar] [CrossRef]
- Carter, A.M. Placental gas exchange and the oxygen supply to the fetus. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2015; Volume 5, pp. 1381–1403. [Google Scholar]
- James, J.L.; Stone, P.R.; Chamley, L.W. The regulation of trophoblast differentiation by oxygen in the first trimester of pregnancy. Hum. Reprod. Update 2006, 12, 137–144. [Google Scholar] [CrossRef]
- Brosens, I.A. The utero-placental vessels at term—The distribution and extent of physiological changes. In Placental Vascularization and Blood Flow; Springer: Boston, MA, USA, 1988; pp. 61–67. [Google Scholar]
- Karimu, A.L.; Burton, G.J. The effects of maternal vascular pressure on the dimensions of the placental capillaries. BJOG: Int. J. Obstet. Gynaecol. 1994, 101, 57–63. [Google Scholar] [CrossRef]
- Lyall, F.; Robson, S.C.; Bulmer, J.N. Spiral artery remodeling and trophoblast invasion in preeclampsia and fetal growth restriction relationship to clinical outcome. Hypertension 2013, 62, 1046–1054. [Google Scholar] [CrossRef] [Green Version]
- Shchyogolev, A.I.; Dubova, E.A.; Pavlov, K.A.; Lyapin, V.M.; Kulikova, G.V.; Shmakov, R.G. Morphometric characteristics of terminal villi of the placenta in pre-eclampsia. Bull. Exp. Biol. Med. 2012, 154, 92–95. [Google Scholar] [CrossRef]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Burton, G.J.; Woods, A.W.; Jauniaux, E.; Kingdom, J.C.P. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta 2009, 30, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sepulveda, A.; Torres, M.J.; Khoury, M.; Illanes, S.E. Innate immune system and preeclampsia. Front. Immunol. 2014, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Staff, A.C.; Fjeldstad, H.E.; Fosheim, I.K.; Moe, K.; Turowski, G.; Johnsen, G.M.; Alnaes-Katjavivi, P.; Sugulle, M. Failure of physiological transformation and spiral artery atherosis: Their roles in preeclampsia. Am. J. Obstet. Gynecol. 2020, 226, S895–S906. [Google Scholar] [CrossRef]
- Brosens, I.; Renaer, M. On the pathogenesis of placental infarcts in pre-eclampsia. BJOG: Int. J. Obstet. Gynaecol. 1972, 79, 794–799. [Google Scholar] [CrossRef]
- Iriyama, T.; Sun, K.; Parchim, N.F.; Li, J.; Zhao, C.; Song, A.; Hart, L.A.; Blackwell, S.C.; Sibai, B.M.; Chan, L.N.L.; et al. Elevated placental adenosine signaling contributes to the pathogenesis of preeclampsia. Circulation 2015, 131, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Zamudio, S. The placenta at high altitude. High Alt. Med. Biol. 2003, 4, 171–191. [Google Scholar] [CrossRef]
- Jones, M.L.; Mark, P.J.; Lewis, J.L.; Mori, T.A.; Keelan, J.A.; Waddell, B.J. Antioxidant defenses in the rat placenta in late gestation: Increased labyrinthine expression of superoxide dismutases, glutathione peroxidase 3, and uncoupling Protein 21. Biol. Reprod. 2010, 83, 254–260. [Google Scholar] [CrossRef] [Green Version]
- higgins, j.s.; vaughan, o.r.; fernandez de liger, e.; fowden, a.l.; sferruzzi-perri, a.n. placental phenotype and resource allocation to fetal growth are modified by the timing and degree of hypoxia during mouse pregnancy. J. Physiol. 2016, 594, 1341–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illsley, N.P.; Caniggia, I.; Zamudio, S. Placental metabolic reprogramming: Do changes in the mix of energy-generating substrates modulate fetal growth? Int. J. Dev. Biol. 2010, 54, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Zoccal, D.B.; Bonagamba, L.G.H.; Antunes-Rodrigues, J.; Machado, B.H. Plasma corticosterone levels is elevated in rats submitted to chronic intermittent hypoxia. Auton. Neurosci. Basic Clin. 2007, 134, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Alfaidy, N.; Gupta, S.; DeMarco, C.; Caniggia, I.; Challis, J.R.G. Oxygen regulation of placental 11β-hydroxysteroid dehydrogenase 2: Physiological and pathological implications. J. Clin. Endocrinol. Metab. 2002, 87, 4797–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benediktsson, R.; Lindsay, R.S.; Noble, J.; Seckl, J.R.; Edwards, C.R.W. Glucocorticoid exposure in utero: New model for adult hypertension. Lancet 1993, 341, 339–341. [Google Scholar] [CrossRef]
- Jellyman, J.K.; Fletcher, A.J.W.; Fowden, A.L.; Giussani, D.A. Glucocorticoid maturation of fetal cardiovascular function. Trends Molec. Med. 2020, 26, 170–184. [Google Scholar] [CrossRef]
- Julian, C.G.; Wilson, M.J.; Lopez, M.; Yamashiro, H.; Tellez, W.; Rodriguez, A.; Bigham, A.W.; Shriver, M.D.; Rodriguez, C.; Vargas, E.; et al. Augmented uterine artery blood flow and oxygen delivery protect andeans from altitude-associated reductions in fetal growth. Am. J. Physiol.-Regulat. Integr. Comp. Physiol. 2009, 296, R1564–R1575. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.; Bock, C.; Wetzel, M.; Maul, H.; Loerbroks, A. The prevalence of gestational diabetes in advanced economies. J. Perinat. Med. 2012, 40, 511–520. [Google Scholar] [CrossRef]
- Ngala, R.A.; Fondjo, L.A.; Gmagna, P.; Ghartey, F.N.; Awe, M.A. Placental peptides metabolism and maternal factors as predictors of risk of gestational diabetes in pregnant women. A case-control study. PLoS ONE 2017, 12, e0181613. [Google Scholar] [CrossRef] [Green Version]
- MacFarlane, C.M.; Tsakalakos, N. Relative fetal hypoxia as a contributing factor to fetal macrosomia in diabetic pregnancy. Med. Hypotheses 1983, 11, 365–374. [Google Scholar] [CrossRef]
- Philipps, A.F.; Dubin, J.W.; Matty, P.J.; Raye, J.R. Arterial hypoxemia and hyperinsulinemia in the chronically hyperglycemic fetal lamb. Pediatr. Res. 1982, 16, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Taricco, E.; Radaelli, T.; Rossi, G.; Nobile De Santis, M.S.; Bulfamante, G.P.; Avagliano, L.; Cetin, I. Effects of gestational diabetes on fetal oxygen and glucose levels in vivo. BJOG: Int. J. Obstet. Gynaecol. 2009, 116, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Tarvonen, M.; Hovi, P.; Sainio, S.; Vuorela, P.; Andersson, S.; Teramo, K. Intrapartal cardiotocographic patterns and hypoxia-related perinatal outcomes in pregnancies complicated by gestational diabetes mellitus. Acta Diabetol. 2021, 58, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, M.; Öztürk, Ö.; Ulubay, M.; Karaşahin, E.; Özgürtaş, T.; Yenen, M.; Aydın, A.; Fıratlıgil, F.; Bodur, S. Gebeliğin Tanısı Ile Birlikte Saptanan Anemi Prevalansı. Turk Jinekoloji ve Obstetrik Dernegi Dergisi 2017, 14, 176–180. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, K.M.; Ingardia, C.J.; Borgida, A.F. Anemia in pregnancy. Clin. Lab. Med. 2013, 33, 281–291. [Google Scholar] [CrossRef]
- Breymann, C. Iron deficiency anemia in pregnancy. Semin. Hematol. 2015, 52, 339–347. [Google Scholar] [CrossRef]
- Watkins, V.Y.; Frolova, A.I.; Stout, M.J.; Carter, E.B.; Macones, G.A.; Cahill, A.G.; Raghuraman, N. The relationship between maternal anemia and umbilical cord oxygen content at delivery. Am. J. Obstet. Gynecol. MFM 2021, 3, 100270. [Google Scholar] [CrossRef]
- Pien, G.W.; Pack, A.I.; Jackson, N.; Maislin, G.; Macones, G.A.; Schwab, R.J. Risk factors for sleep-disordered breathing in pregnancy. Thorax 2014, 69, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.; White, D.P. Obstructive sleep apnoea. Lancet 2002, 360, 237–245. [Google Scholar] [CrossRef]
- Almendros, I.; Martínez-Ros, P.; Farré, N.; Rubio-Zaragoza, M.; Torres, M.; Gutiérrez-Bautista, Á.J.; Carrillo-Poveda, J.M.; Sopena-Juncosa, J.J.; Gozal, D.; Gonzalez-Bulnes, A.; et al. Placental oxygen transfer reduces hypoxia-reoxygenation swings in fetal blood in a sheep model of gestational sleep apnea. J. Appl. Physiol. 2019, 127, 745–752. [Google Scholar] [CrossRef]
- Warland, J.; Dorrian, J.; Morrison, J.L.; O’Brien, L.M. Maternal sleep during pregnancy and poor fetal outcomes: A scoping review of the literature with meta-analysis. Sleep Med. Rev. 2018, 41, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Bonham, C.A.; Patterson, K.C.; Strek, M.E. Asthma outcomes and management during pregnancy. Chest 2018, 153, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Waddell, J.A.; Emerson, P.A.; Gunstone, R.F. Hypoxia in bronchial asthma. Br. Med. J. 1967, 2, 402–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foeller, M.E.; Foeller, T.M.; Druzin, M. Maternal congenital heart disease in pregnancy. Obstet. Gynecol. Clin. N. Am. 2018, 45, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Meah, V.L.; Cockcroft, J.R.; Backx, K.; Shave, R.; Stöhr, E.J. Cardiac output and related haemodynamics during pregnancy: A series of meta-analyses. Heart 2016, 102, 518–526. [Google Scholar] [CrossRef]
- Poole, J.H.; Spreen, D.T. Acute pulmonary edema in pregnancy. J. Perinatal Neonatal Nurs. 2005, 19, 316–331. [Google Scholar] [CrossRef]
- Balci, A.; Sollie-Szarynska, K.M.; van der Bijl, A.G.L.; Ruys, T.P.E.; Mulder, B.J.M.; Roos-Hesselink, J.W.; van Dijk, A.P.J.; Wajon, E.M.C.J.; Vliegen, H.W.; Drenthen, W.; et al. Prospective validation and assessment of cardiovascular and offspring risk models for pregnant women with congenital heart disease. Heart 2014, 100, 1373–1381. [Google Scholar] [CrossRef]
- Frias, A.E.; Morgan, T.K.; Evans, A.E.; Rasanen, J.; Oh, K.Y.; Thornburg, K.L.; Grove, K.L. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 2011, 152, 2456–2464. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, J.E.; Krystal, A.D.; Habib, A.S. Obstructive sleep apnea in pregnant women. Anesth. Analg. 2018, 127, 1167–1177. [Google Scholar] [CrossRef]
- Quigley, M.E.; Sheehan, K.L.; Wilkes, M.M.; Yen, S.S.C. Effects of maternal smoking on circulating catecholamine levels and fetal heart rates. Am. J. Obstet. Gynecol. 1979, 133, 685–690. [Google Scholar] [CrossRef]
- Levy, M.; Kovo, M.; Ben-Ezry, E.; Torem, M.; Shahaf, H.; Anchel, N.; Bar, J.; Schreiber, L.; Weiner, E. Passively inhaled tobacco smoke—Pregnancy and neonatal outcomes in correlation with placental histopathology. Placenta 2021, 112, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Bosco, C.; Diaz, E. Placental hypoxia and foetal development versus alcohol exposure in pregnancy. Alcohol Alcoholism 2012, 47, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhou, S.; Ping, J.; Pan, X.; Liang, G.; Xu, D.; Kou, H.; Bao, C.; Wang, H. Role of P53-dependent placental apoptosis in the reproductive and developmental toxicities of caffeine in rodents. Clin. Exp. Pharmacol. Physiol. 2012, 39, 357–363. [Google Scholar] [CrossRef] [PubMed]
- James, J.E. Maternal caffeine consumption and pregnancy outcomes: A narrative review with implications for advice to mothers and mothers-to-be. BMJ Evid. Based Med. 2021, 26, 114–115. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.; Okuliarova, M. Sex-specific cardiovascular susceptibility to ischaemic myocardial injury following exposure to prenatal hypoxia. Clin. Sci. 2017, 131, 2791–2794. [Google Scholar] [CrossRef]
- Shah, A.; Matsumura, N.; Quon, A.; Morton, J.S.; Dyck, J.R.B.; Davidge, S.T. Cardiovascular susceptibility to in vivo ischemic myocardial injury in male and female rat offspring exposed to prenatal hypoxia. Clin. Sci. 2017, 131, 2303–2317. [Google Scholar] [CrossRef]
- Xue, Q.; Zhang, L. Prenatal hypoxia causes a sex-dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: Role of protein kinase Cε. J. Pharmacol. Exp. Ther. 2009, 330, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, E.; Aljunaidy, M.M.; Kirschenman, R.; Spaans, F.; Morton, J.S.; Phillips, T.E.J.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Sex-specific effects of nanoparticle-encapsulated MitoQ (NMitoQ) delivery to the placenta in a rat model of fetal hypoxia. Front. Physiol. 2019, 10, 562. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Beltz, T.G.; Guo, F.; Johnson, A.K. Sex differences in maternal gestational hypertension-induced sensitization of angiotensin II hypertension in rat offspring: The protective effect of estrogen. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2018, 314, R274–R281. [Google Scholar] [CrossRef]
- Davis, G.K.; Newsome, A.D.; Ojeda, N.B.; Alexander, B.T. Effects of intrauterine growth restriction and female sex on future blood pressure and cardiovascular disease. Curr. Hypertens. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Huang, X.; Xue, Q.; Zhang, L. Antenatal hypoxia induces programming of reduced arterial blood pressure response in female rat offspring: Role of ovarian function. PLoS ONE 2014, 9, e98743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Xiao, D.; Zhang, L. Estrogen regulates angiotensin II receptor expression patterns and protects the heart from ischemic injury in female rats1. Biol. Reprod. 2015, 93, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasinger, J.H.; Intapad, S.; Rudsenske, B.R.; Davis, G.K.; Newsome, A.D.; Alexander, B.T. Chronic blockade of the androgen receptor abolishes age-dependent increases in blood pressure in female growth-restricted rats. Hypertension 2016, 67, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojeda, N.B.; Grigore, D.; Yanes, L.L.; Iliescu, R.; Robertson, E.B.; Zhang, H.; Alexander, B.T. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am. J. Physiol. Regulat. Integr. Comp. Physiol. 2007, 292, R758–R763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.; Mishra, J.S.; Dangudubiyyam, S.V.; Antony, K.M.; Baker, T.L.; Watters, J.J.; Kumar, S. Gestational intermittent hypoxia induces sex-specific impairment in endothelial mechanisms and sex steroid hormone levels in male rat offspring. Reprod. Sci. 2021. [Google Scholar] [CrossRef]
- Astiz, M.; Oster, H. Feto-maternal crosstalk in the development of the circadian clock system. Front. Neurosci. 2021, 14, 631687. [Google Scholar] [CrossRef]
- Crnko, S.; du Pré, B.C.; Sluijter, J.P.G.; van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Maywood, E.S. Synchronization and maintenance of circadian timing in the mammalian clockwork. Eur. J. Neurosci. 2020, 51. [Google Scholar] [CrossRef] [Green Version]
- Sato, F.; Kohsaka, A.; Bhawal, U.; Muragaki, Y. Potential roles of Dec and Bmal1 genes in interconnecting circadian clock and energy metabolism. Int. J. Molec. Sci. 2018, 19, 781. [Google Scholar] [CrossRef] [Green Version]
- Stokkan, K.-A. Entrainment of the circadian clock in the liver by feeding. Science 2001, 291, 490–493. [Google Scholar] [CrossRef] [Green Version]
- Storch, K.-F.; Lipan, O.; Leykin, I.; Viswanathan, N.; Davis, F.C.; Wong, W.H.; Weitz, C.J. Extensive and divergent circadian gene expression in liver and heart. Nature 2002, 417, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Wharfe, M.D.; Mark, P.J.; Wyrwoll, C.S.; Smith, J.T.; Yap, C.; Clarke, M.W.; Waddell, B.J. Pregnancy-induced adaptations of the central circadian clock and maternal glucocorticoids. J. Endocrinol. 2016, 228, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kováčiková, Z.; Sládek, M.; Bendová, Z.; Illnerová, H.; Sumová, A. Expression of clock and clock-driven genes in the rat suprachiasmatic nucleus during late fetal and early postnatal development. J. Biol. Rhythm. 2006, 21, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Čečmanová, V.; Houdek, P.; Šuchmanová, K.; Sládek, M.; Sumová, A. Development and entrainment of the fetal clock in the suprachiasmatic nuclei: The role of glucocorticoids. J. Biol. Rhythm. 2019, 34, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Lamadé, E.K.; Hendlmeier, F.; Wudy, S.A.; Witt, S.H.; Rietschel, M.; Coenen, M.; Gilles, M.; Deuschle, M. Rhythm of fetoplacental 11β-hydroxysteroid dehydrogenase type 2-fetal protection from morning maternal glucocorticoids. J. Clin. Endocrinol. Metab. 2021, 106, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Waddell, B.J.; Wharfe, M.D.; Crew, R.C.; Mark, P.J. A rhythmic placenta? Circadian variation, clock genes and placental function. Placenta 2012, 33, 533–539. [Google Scholar] [CrossRef]
- Sletten, J.; Cornelissen, G.; Assmus, J.; Kiserud, T.; Albrechtsen, S.; Kessler, J. Maternal exercise, season and sex modify the daily fetal heart rate rhythm. Acta Physiol. 2018, 224, e13093. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Hou, Y.; Huang, L.; Bian, Y.; Song, G.; Qiao, C. Circadian clock gene clock is involved in the pathogenesis of preeclampsia through hypoxia. Life Sci. 2020, 247, 117441. [Google Scholar] [CrossRef]
- Manella, G.; Aviram, R.; Bolshette, N.; Muvkadi, S.; Golik, M.; Smith, D.F.; Asher, G. Hypoxia induces a time- and tissue-specific response that elicits intertissue circadian clock misalignment. Proc. Natl. Acad. Sci. USA 2020, 117, 779–786. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Molec. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Dimova, E.Y.; Jakupovic, M.; Kubaichuk, K.; Mennerich, D.; Chi, T.F.; Tamanini, F.; Oklejewicz, M.; Hänig, J.; Byts, N.; Mäkelä, K.A.; et al. The circadian clock protein CRY1 is a negative regulator of HIF-1α. iScience 2019, 13, 284–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, T.; Morrell, M.; Simonds, A.; Adcock, I.; Durham, A. The role of hypoxia and the circadian rhythm in sleep apnoea. In Proceedings of the 4.2 Sleep and Control of Breathing, Lausanne, Switzerland, 30 September 2015; European Respiratory Society: Lausanne, Switzerland, 2015; p. OA298. [Google Scholar]
- Kobayashi, M.; Morinibu, A.; Koyasu, S.; Goto, Y.; Hiraoka, M.; Harada, H. A Circadian clock gene, PER2, activates HIF-1 as an effector molecule for recruitment of HIF-1α to promoter regions of its downstream genes. FEBS J. 2017, 284, 3804–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, E.J.; Martinez, C.A.; Liang, Y.G.; Cistulli, P.A.; Cook, K.M. Out of Breath, out of time: Interactions between HIF and circadian rhythms. Am. J. Physiol. Cell Physiol. 2020, 319, C533–C540. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, Y.; Ladeuix, B.; Golik, M.; Koeners, M.P.; Asher, G. Rhythmic oxygen levels reset circadian clocks through HIF1α. Cell Metab. 2017, 25, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Koritala, B.S.C.; Lee, Y.Y.; Bhadri, S.S.; Gaspar, L.S.; Stanforth, C.; Wu, G.; Ruben, M.D.; Francey, L.J.; Smith, D.F. Intermittent hypoxia alters the circadian expression of clock genes in mouse brain and liver. Genes 2021, 12, 1627. [Google Scholar] [CrossRef]
- Yun, S.; Lee, E.J.; Choe, H.K.; Son, G.H.; Kim, K.; Chung, S. Programming effects of maternal stress on the circadian system of adult offspring. Exp. Molec. Med. 2020, 52, 473–484. [Google Scholar] [CrossRef]
- Reh, R.K.; Dias, B.G.; Nelson, C.A.; Kaufer, D.; Werker, J.F.; Kolb, B.; Levine, J.D.; Hensch, T.K. Critical period regulation across multiple timescales. Proc. Natl. Acad. Sci. USA 2020, 117, 23242–23251. [Google Scholar] [CrossRef]
- Sengupta, S.; Tang, S.Y.; Devine, J.C.; Anderson, S.T.; Nayak, S.; Zhang, S.L.; Valenzuela, A.; Fisher, D.G.; Grant, G.R.; López, C.B.; et al. Circadian control of lung inflammation in influenza infection. Nat. Commun. 2019, 10, 4107. [Google Scholar] [CrossRef] [Green Version]
- Issah, Y.; Naik, A.; Tang, S.Y.; Forrest, K.; Brooks, T.G.; Lahens, N.; Theken, K.N.; Mermigos, M.; Sehgal, A.; Worthen, G.S.; et al. Loss of circadian protection against influenza infection in adult mice exposed to hyperoxia as neonates. eLife 2021, 10, e61241. [Google Scholar] [CrossRef]
- Obayashi, K.; Yamagami, Y.; Tatsumi, S.; Kurumatani, N.; Saeki, K. Indoor light pollution and progression of carotid atherosclerosis: A longitudinal study of the HEIJO-KYO cohort. Environ. Intern. 2019, 133, 105184. [Google Scholar] [CrossRef]
- Giachello, C.N.G.; Baines, R.A. Inappropriate neural activity during a sensitive period in embryogenesis results in persistent seizure-like behavior. Curr. Biol. 2015, 25, 2964–2968. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sutovska, H.; Babarikova, K.; Zeman, M.; Molcan, L. Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review. Int. J. Mol. Sci. 2022, 23, 2885. https://doi.org/10.3390/ijms23052885
Sutovska H, Babarikova K, Zeman M, Molcan L. Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review. International Journal of Molecular Sciences. 2022; 23(5):2885. https://doi.org/10.3390/ijms23052885
Chicago/Turabian StyleSutovska, Hana, Katarina Babarikova, Michal Zeman, and Lubos Molcan. 2022. "Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review" International Journal of Molecular Sciences 23, no. 5: 2885. https://doi.org/10.3390/ijms23052885
APA StyleSutovska, H., Babarikova, K., Zeman, M., & Molcan, L. (2022). Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review. International Journal of Molecular Sciences, 23(5), 2885. https://doi.org/10.3390/ijms23052885