
Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach


Abstract
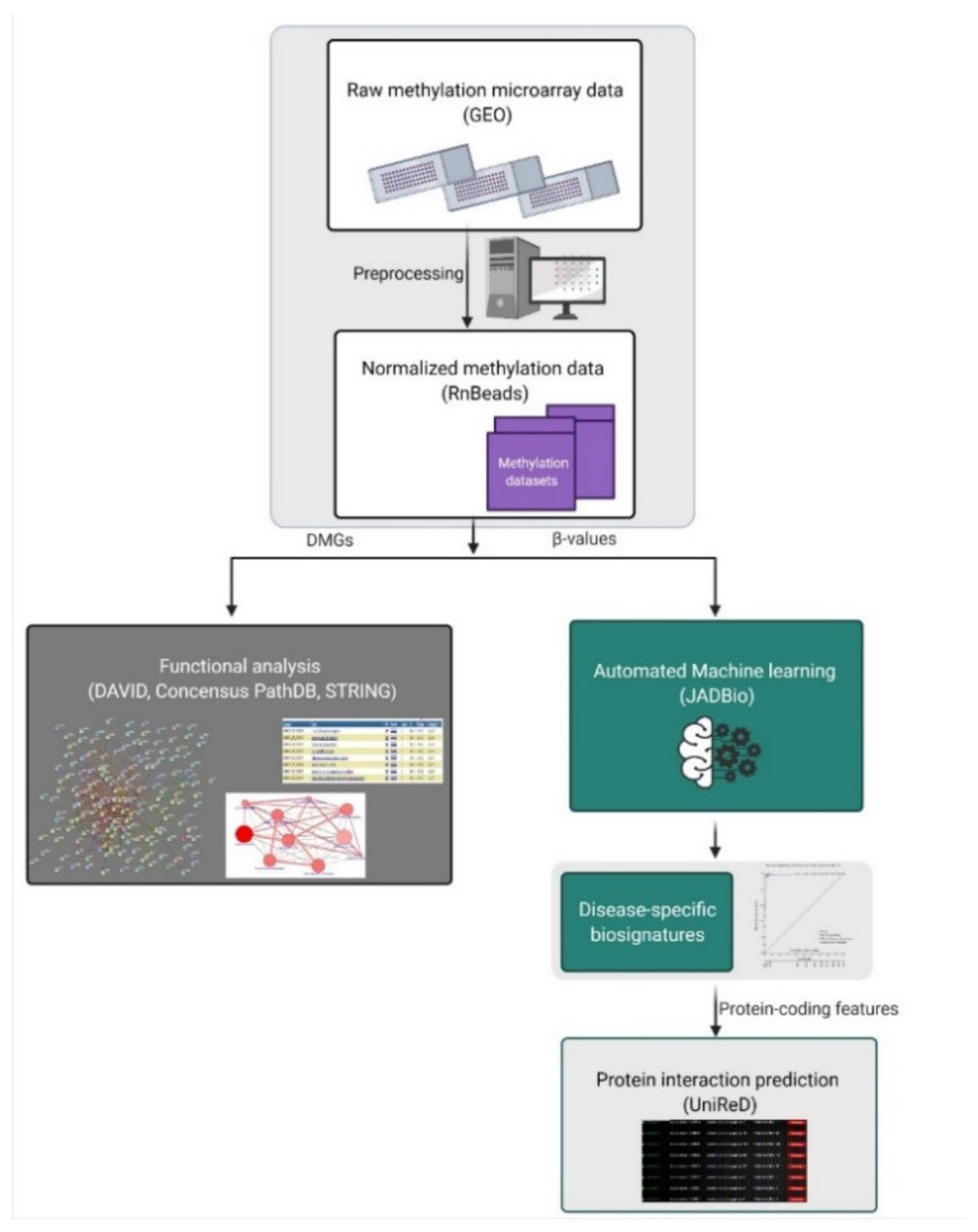
:1. Introduction
2. Results
2.1. Breast Cancer
2.1.1. Differential Methylation Analysis Comparing BrCa and Healthy Tissues
2.1.2. Functional Analysis of DMGs Comparing BrCa and Healthy Tissues
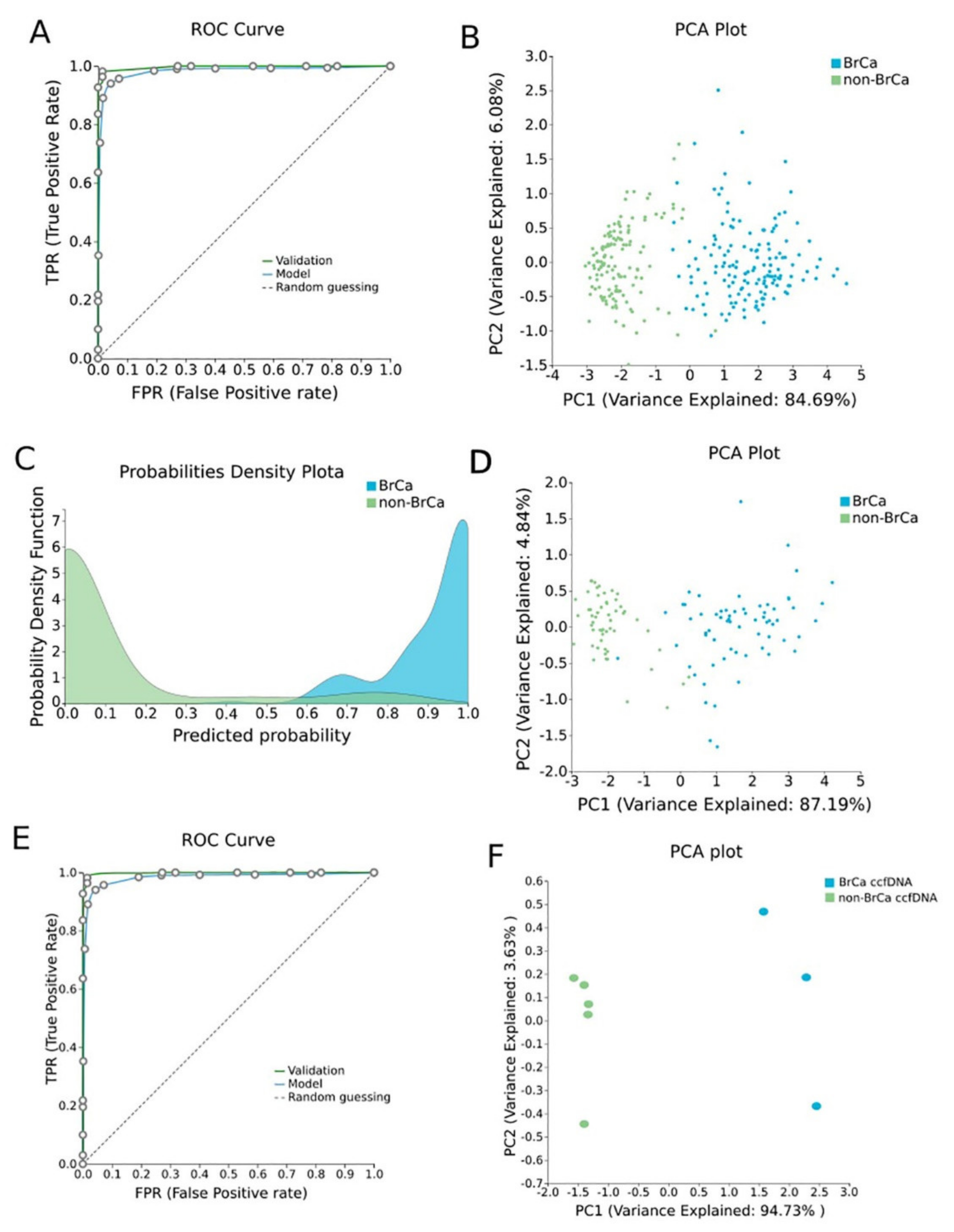
2.1.3. BrCa-Specific Methylation Biosignature through AutoML
2.1.4. Validation and Applicability of BrCa-Specific Methylation Biosignature on ccfDNA
2.1.5. Biological Relevance of Genes Selected in the BrCa-Specific Methylation Biosignature
2.2. Osteoarhtitis
2.2.1. Differential Methylation Analysis Comparing OA and Healthy Tissues
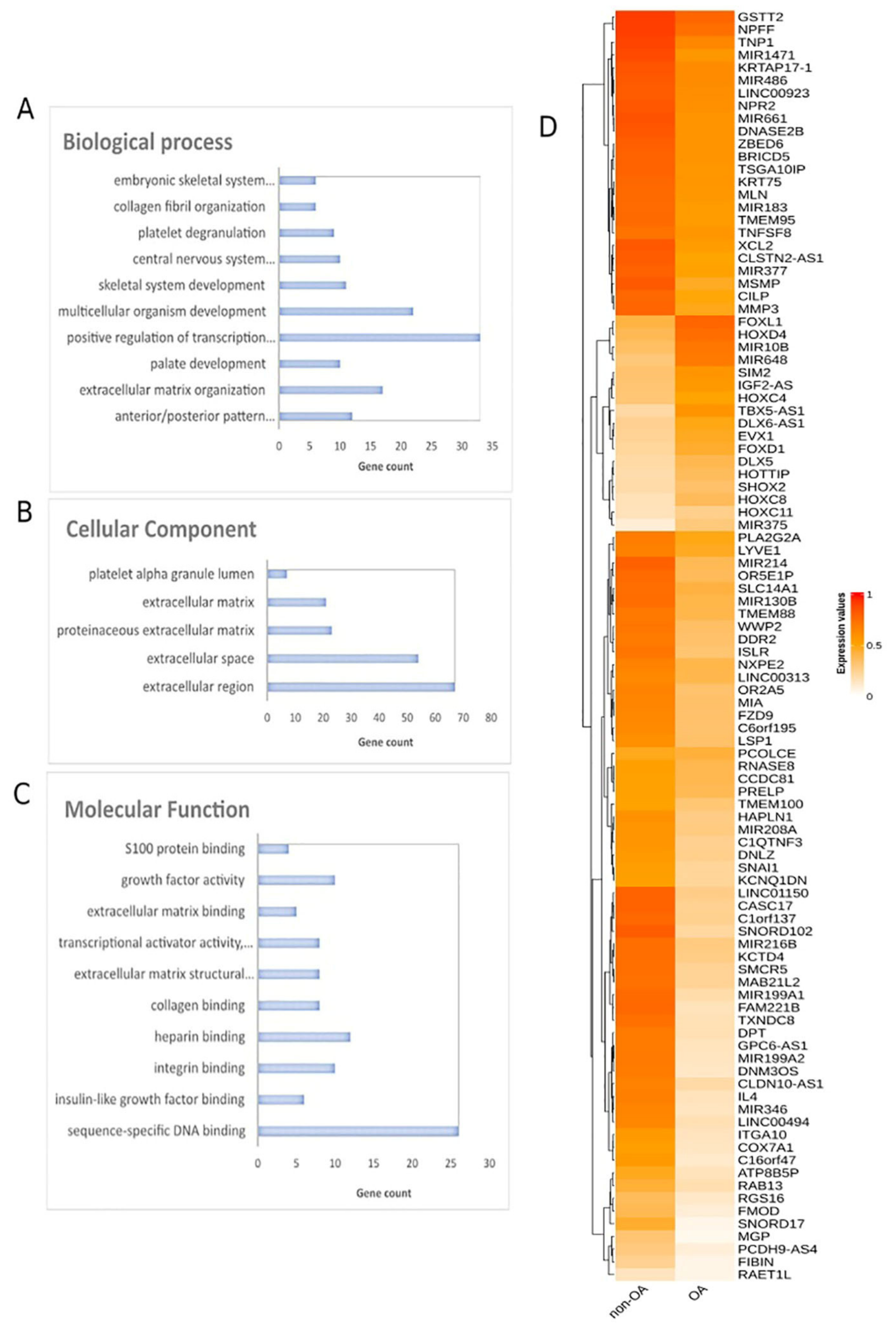
2.2.2. Functional Analysis of DMGs Comparing OA and Healthy Tissues
2.2.3. OA Specific Methylation Biosignature through AutoML
2.2.4. Biological Relevance of Genes Selected in the OA-Specific Methylation Biosignature
2.3. Diabetes
2.3.1. Differential Methylation Analysis Comparing Pancreatic β-Cells and Other Tissues
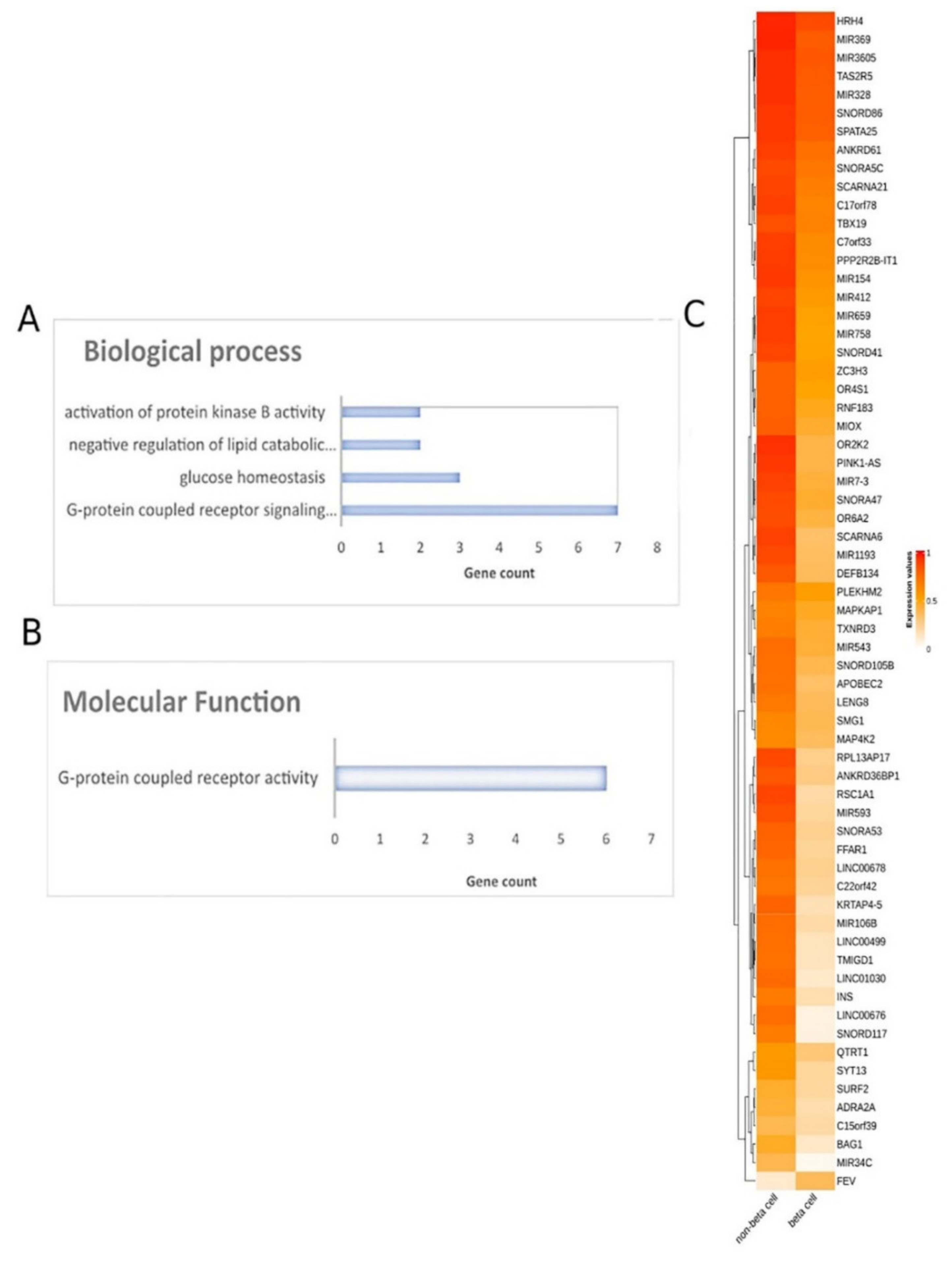
2.3.2. Functional Analysis of DMGs Comparing Pancreatic β-Cells and Other Tissues
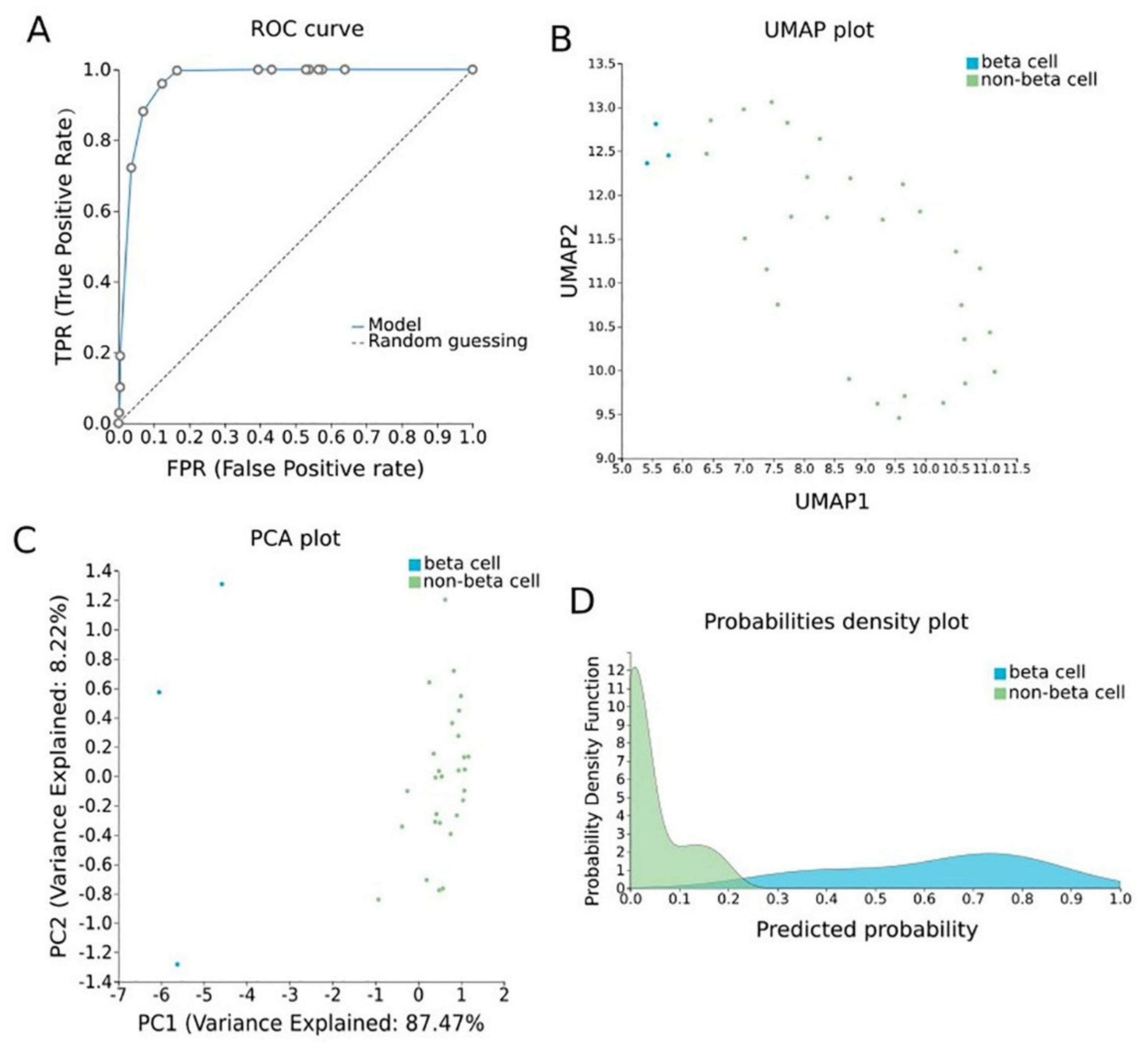
2.3.3. Pancreatic β-Cell Specific Methylation Biosignature Using AutoML
2.3.4. Biological Relevance of Genes Selected in the β-Cell-Specific Methylation Biosignature
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.2. Data Preprocessing and Differential Methylation Analysis
4.3. Automated Machine Learning Analysis (AutoML)
4.4. Biological Association Analysis through Text Mining
4.5. Functional Analysis of DMGs
4.6. Evaluation of Biosignatures on Liquid Biopsy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. 2–DNA Methylation and Cancer. In Advances in Genetics; Herceg, Z., Ushijima, T., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 70, pp. 27–56. [Google Scholar]
- Richardson, B. DNA methylation and autoimmune disease. Clin. Immunol. 2003, 109, 72–79. [Google Scholar] [CrossRef]
- Bansal, A.; Pinney, S.E. DNA methylation and its role in the pathogenesis of diabetes. Pediatr. Diabetes 2017, 18, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ammal Kaidery, N.; Tarannum, S.; Thomas, B. Epigenetic Landscape of Parkinson’s Disease: Emerging Role in Disease Mechanisms and Therapeutic Modalities. Neurotherapeutics 2013, 10, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.; Tiirikainen, M. Diagnostic Power of DNA Methylation Classifiers for Early Detection of Cancer. Trends Cancer 2020, 6, 78–81. [Google Scholar] [CrossRef]
- Klughammer, J.; Kiesel, B.; Roetzer, T.; Fortelny, N.; Nemc, A.; Nenning, K.-H.; Furtner, J.; Sheffield, N.C.; Datlinger, P.; Peter, N.; et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat. Med. 2018, 24, 1611–1624. [Google Scholar] [CrossRef]
- Henderson-Smith, A.; Fisch, K.M.; Hua, J.; Liu, G.; Ricciardelli, E.; Jepsen, K.; Huentelman, M.; Stalberg, G.; Edland, S.D.; Scherzer, C.R.; et al. DNA methylation changes associated with Parkinson’s disease progression: Outcomes from the first longitudinal genome-wide methylation analysis in blood. Epigenetics 2019, 14, 365–382. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.T.; Narayan, P.; Grant, M.J.; Langfelder, P.; Wang, N.; Kwak, S.; Wilkinson, H.; Chen, R.Z.; Chen, J.; Bawden, C.S.; et al. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun. 2020, 11, 4529. [Google Scholar] [CrossRef]
- Goud Alladi, C.; Etain, B.; Bellivier, F.; Marie-Claire, C. DNA Methylation as a Biomarker of Treatment Response Variability in Serious Mental Illnesses: A Systematic Review Focused on Bipolar Disorder, Schizophrenia, and Major Depressive Disorder. Int. J. Mol. Sci. 2018, 19, 3026. [Google Scholar] [CrossRef] [Green Version]
- Marie-Claire, C.; Lejeune, F.X.; Mundwiller, E.; Ulveling, D.; Moszer, I.; Bellivier, F.; Etain, B. A DNA methylation signature discriminates between excellent and non-response to lithium in patients with bipolar disorder type 1. Sci. Rep. 2020, 10, 12239. [Google Scholar] [CrossRef]
- Sigin, V.O.; Kalinkin, A.I.; Kuznetsova, E.B.; Simonova, O.A.; Chesnokova, G.G.; Litviakov, N.V.; Slonimskaya, E.M.; Tsyganov, M.M.; Ibragimova, M.K.; Volodin, I.V.; et al. DNA methylation markers panel can improve prediction of response to neoadjuvant chemotherapy in luminal B breast cancer. Sci. Rep. 2020, 10, 9239. [Google Scholar] [CrossRef]
- Chatzaki, E.; Tsamardinos, I. Somatic copy number aberrations detected in circulating tumor DNA can hold diagnostic value for early detection of hepatocellular carcinoma. EBioMedicine 2020, 57, 102851. [Google Scholar] [CrossRef]
- Lai, H.; Huang, H.; Keshavjee, K.; Guergachi, A.; Gao, X. Predictive models for diabetes mellitus using machine learning techniques. BMC Endocr. Disord. 2019, 19, 101. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Xu, D.; Zhang, Q.; Zhang, J.; Ngai, S.M.; Shao, J. Classification of lung cancer using ensemble-based feature selection and machine learning methods. Mol. Biosyst. 2015, 11, 791–800. [Google Scholar] [CrossRef]
- Aref-Eshghi, E.; Schenkel, L.C.; Ainsworth, P.; Lin, H.; Rodenhiser, D.I.; Cutz, J.-C.; Sadikovic, B. Genomic DNA Methylation-Derived Algorithm Enables Accurate Detection of Malignant Prostate Tissues. Front Oncol. 2018, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, M.; Karaglani, M.; Manolopoulos, V.G.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Deciphering the Methylation Landscape in Breast Cancer: Diagnostic and Prognostic Biosignatures through Automated Machine Learning. Cancers 2021, 13, 1677. [Google Scholar] [CrossRef]
- Karaglani, M.; Gourlia, K.; Tsamardinos, I.; Chatzaki, E. Accurate Blood-Based Diagnostic Biosignatures for Alzheimer’s Disease via Automated Machine Learning. J. Clin. Med. 2020, 9, 3016. [Google Scholar] [CrossRef]
- Dogan, M.V.; Grumbach, I.M.; Michaelson, J.J.; Philibert, R.A. Integrated genetic and epigenetic prediction of coronary heart disease in the Framingham Heart Study. PLoS ONE 2018, 13, e0190549. [Google Scholar] [CrossRef] [Green Version]
- Tiulpin, A.; Klein, S.; Bierma-Zeinstra, S.M.A.; Thevenot, J.; Rahtu, E.; Meurs, J.v.; Oei, E.H.G.; Saarakkala, S. Multimodal Machine Learning-based Knee Osteoarthritis Progression Prediction from Plain Radiographs and Clinical Data. Sci. Rep. 2019, 9, 20038. [Google Scholar] [CrossRef]
- Enríquez, J.G.; Martínez-Rojas, A.; Lizcano, D.; Jiménez-Ramírez, A. A Unified Model Representation of Machine Learning Knowledge. J. Web Eng. 2020, 19, 2. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Charonyktakis, P.; Lakiotaki, K.; Borboudakis, G.; Zenklusen, J.C.; Juhl, H.; Chatzaki, E.; Lagani, V. Just Add Data: Automated Predictive Modeling and BioSignature Discovery. bioRxiv, 2020; in press. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nature Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ren, J.; Luo, N.; Guo, H.; Zheng, Y.; Li, J.; Tang, F.; Wen, L.; Peng, J. Comprehensive DNA methylation analysis of tissue of origin of plasma cell-free DNA by methylated CpG tandem amplification and sequencing (MCTA-Seq). Clin. Epigenetics 2019, 11, 93. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Pantazi, C.; Kolios, G.; Kakolyris, S.; Chatzaki, E. Circulating cell-free DNA release in vitro: Kinetics, size profiling, and cancer-related gene methylation. J. Cell. Physiol. 2019, 234, 14079–14089. [Google Scholar] [CrossRef]
- Narod, S.A.; Salmena, L. BRCA1 and BRCA2 mutations and breast cancer. Discov. Med. 2011, 12, 445–453. [Google Scholar]
- Li, M.; Wang, C.; Yu, B.; Zhang, X.; Shi, F.; Liu, X. Diagnostic value of RASSF1A methylation for breast cancer: A meta-analysis. Biosci. Rep. 2019, 39, BSR20190923. [Google Scholar] [CrossRef] [Green Version]
- Dustin, D.; Gu, G.; Fuqua, S.A.W. ESR1 mutations in breast cancer. Cancer 2019, 125, 3714–3728. [Google Scholar] [CrossRef]
- Li, X.; Chen, X.; Wen, L.; Wang, Y.; Chen, B.; Xue, Y.; Guo, L.; Liao, N. Impact of TP53 mutations in breast cancer: Clinicopathological features and prognosisImpact of TP53 mutations in breast CA. Thorac. Cancer 2020, 11, 1861–1868. [Google Scholar] [CrossRef]
- Arsenic, R.; Lehmann, A.; Budczies, J.; Koch, I.; Prinzler, J.; Kleine-Tebbe, A.; Schewe, C.; Loibl, S.; Dietel, M.; Denkert, C. Analysis of PIK3CA mutations in breast cancer subtypes. Appl. Immunohistochem. Mol. Morphol. AIMM 2014, 22, 50–56. [Google Scholar] [CrossRef]
- Zhang, Y.; Ye, L.; Tan, Y.; Sun, P.; Ji, K.; Jiang, W.G. Expression of breast cancer metastasis suppressor-1, BRMS-1, in human breast cancer and the biological impact of BRMS-1 on the migration of breast cancer cells. Anticancer. Res. 2014, 34, 1417–1426. [Google Scholar]
- Corso, G.; Veronesi, P.; Sacchini, V.; Galimberti, V. Prognosis and outcome in CDH1-mutant lobular breast cancer. Eur. J. Cancer Prev. 2018, 27, 237–238. [Google Scholar] [CrossRef] [PubMed]
- Chimonidou, M.; Tzitzira, A.; Strati, A.; Sotiropoulou, G.; Sfikas, C.; Malamos, N.; Georgoulias, V.; Lianidou, E. CST6 promoter methylation in circulating cell-free DNA of breast cancer patients. Clin. Biochem. 2013, 46, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keen, R.W.; Hart, D.J.; Lanchbury, J.S.; Spector, T.D. Association of early osteoarthritis of the knee with a Taq I polymorphism of the vitamin D receptor gene. Arthritis Rheum. 1997, 40, 1444–1449. [Google Scholar] [CrossRef]
- Gleghorn, L.; Ramesar, R.; Beighton, P.; Wallis, G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 2005, 77, 484–490. [Google Scholar] [CrossRef]
- Wei, F.Y.; Lee, J.K.; Wei, L.; Qu, F.; Zhang, J.Z. Correlation of insulin-like growth factor 1 and osteoarthritic cartilage degradation: A spontaneous osteoarthritis in guinea-pig. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4493–4500. [Google Scholar]
- Verma, P.; Dalal, K. ADAMTS-4 and ADAMTS-5: Key enzymes in osteoarthritis. J. Cell. Biochem. 2011, 112, 3507–3514. [Google Scholar] [CrossRef]
- Shen, J.; Li, S.; Chen, D. TGF-β signaling and the development of osteoarthritis. Bone Res. 2014, 2, 14002. [Google Scholar] [CrossRef] [Green Version]
- Pullig, O.; Tagariello, A.; Schweizer, A.; Swoboda, B.; Schaller, P.; Winterpacht, A. MATN3 (matrilin-3) sequence variation (pT303M) is a risk factor for osteoarthritis of the CMC1 joint of the hand, but not for knee osteoarthritis. Ann. Rheum. Dis. 2007, 66, 279–280. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.-J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef] [Green Version]
- Vikkula, M.; Palotie, A.; Ritvaniemi, P.; Ott, J.; Ala-Kokko, L.; Sievers, U.; Aho, K.; Peltonen, L. Early-onset osteoarthritis linked to the type ii procollagen gene. detailed clinical phenotype and further analyses of the gene. Arthritis Rheum. 1993, 36, 401–409. [Google Scholar] [CrossRef]
- Raine, E.V.; Dodd, A.W.; Reynard, L.N.; Loughlin, J. Allelic expression analysis of the osteoarthritis susceptibility gene COL11A1 in human joint tissues. BMC Musculoskelet. Disord. 2013, 14, 85. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, Z.; Chapman, K.; Irven, C.; Carr, A.J.; Clipsham, K.; Chitnavis, J.; Sinsheimer, J.S.; Bloomfield, V.A.; McCartney, M.; Cox, O.; et al. Linkage analysis of candidate genes as susceptibility loci for osteoarthritis—Suggestive linkage of COL9A1 to female hip osteoarthritis. Rheumatology 2000, 39, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Laukkanen, O.; Lindström, J.; Eriksson, J.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Tuomilehto, J.; Uusitupa, M.; Laakso, M. Polymorphisms in the SLC2A2 (GLUT2) gene are associated with the conversion from impaired glucose tolerance to type 2 diabetes: The Finnish Diabetes Prevention Study. Diabetes 2005, 54, 2256–2260. [Google Scholar] [CrossRef] [Green Version]
- Kanatsuka, A.; Kou, S.; Makino, H. IAPP/amylin and β-cell failure: Implication of the risk factors of type 2 diabetes. Diabetol. Int. 2018, 9, 143–157. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Kazemi, B.; Seyed, N.; Moslemi, E.; Bandehpour, M.; Bikhof Torbati, M.; Saadat, N.; Eidi, A.; Ghayoor, E.; Azizi, F. Insulin receptor gene mutations in iranian patients with type II diabetes mellitus. Iran. Biomed. J. 2009, 13, 161–168. [Google Scholar]
- Zeggini, E.; Parkinson, J.; Halford, S.; Owen, K.R.; Frayling, T.M.; Walker, M.; Hitman, G.A.; Levy, J.C.; Sampson, M.J.; Feskens, E.J.M.; et al. Association Studies of Insulin Receptor Substrate 1 Gene (IRS1) Variants in Type 2 Diabetes Samples Enriched for Family History and Early Age of Onset. Diabetes 2004, 53, 3319–3322. [Google Scholar] [CrossRef] [Green Version]
- Stumvoll, M.; Häring, H. The Peroxisome Proliferator-Activated Receptor-γ2 Pro12Ala Polymorphism. Diabetes 2002, 51, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Karaglani, M.; Ragia, G.; Panagopoulou, M.; Balgkouranidou, I.; Nena, E.; Kolios, G.; Papanas, N.; Manolopoulos, V.G.; Chatzaki, E. Search for Pharmacoepigenetic Correlations in Type 2 Diabetes Under Sulfonylurea Treatment. Exp. Clin. Endocrinol. Diabetes 2019, 127, 226–233. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, C.; Yin, D.; Zhao, F.; Bao, Z.; Zhao, Y.; Wang, X.; Li, W.; Wang, T.; Jin, Y.; et al. A Variation in the ABCC8 Gene Is Associated with Type 2 Diabetes Mellitus and Repaglinide Efficacy in Chinese Type 2 Diabetes Mellitus Patients. Intern. Med. 2019, 58, 2341–2347. [Google Scholar] [CrossRef] [Green Version]
- Hattersley, A.T. Prime suspect: The TCF7L2 gene and type 2 diabetes risk. J. Clin. Investig. 2007, 117, 2077–2079. [Google Scholar] [CrossRef]
- Chauhan, G.; Tabassum, R.; Mahajan, A.; Dwivedi, O.P.; Mahendran, Y.; Kaur, I.; Nigam, S.; Dubey, H.; Varma, B.; Madhu, S.V.; et al. Common variants of FTO and the risk of obesity and type 2 diabetes in Indians. J. Hum. Genet. 2011, 56, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Lappano, R.; Jacquot, Y.; Maggiolini, M. GPCR Modulation in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 3840. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.-L.; Wang, D.-Y.; Ju, L.-G.; Yao, J.; Gao, C.; Lei, P.-J.; Li, L.-Y.; Zhao, X.-L.; Wu, M. The hyper-activation of transcriptional enhancers in breast cancer. Clin. Epigenetics 2019, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Gururaj, A.E.; Singh, R.R.; Rayala, S.K.; Holm, C.; den Hollander, P.; Zhang, H.; Balasenthil, S.; Talukder, A.H.; Landberg, G.; Kumar, R. MTA1, a transcriptional activator of breast cancer amplified sequence 3. Proc. Natl. Acad. Sci. USA 2006, 103, 6670–6675. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, W.A.; Mohamed, M.A.E.-R.; Elgharieb, A.G. Assessment of sensory perception alterations for patients receiving Antineoplastic chemotherapy. Port Said Sci. J. Nurs. 2020, 7, 94–118. [Google Scholar]
- Liu, K.; Dong, F.; Gao, H.; Guo, Y.; Li, H.; Yang, F.; Zhao, P.; Dai, Y.; Wang, J.; Zhou, W.; et al. Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol. Int. 2020, 44, 603–609. [Google Scholar] [CrossRef]
- Yu, J.; Zhu, T.; Wang, Z.; Zhang, H.; Qian, Z.; Xu, H.; Gao, B.; Wang, W.; Gu, L.; Meng, J.; et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin. Cancer Res. 2007, 13, 7296–7304. [Google Scholar] [CrossRef] [Green Version]
- Moribe, T.; Iizuka, N.; Miura, T.; Kimura, N.; Tamatsukuri, S.; Ishitsuka, H.; Hamamoto, Y.; Sakamoto, K.; Tamesa, T.; Oka, M. Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma. Int. J. Cancer 2009, 125, 388–397. [Google Scholar] [CrossRef]
- Schulz, H.; Tator, M.; Spillner, J.; Dreher, M.; Knüchel-Clarke, R.; Kloten, V.; Dahl, E. Liquid biopsy in human non-small-cell lung cancer: Blood-based analysis of ctDNA methylation. Pathologe 2018, 39 (Suppl. S2), 193–198. [Google Scholar] [CrossRef] [PubMed]
- Strand, S.H.; Orntoft, T.F.; Sorensen, K.D. Prognostic DNA methylation markers for prostate cancer. Int. J. Mol. Sci. 2014, 15, 16544–16576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Zhuang, J.; Li, H.; Liu, C.; Zhou, C.; Liu, L.; Sun, C. Exploration of methylation-driven genes for monitoring and prognosis of patients with lung adenocarcinoma. Cancer Cell Int. 2018, 18, 194. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, H.; Yamashita, S.; Fujii, S.; Tanabe, K.; Mukai, H.; Ushijima, T. DNA methylation marker to estimate the breast cancer cell fraction in DNA samples. Med. Oncol. 2018, 35, 147. [Google Scholar] [CrossRef]
- Makabe, T.; Arai, E.; Hirano, T.; Ito, N.; Fukamachi, Y.; Takahashi, Y.; Hirasawa, A.; Yamagami, W.; Susumu, N.; Aoki, D.; et al. Genome-wide DNA methylation profile of early-onset endometrial cancer: Its correlation with genetic aberrations and comparison with late-onset endometrial cancer. Carcinogenesis 2019, 40, 611–623. [Google Scholar] [CrossRef]
- Panagopoulou, M.; Cheretaki, A.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Amarantidis, K.; Xenidis, N.; Kakolyris, S.; Baritaki, S.; Chatzaki, E. Methylation Status of Corticotropin-Releasing Factor (CRF) Receptor Genes in Colorectal Cancer. J. Clin. Med. 2021, 10, 2680. [Google Scholar] [CrossRef]
- Sharma, G.; Mirza, S.; Parshad, R.; Srivastava, A.; Gupta, S.D.; Pandya, P.; Ralhan, R. Clinical significance of promoter hypermethylation of DNA repair genes in tumor and serum DNA in invasive ductal breast carcinoma patients. Life Sci. 2010, 87, 83–91. [Google Scholar] [CrossRef]
- Li, Z.; Guo, X.; Tang, L.; Peng, L.; Chen, M.; Luo, X.; Wang, S.; Xiao, Z.; Deng, Z.; Dai, L.; et al. Methylation analysis of plasma cell-free DNA for breast cancer early detection using bisulfite next-generation sequencing. Tumor Biol. 2016, 37, 13111–13119. [Google Scholar] [CrossRef]
- Salta, S.; Nunes, S.P.; Fontes-Sousa, M.; Lopes, P.; Freitas, M.; Caldas, M.; Antunes, L.; Castro, F.; Antunes, P.; Palma de Sousa, S.; et al. A DNA Methylation-Based Test for Breast Cancer Detection in Circulating Cell-Free DNA. J. Clin. Med. 2018, 7, 420. [Google Scholar] [CrossRef] [Green Version]
- Panagopoulou, M.; Karaglani, M.; Balgkouranidou, I.; Biziota, E.; Koukaki, T.; Karamitrousis, E.; Nena, E.; Tsamardinos, I.; Kolios, G.; Lianidou, E.; et al. Circulating cell-free DNA in breast cancer: Size profiling, levels, and methylation patterns lead to prognostic and predictive classifiers. Oncogene 2019, 38, 3387–3401. [Google Scholar] [CrossRef]
- Schouten, J.S.A.G.; Van Den Ouweland, F.A.; Valkenburg, H.A.; Lamberts, S.W.J. Insulin-Like Growth Factor-1: A Prognostic Factor of Knee Osteoarthritis. Rheumatology 1993, 32, 274–280. [Google Scholar] [CrossRef]
- Jin, H.; Jiang, S.; Wang, R.; Zhang, Y.; Dong, J.; Li, Y. Mechanistic Insight into the Roles of Integrins in Osteoarthritis. Front Cell Dev. Biol. 2021, 9, 693484. [Google Scholar] [CrossRef]
- Poole, A.R.; Kobayashi, M.; Yasuda, T.; Laverty, S.; Mwale, F.; Kojima, T.; Sakai, T.; Wahl, C.; El-Maadawy, S.; Webb, G.; et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. S2), ii78. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, P.; Bayliss, M.T.; Heinegård, D. Altered patterns and synthesis of extracellular matrix macromolecules in early osteoarthritis. Matrix Biol. 2004, 23, 381–391. [Google Scholar] [CrossRef]
- Lambrecht, S.; Verbruggen, G.; Verdonk, P.C.M.; Elewaut, D.; Deforce, D. Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthr. Cartil. 2008, 16, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Riddy, D.M.; Delerive, P.; Summers, R.J.; Sexton, P.M.; Langmead, C.J. G Protein—Coupled Receptors Targeting Insulin Resistance, Obesity, and Type 2 Diabetes Mellitus. Pharmacol. Rev. 2018, 70, 39. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.-P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef]
- Schuit, F.C.; Huypens, P.; Heimberg, H.; Pipeleers, D.G. Glucose Sensing in Pancreatic β-Cells: A Model for the Study of Other Glucose-Regulated Cells in Gut, Pancreas, and Hypothalamus. Diabetes 2001, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hauffe, R.; Stein, V.; Chudoba, C.; Flore, T.; Rath, M.; Ritter, K.; Schell, M.; Wardelmann, K.; Deubel, S.; Kopp, J.F.; et al. GPx3 dysregulation impacts adipose tissue insulin receptor expression and sensitivity. JCI Insight 2020, 5, e136283. [Google Scholar] [CrossRef]
- Stancill, J.S.; Broniowska, K.A.; Oleson, B.J.; Naatz, A.; Corbett, J.A. Pancreatic β-cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J. Biol. Chem. 2019, 294, 4843–4853. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, S.M.; Ross, J.P.; Drew, H.R.; Ho, T.; Brown, G.S.; Saunders, N.F.W.; Duesing, K.R.; Buckley, M.J.; Dunne, R.; Beetson, I.; et al. A panel of genes methylated with high frequency in colorectal cancer. BMC Cancer 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranova, I.; Kovarikova, H.; Laco, J.; Sedlakova, I.; Vrbacky, F.; Kovarik, D.; Hejna, P.; Palicka, V.; Chmelarova, M. Identification of a four-gene methylation biomarker panel in high-grade serous ovarian carcinoma. Clin. Chem. Lab. Med. 2020, 58, 1332–1340. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Dybska, E.; Barciszewska, A.-M. DNA methylation analysis with methylation-sensitive high-resolution melting (MS-HRM) reveals gene panel for glioma characteristics. CNS Neurosci. Ther. 2020, 26, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Zick, A.; Grinshpun, A.; Carmon, E.; Maoz, M.; Ochana, B.L.; Abraham, O.; Arieli, O.; Germansky, L.; Meir, K.; et al. Circulating breast-derived DNA allows universal detection and monitoring of localized breast cancer. Ann. Oncol. 2020, 31, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Zemmour, H.; Planer, D.; Magenheim, J.; Moss, J.; Neiman, D.; Gilon, D.; Korach, A.; Glaser, B.; Shemer, R.; Landesberg, G.; et al. Non-invasive detection of human cardiomyocyte death using methylation patterns of circulating DNA. Nat. Commun. 2018, 9, 1443. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Magenheim, J.; Moss, J.; Neiman, D.; Abraham, O.; Piyanzin, S.; Zemmour, H.; Fox, I.; Dor, T.; Grompe, M.; et al. Monitoring liver damage using hepatocyte-specific methylation markers in cell-free circulating DNA. JCI Insight 2018, 3, e120687. [Google Scholar] [CrossRef] [Green Version]
- Papoutsoglou, G.; Karaglani, M.; Lagani, V.; Thomson, N.; Røe, O.D.; Tsamardinos, I.; Chatzaki, E. Automated machine learning optimizes and accelerates predictive modeling from COVID-19 high throughput datasets. Sci. Rep. 2021, 11, 15107. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Müller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. RnBeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef] [Green Version]
- Lagani, V.; Athineou, G.; Farcomeni, A.; Tsagris, M.; Tsamardinos, I. Feature Selection with the R Package MXM: Discovering Statistically Equivalent Feature Subsets. J. Stat. Softw. 2017, 80, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Tsamardinos, I.; Greasidou, E.; Borboudakis, G. Bootstrapping the out-of-sample predictions for efficient and accurate cross-validation. Mach. Learn. 2018, 107, 1895–1922. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Theodosiou, T.; Papanikolaou, N.; Savvaki, M.; Bonetto, G.; Maxouri, S.; Fakoureli, E.; Eliopoulos, A.G.; Tavernarakis, N.; Amoutzias, G.D.; Pavlopoulos, G.A.; et al. UniProt-Related Documents (UniReD): Assisting wet lab biologists in their quest on finding novel counterparts in a protein network. NAR Genom. Bioinform. 2020, 2, lqaa005. [Google Scholar] [CrossRef]
- Papanikolaou, N.; Pavlopoulos, G.A.; Pafilis, E.; Theodosiou, T.; Schneider, R.; Satagopam, V.P.; Ouzounis, C.A.; Eliopoulos, A.G.; Promponas, V.J.; Iliopoulos, I. BioTextQuest(+): A knowledge integration platform for literature mining and concept discovery. Bioinformatics 2014, 30, 3249–3256. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Kamburov, A.; Stelzl, U.; Lehrach, H.; Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2012, 41, D793–D800. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signature Genes | Gene Type | Description | Pathway | GO—Molecular Function | GO—Cellular Components | GO—Biological Process | UniReD Score | Methylation in BrCa in Relation to Healthy Tissues |
|---|---|---|---|---|---|---|---|---|
| CCDC181 | Protein Coding | Coiled-Coil Domain Containing 181 | NA | microtubule binding | manchette, cytoplasm, cytoskeleton, microtubule, cilium | NA | 5 | Hypermethylation |
| HIST2H3PS2 | Protein Coding | Histone Cluster 2, H3, Pseudogene 2 | NA | DNA binding, protein heterodimerization activity | Nucleus, Chromosome | NA | 1 | Hypermethylation |
| RUVBL1-AS1 | RNA Gene | RUVBL1 Antisense RNA 1 | NA | NA | NA | NA | NA | Hypermethylation |
| CFTR | Protein Coding | CF Transmembrane Conductance Regulator | CDK-mediated phosphorylation and removal of Cdc6, bacterial infections in CF airways, regulation of CFTR activity, salivary secretion | nucleotide binding, chloride channel activity, intracellularly ATP-gated chloride channel activity | nucleus, cytoplasm, lysosomal membrane, endsome, early endsome | cholesterol biosynthetic process, ion transport, chloride transport, vesicle docking involved in exocytes | 7 | Hypermethylation |
| AL161908.1 | RNA Gene | Novel Transcript, Antisense To LIM1B | NA | NA | NA | NA | NA | Hypermethylation |
| Signature Genes | Gene Type | Description | Pathway | GO—Molecular Function | GO—Cellular Components | GO—Biological Process | UniReD Score | Methylation in OA in Relation to Other Tissues |
|---|---|---|---|---|---|---|---|---|
| CASD1 | Protein Coding | CAS1 Domain Containing 1 | NA | acetyltransferase activity, transferase activity, transferring acyl groups | Golgi membrane, Golgi apparatus, membrane, integral component of membrane, integral component of Golgi membrane | Carbohydrate metabolic process | 0 | Hypomethylation |
| LINC01350 | LncRNA | Long Intergenic Non-Protein Coding RNA 1350 | NA | NA | NA | NA | NA | Hypomethylation |
| RP11-515E23.2 | NA | NA | NA | NA | NA | NA | NA | Hypomethylation |
| STOML1 | Protein Coding | Stomatin-Like 1 | NA | protein binding | endosome, plasma membrane, membrane, integral component of membrane | lipid transport | 2.5 | Hypomethylation |
| CARMAL | RNA Gene | Coronary Artery Disease Region-Linked MFGE8 Regulatory LncRNA | NA | NA | NA | NA | NA | Hypomethylation |
| RP11-272L13.3 | LncRNA | NA | NA | NA | NA | NA | NA | Hypomethylation |
| Signature Genes | Gene Type | Description | Pathway | GO—Molecular Function | GO—Cellular Components | GO—Biological Process | UniReD Score | Methylation in Pancreatic β Cells in Relation to Other Healthy Tissues |
|---|---|---|---|---|---|---|---|---|
| SCARNA6 | snoRNA | Small Cajal Body-Specific RNA 6 | NA | NA | nucleolus | RNA processing | ΝA | Hypomethylation |
| TXNRD3 | Protein Coding | Thioredoxin Reductase 3 | folate metabolism and mechanisms of CFTR activation by S-nitrosoglutathione | nucleotide binding, thioredoxin disulfide reductase activity, electron transfer activity, protein disulfide oxidoreductase activity | cell, nucleoplasm, cytoplasm, endoplasmic reticulum, cytosol | multicellular organism development, spermatogenesis, electron transport chain, cell differentiation | 5.5 | Hypomethylation |
| AC008741.1 | lncRNA | Novel Transcript, Antisense To ZKSCAN2 | NA | NA | NA | NA | ΝA | Hypomethylation |
| LENG8 | Protein Coding | Leukocyte Receptor Cluster Member | NA | protein binding | nucleus | NA | NA | Hypomethylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karaglani, M.; Panagopoulou, M.; Baltsavia, I.; Apalaki, P.; Theodosiou, T.; Iliopoulos, I.; Tsamardinos, I.; Chatzaki, E. Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach. Int. J. Mol. Sci. 2022, 23, 2959. https://doi.org/10.3390/ijms23062959
Karaglani M, Panagopoulou M, Baltsavia I, Apalaki P, Theodosiou T, Iliopoulos I, Tsamardinos I, Chatzaki E. Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach. International Journal of Molecular Sciences. 2022; 23(6):2959. https://doi.org/10.3390/ijms23062959
Chicago/Turabian StyleKaraglani, Makrina, Maria Panagopoulou, Ismini Baltsavia, Paraskevi Apalaki, Theodosis Theodosiou, Ioannis Iliopoulos, Ioannis Tsamardinos, and Ekaterini Chatzaki. 2022. "Tissue-Specific Methylation Biosignatures for Monitoring Diseases: An In Silico Approach" International Journal of Molecular Sciences 23, no. 6: 2959. https://doi.org/10.3390/ijms23062959