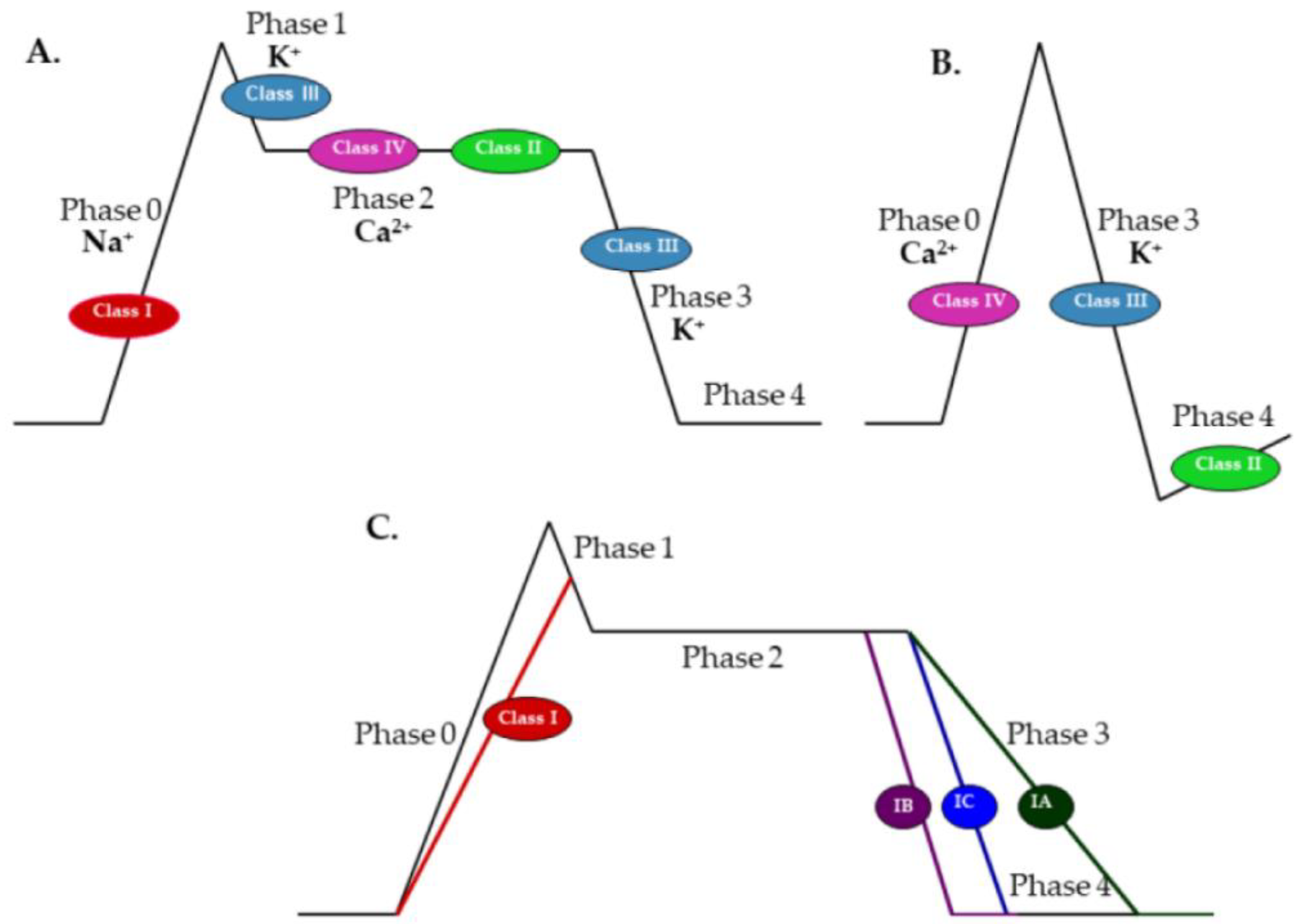
4.2. Probable Mechanisms of Pharmacodynamic Interactions between Antiarrhythmic and Antiepileptic Drugs
According to Deckers et al. [
74], if the two drugs applied in combination have different mechanisms of action, synergistic interactions between them are more likely; otherwise, additivity probably occurs. Therefore, in the case of positive interaction between two drugs, their mechanisms of action should be analyzed. Mechanisms of action of antiepileptic drugs have been presented in
Table 5.
Antiseizure drugs taken into consideration in this review are: valproate, carbamazepine, phenytoin, phenobarbital, diazepam, oxcarbazepine, topiramate, pregabalin, lamotrigine, and lacosamide. The leading action of valproate, carbamazepine, phenytoin, oxcarbazepine, and lamotrigine is a blockade of VDSCs. Carbamazepine, phenytoin, lamotrigine, and oxcarbazepine block VDSCs during high-frequency discharges, but in therapeutic concentrations, they have no effect on physiological synaptic transmission [
75]. Moreover, phenytoin, lamotrigine, and valproate also inhibit the persistent sodium currents [
76]. Phenytoin, carbamazepine, oxcarbazepine, lamotrigine, and eslicarbazepine block VDSCs in the nonconducting fast inactivated state. On the other hand, lacosamide was shown to act on VDSCs channels during the slow activation phase [
50,
75,
77]. Blocking of VDSCs in their inactivated conformation stabilizes this inactive form and prevents the return of the channel to the active state. In light of the above, phenytoin and carbamazepine seem to have very similar mechanisms of action on VDSCs. Both drugs limit high-frequency repetitive firing in a use-dependent manner, i.e., the higher the frequency of channel openings, the better the inhibitory effect of phenytoin and carbamazepine. Inhibiting high-frequency repetitive firing correlates well with a slower reactivation of VDSCs. Interestingly, both antiepileptics also exhibit membrane-stabilizing properties. On the other hand, phenytoin and carbamazepine at therapeutic doses affect neither GABA-ergic nor glutamatergic neurotransmission. However, there are also subtle differences between their action on VDSCs. Phenytoin requires a prolonged depolarization (around 100 ms) to start its action on sodium currents. Much shorter depolarization is enough for carbamazepine [
58,
75].
Regarding VDSCs subtypes located within α-subunit, valproate, carbamazepine, oxcarbazepine, lamotrigine, and topiramate block receptor subpopulations from Nav1.1 to Nav1.9. Phenytoin inhibits currents conducted through Nav1.1, 1.2, 1.5, and 1.8, while lacosamide shows an affinity for Nav1.3, 1.7, and 1.8 (data from DrugBank,
https://go.drugbank.com).
Numerous antiepileptic drugs present more complex mechanisms of action. For instance, carbamazepine is an agonist of adenosine A
1 and A
2 receptors. It also blocks L-type voltage-dependent calcium channels (VDCCs) [
75,
78]. Anticonvulsant effects of oxcarbazepine may to some extent result from the enhancement of the outward potassium currents and blockade of high-voltage activated calcium channels of N/P and R types [
78]. Interestingly, the blockade of N or P/Q channels inhibits the presynaptic release of excitatory amino acids [
75,
79].
In addition to the limitation of high-frequency repetitive firing, lamotrigine reduces the synaptic release of excitatory amino acids. In vitro studies revealed that lamotrigine enhances currents conducted through HCN channels and increases activity of GABA-ergic interneurons [
75,
80].
Valproate elevates GABA concentrations in the brain through three mechanisms: 1. increased synthesis of GABA by activating glutamic acid decarboxylase, a GABA synthesizing enzyme; 2. the increased potassium-induced release of GABA to the synaptic cleft; and 3. decreased activity of GABA-transaminase, an enzyme catalyzing GABA degradation. Moreover, valproate activates potassium conductance and inhibits low-threshold T-type calcium channels. The latter is crucial in inhibiting absence seizures [
77,
79,
81,
82].
Additionally, topiramate shows a complex mode of action. In addition to blocking VDSCs, the drug reduces excitatory neurotransmission through a negative modulatory effect on calcium-permeable AMPA/kainate receptors; potentiates GABA-mediated inhibitory neurotransmission through binding to a novel site within the GABA
A receptor complex; inhibits neuronal L-type high-voltage-activated calcium channels; weakly inhibits carbonic anhydrase; and activates potassium currents [
75,
79,
83].
Phenobarbital is an agonist of the barbiturate recognition site, while diazepam binds to the benzodiazepine site within the GABA
A receptor complex. Barbiturates increase the time of GABA
A-receptor-dependent chloride channel openings. In contrast, benzodiazepines increase the frequency but not the conductance or the time of opening of GABA
A-related chloride channels. In addition, barbiturates can block VDSCs (but at a 10-times higher concentration than carbamazepine or phenytoin), activate voltage-dependent potassium channels, and inhibit AMPA-related glutamatergic transmission. Regarding benzodiazepines, other mechanisms of action include inhibition of adenosine uptake and, at higher doses, blocking VDSCs and voltage-dependent calcium channels [
75,
79,
81].
Pregabalin acts presynaptically by binding to the alpha2-delta auxiliary subunit of voltage-dependent calcium channels. The drug reduces the calcium release, and, in consequence, several neurotransmitters, such as glutamate, substance P, and norepinephrine. Moreover, pregabalin blocks high-voltage-dependent calcium channels [
75,
79,
84,
85].
Lacosamide, in addition to its effect on slow activation of sodium channels, acts as an antagonist of the glycine-binding site on NMDA receptors [
75].
Careful analysis of the combined treatment with antiarrhythmic and antiepileptic drugs (
Table 1,
Table 2 and
Table 3) supports the widespread opinion that newer antiepileptic drugs are less likely to interact with other medications. Among the antiarrhytmic drugs, propafenone and mexiletine were the most active in interactions with antiseizure drugs. Both antiarrhythmic drugs block Nav1.5 currents and KCNH
2 receptors and present membrane-stabilizing activity. Propafenone potentiated the antielectroshock action of valproate (
p < 0.001), carbamazepine (
p < 0.001), phenytoin (
p < 0.01), phenobarbital (
p < 0.01), pregabalin (
p < 0.01), and topiramate (
p < 0.05), but not that of lamotrigine [
22,
23]. Even considering that increased action of valproate, carbamazepine, and pregabalin was due in part to the increased brain concentration of the two drugs, it seems to be clear that the strongest interactions were observed between propafenone and antiepileptics blocking sodium channels. All of them block a wide range of sodium channels from Nav1.1 to Nav1.9. One could assume that in the case of identified drug-drug interactions it comes down to complementary effects of drug components on sodium channels. A reasoning problem arises with lamotrigine, which also blocks the whole range of sodium channels, and pregabalin reducing the release of excitatory amino acids to the synaptic cleft. At the present level of knowledge, the reason for the lack of interaction between the two antiepileptics with propafenone remains unclear.
In the case of mexiletine (
Table 3), isobolographic analysis showed synergism with topiramate and pregabalin, which was independent of mexiletine-induced changes in brain levels of both antiepileptics. Surprisingly, a tendency towards antagonism was revealed for the combination of mexiletine and valproate in proportions of 1:1 and 3:1. Although the lowered brain concentration of valproate could be in part responsible for this result in the 1:1 proportion, no pharmacokinetic events were observed for the fixed ratios of 3:1 [
24,
25]. It can be suggested that a synergistic interaction occurred between mexiletine, a Nav1.5 and KCNH
2 antagonist and two antiepileptic drugs with different mexiletine mechanisms of action. Additivity was observed between the antiarrhythmic drug and antiepileptics blocking VDSCs. However, this line of reasoning does not explain why additivity, not synergism, was shown in the case of phenobarbital (a strong enhancer of GABA-ergic transmission). Additionally, the reasons for the antagonism between mexiletine and valproate (in the 3:1 proportion) seem to be unclear. In this case, mexiletine did not lower the brain concentration of valproate.
Ivabradine, an inhibitor of HCN
2 channels, enhanced the antielectroshock action of valproate but decreased that of phenytoin and lamotrigine. Pharmacokinetic interactions were only relevant in the case of phenytoin [
20,
21]. It is not clear why ivabradine exhibited the opposite effect on antiepileptic drugs, whose main mechanism of action is sodium channel blocking. The observed decrease in the anticonvulsant potency of lamotrigine in the presence of ivabradine could be explained by the common action of the two drugs on HCN channels [
80]. Ivabradine potentially has a higher affinity for HCN channels and does not allow lamotrigine to express its full antiseizure properties.
Numerous β-blockers were examined in the MES test [
28,
30]. Their pharmacological properties are gathered in
Table 4. Propranolol, pindolol, acebutolol, metoprolol, alprenolol, and nebivolol interacted with antiepileptic drugs. Propranolol, metoprolol, and acebutolol enhanced the action of valproate in the MES test. Furthermore, propranolol, pindolol, and alprenolol potentiated the antielectroshock effect of phenobarbital. Propranolol and metoprolol enhanced the action of diazepam. This indicates that β-blockers readily interact with antiepileptics enhancing GABA-ergic neurotransmission [
30]. The main property of all β-receptor antagonists interacting with antiepileptic drugs is their membrane-stabilizing activity. The same can be said about nevivolol, but this drug behaved differently in the MES test, decreasing the action of carbamazepine. At present, there is no plausible explanation for this phenomenon.
Amiodarone potentiated the antielectroshock action of carbamazepine, oxcarbazepine, and pregabalin, whereas dronedarone potentiated that of lamotrigine. Surprisingly, dronedarone diminished the effect of phenytoin in the MES test in mice. The two pure multichannel blockers did not produce any pharmacokinetic interactions [
32,
33,
34,
35]. Surprisingly, amiodarone and dronedarone, although structurally related to each other, enhanced the action of different antiepileptic drugs. Additionally, processes underlying the dronedarone-induced decrease in the action of phenytoin remain incomprehensible. It should be remembered that the antielectroshock effect of phenytoin was not influenced by amiodarone and increased by sotalol. Even subtle differences in the mechanism of action of the component drugs are probably important for the resultant interactions between them. Undoubtedly, further molecular studies are necessary to explain the whole background of the above findings.
The effect of either valproate or phenytoin was potentiated by sotalol [
36]. Additionally, these interactions seem to be pharmacodynamic. The interaction observed between sotalol and valproate could possibly be due to β-adrenolytic properties of sotalol, as pure β-blockers also enhanced the action of valproate.
Class IV antiarrhythmics, verapamil and diltiazem, block L-type calcium channels. In addition, verapamil moderately inhibits N, P, Q, and T calcium currents. It also slows conduction through KCNH
2 channels and presents α1 antagonistic activity (DrugBank,
https://go.drugbank.com). Because verapamil does not pass through the blood–brain barrier, it was ineffective against electrically induced seizures in mice. In contrast, diltiazem increased the ECT and enhanced the antielectroshock action of carbamazepine, phenytoin, and topiramate [
38,
40]. This effect may result from the complementary blockade of calcium and sodium channels. It does not explain, however, why diltiazem did not affect the action of other antiepileptic drugs inhibiting sodium currents.
4.4. Considerations Resulting from Analyzed Data
According to Raegan-Shaw et al. [
86], maximal single doses of antiarrhythmic drugs used in patients have been converted to mouse doses (in mg/kg) and presented in
Table 6 and
Table 7. This allows us to find out whether doses applied in experimental studies are comparable and able to be interpolated to those used in clinical practice.
It appears that only diltiazem increased the ECT at doses comparable to human ranges; remaining antiarrhythmics affected this parameter at much higher doses than those applied in patients. More antiarrhythmics interacted with antiepileptic drugs at dosages close to clinical ranges, including propafenone, dronedarone, diltiazem, propranolol, metoprolol, alprenolol, and ivabradine. This may be of particular clinical importance in the case of dronedarone, ivabradine, and nebivolol. Dronedarone diminished the action of phenytoin, and ivabradine decreased the effect of phenytoin and lamotrigine, while nebivolol reduced the anti-MES action of carbamazepine.
In general, only some antiarrhythmic drugs exhibited their own anti-MES action or increased the ECT in mice. Moreover, in the case of propranolol and diltiazem, the available data remain inconsistent. The antielectroshock action of antiepileptic drugs was enhanced by all four classes of antiarrhythmic drugs and ivabradine. However, propafenone and mexiletine, i.e., sodium channel blockers, were the most active in this respect. On the other hand, the three mostly potentiated antiepileptics were valproate, carbamazepine, and phenytoin, whose mechanism of action is based on sodium channel blockades. This may contradict the theory that synergism happens only when the component drugs differ in their mechanisms of action. However, it should be remembered that sodium channels can be blocked in distinct target points and in divergent physiological states. On the other hand, most antiarrhythmics interacting with antiepileptic drugs, including propafenone, mexiletine, diltiazem, propranolol, pindolol, metoprolol, acebutolol, alprenolol, and nebivolol, present membrane-stabilizing activity. Additionally, antiepileptic drugs, whose antiseizure action is most often affected (valproate, carbamazepine, phenytoin), stabilize the cell membrane [
87].
Interestingly, “pure” class III antiarrhythmics, amiodarone and dronedarone, did not interact pharmacokinetically with antiepileptic drugs. Remaining antiarrhythmics and ivabradine were able to increase or decrease brain concentrations of antiepileptic drugs; however, no regularity was found.




{kind=link}