Abstract
The peripheral nervous system (PNS) has a remarkable regenerative capacity in comparison to the central nervous system (CNS), a phenomenon that is impaired during ageing. The ability of PNS axons to regenerate after injury is due to Schwann cells (SC) being reprogrammed into a repair phenotype called Repair Schwann cells. These repair SCs are crucial for supporting axonal growth after injury, myelin degradation in a process known as myelinophagy, neurotropic factor secretion, and axonal growth guidance through the formation of Büngner bands. After regeneration, repair SCs can remyelinate newly regenerated axons and support nonmyelinated axons. Increasing evidence points to an epigenetic component in the regulation of repair SC gene expression changes, which is necessary for SC reprogramming and regeneration. One of these epigenetic regulations is histone acetylation by histone acetyl transferases (HATs) or histone deacetylation by histone deacetylases (HDACs). In this review, we have focused particularly on three HDAC classes (I, II, and IV) that are Zn2+-dependent deacetylases. These HDACs are important in repair SC biology and remyelination after PNS injury. Another key aspect explored in this review is HDAC genetic compensation in SCs and novel HDAC inhibitors that are being studied to improve nerve regeneration.
1. Introduction
Epigenetic markers such as acetylation or methylation can change gene expression. Acetylation depends on the opposite action of Histone acetyl transferases (HATs) and Histone deacetylases (HDACs), which acetylate and deacetylate histones, respectively.
HDACs have been shown to play a critical role in peripheral nerve myelination and remyelination following injury. Injured nerves in the peripheral nervous system (PNS) have the capacity to regenerate and reinnervate their target organs, which does not occur in the central nervous system (CNS), due to a lower expression of growth-promoting factors and inhibition by glial cells (reactive astrocytes and oligodendrocytes) [1]. After an injury in the PNS, Schwann cells (SCs) can differentiate into a repair phenotype. This change in the phenotype is controlled by JUN, a transcription factor that is upregulated after injury. In the absence of JUN, SCs are not able to differentiate into this repair phenotype, a phenomenon which also occurs in ageing or chronic denervation [2,3].
HDACs have been implicated in SC development, myelination, and remyelination after injury. Specifically, HDACs are needed to repress the Jun promoter and, therefore, allow myelination to proceed [4]. HDAC downregulation is also important in repair SCs, as it has been shown that when certain HDACs are depleted in SCs, remyelination, as well as the induction of the repair program, are affected [5,6].
Furthermore, there is redundancy in HDAC expression, suggesting that there is genetic compensation of HDACs in the PNS: when one HDAC is depleted, another is upregulated, and consequently peripheral nerve myelination and maintenance is conserved [6].
HDACs have also been implicated in several PNS and CNS diseases, such as Charcot Marie-Tooth (CMT), Amyotrophic lateral sclerosis (ALS), or Alzheimer’s disease (AD). HDAC therapies are being studied to improve nerve regeneration, and several studies suggest that HDAC inhibitors (HDACi) can improve disease outcome.
This review focuses on the role of HDACs in SC biology in both development and repair, as well as on genetic compensation between HDACs, a genetic mechanism that is gaining relevance as suggested by recent studies. Finally, we will discuss ongoing clinical trials involving HDACis in the PNS.
2. Histone Deacetylases (HDACs)
Chromatin in eukaryotic cells is organized in nucleosomes. DNA is wrapped around histone proteins to form compact nucleosomes, which are defined by an octamer core of histone protein wrapped by 147 base pairs (bp) of DNA. The state of chromatin and, therefore, the accessibility of the genetic code depends on DNA and histone modifications. Methyl residues can be added to DNA, and acetyl groups can be added to histones. The complex formed by DNA, histone proteins, and RNA that packages the genetic information of the cell is called the epigenome [7,8].
Histone acetylation is regulated by two groups of enzymes with opposing functions: Histones Acetyltransferases (HATs), which add an acetyl group, and Histone Deacetylases (HDACs), which remove these acetyl groups. HATs and HDACs control acetylation levels in eukaryotic cells [9]. HDACs most frequently repress gene transcription by forming a complex with transcription factors and other proteins to regulate fundamental cellular processes such as cell cycle progression, survival, and differentiation [10].
Until now, 18 human HDACs have been described, all of which have a highly conserved deacetylase domain [10]. They are divided into four classes based on their homology with yeast deacetylases: class I, class II, class III, and class IV (Figure 1). Class I is composed of HDAC1, HDAC2, HDAC3, and HDAC8. Class II is divided into two groups: class IIa, which includes HDAC4, HDAC5, HDAC7, and HDAC9, and class IIb, composed of HDAC6 and HDAC10. Class III includes Sirtuins (SIRT) 1–7. Class IV is formed only by HDAC11. The enzymatic activity of class I, class II, and class IV HDACs depends on Zn2+ ions. In contrast, Class III enzymatic activity depends on nicotinamide adenine dinucleotide (NAD)+ levels [11,12]. For this review, we will further describe only HDAC classes I, II, and IV, and discuss the growing evidence for their roles in SC biology and nerve regeneration.
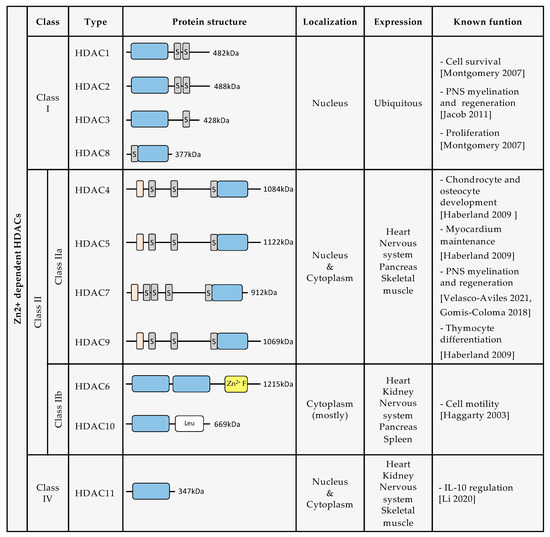
Figure 1.
Description of protein structure, localization, expression, and known biological functions of Zn2+ dependent HDACs [4,6,13,14,15,16,17]. (Dark grey rectangles containing an S indicate serine residues. Orange rectangles indicate a MEF2 binding site. Blue rectangles indicate the deacetylase domain. Yellow rectangle indicates a Zn2+ finger. White rectangle indicates leucine rich domain).
2.1. Class I HDACs
HDAC1, HDAC2, HDAC3, and HDAC8 belong to class I HDACs. Class I HDAC expression is ubiquitous, and regulates fundamental cellular mechanisms such as cell proliferation, survival, and migration [10].
Class I HDACs share a structural similarity to yeast RPD3. They have a deacetylase domain with short amino- and carboxy-extensions and are predominantly confined to the nucleus. HDAC1, HDAC2, and HDAC3 form multiprotein complexes, more specifically co-repressor complexes that are regulated by inositol phosphate. Often, HDAC1 and HDAC2 are recruited together, forming the same co-repressor complex, such as the nucleosome remodeling and deacetylase complex (NuRD), Sin3 complex, CoREST complex, or the mitotic deacetylase complex (MiDAC). HDAC3’s enzymatic activity is switched on when it interacts with the deacetylase activation domain (DAD) of NCOR1 and SMRT (also called NCOR2), forming the NCOR/SMRT complex [7,16,18].
2.2. Class II HDACs
Class II HDACs have structural homology with yeast HDA1 and have a conserved, inactive deacetylase domain in their C-terminus. They are classified into two subclasses: class IIa, which consists of HDAC4, HDAC5, HDAC7, and HDAC9, and class IIb, which consists of HDAC6 and HDAC10 [19,20].
Class IIa HDACs have a nuclear localization sequence (NLS) which allows them to shuttle to the nucleus, where they exhibit poor enzymatic activity due to the substitution of a catalytically important tyrosine for a histidine [21]. They are only considered to be active when forming a complex with NCOR/SMRT and HDAC3 (which provides the catalytic activity) [18]. All Class IIa HDACs also have conserved serine residues in their N-terminal end, which are important for signal dependent phosphorylation and, therefore, for their localization and function [16,22]. Class IIa HDACs are expressed in the heart, skeletal muscle, brain, and pancreas, and play a significant role in chondrocyte and osteocyte development. They are also relevant for thymocyte differentiation and myocardium maintenance [10].
Class IIb HDACs are mainly cytoplasmic and appear to be important for cell motility. HDAC6 has a microtubule binding domain, and some of its major substrates are non-histone proteins such as α-TUBULIN, Contractin, Ku70, and HSP90 [23]. It also has highly conserved domains that are necessary for cytoplasmic localization, as well as for nuclear export. Nuclear export signaling (NES) mediates translocation to the cytoplasm and interaction with cytoskeletal proteins. HDAC6 is the only class II HDAC with two independent catalytic domains. HDAC10 has a leucine rich domain, and like class IIa HDACs, HDAC10 associates with class I HDACs, forming a multiprotein repression complex [16].
HDAC6 and HDAC10 are expressed in the brain, heart, liver, kidney, pancreas, and spleen [10,24].
2.3. Class III HDACs
Class III HDACs are NAD+-dependent and are also known as SIRTs, with seven being currently described (SIRT1–7). They have been found in the nucleus, cytoplasm, and mitochondria, and exhibit deacetylase and ADP ribosyl transferase activities that regulate other proteins [18,25].
2.4. Class IV HDACs
HDAC11 is the only class IV HDAC. It is equivalent to yeast Hos3 and has homology in its catalytic domain with class I and class II HDACs. Its fatty acid acylase activity is more important than its deacetylase activity [26]. It regulates interleukin 10 expression and the DNA replication factor CDT1 [16]. HDAC11 has a ubiquitous subcellular location, but is predominantly confined to the nucleus [16,27]. It is highly expressed in the brain, but is also present in skeletal muscle, heart, kidney, and testis [27].
3. The Role of HDACs in Schwann Cells
Schwann cells (SCs) are the principal glial cells in the Peripheral Nervous System (PNS). They play essential roles in the development, maintenance, function, and regeneration of peripheral nerves.
SCs are derived from neural crest cells, migrate along axons to reach their target tissue, and later differentiate into SC precursors and then into immature SCs between embryonic days 12 and 15 in mice. Around birth, axonal sorting and myelination begin in peripheral nerves. In the mature nervous system, SCs can be classified into two major classes: myelinating and non-myelinating SCs [3,28]. There are, however, also terminal SCs in the neuromuscular junction [29] and nociceptive SCs in the skin [30]. Myelinating SCs provide the myelin ensheathment of all large-diameter peripheral axons or A fibers (relay motor or proprioceptive information, touch, or pressure). Non-myelinating SCs, or Remak SCs, ensheath several small caliber axons or C fibers (respond to pain, thermal, or mechanic stimuli). After axonal sorting, myelinating SCs establish a 1:1 relationship with large diameter axons and wrap around them multiple times to form a thick and compact myelin sheath. This allows the fast conduction of action potentials by insulating the axons [3]. SCs’ decision whether to myelinate or not is dictated by the axon itself, based on the amount of neuregulin 1 type III exposed on its membrane, activating ERBB2 and ERBB3 receptors on SC membranes [31]. The non-myelinating SCs typically associate with several small-diameter axons to form Remak bundles that provide support to the axon. Interestingly, SC differentiation into myelin or non-myelin SCs is reversible during pathology, accounting for the remarkable plasticity of SCs and contributing to the regenerative potential of the PNS [32].
The PNS has a higher regenerative capacity than the CNS. This ability to quickly recover following damage is, to a large extent, due to the plasticity of SCs. Nerve injury triggers the conversion of myelin and non-myelin SCs to a cell phenotype specialized to promote repair, known as repair SCs. These cells provide the necessary signals and spatial cues for the survival of injured neurons, axonal regeneration, and target reinnervation. The conversion into repair SCs involves de-differentiation together with alternative differentiation or activation, a combination that is typical of cell type conversions often referred to as (direct or lineage) reprogramming [33,34]. Thus, injury-induced SC reprogramming involves down-regulation of myelin genes, combined with activation of a set of repair-supportive features, including up-regulation of trophic factors and an increase in cytokines as part of the innate immune response, as well as myelin clearance by activation of myelin autophagy in repair SCs (a process called myelinophagy) [35], and phagocytosis of myelin debris by macrophages and SCs [36,37,38]. Moreover, repair SCs promote macrophage recruitment by LIF (leukemia inhibitory factor) and MCP-1 (macrophage chemoattractant protein-1) secretion, and the formation of regeneration tracks, Büngner bands, that serve as guidance for axons to their targets [32,33,34,35,36,37,38,39]. This repair program is controlled by a transcriptional program involving the transcription factor JUN, which is strongly and rapidly upregulated in SCs after injury. Jun elevation in Schwann cells after nerve injury was first reported by De Felipe and Hunt in 1994 [40]. In the absence of Jun, damage results in the formation of a dysfunctional repair cell, leading to neuronal death and failure of functional recovery. Jun, although not required for SC development, is critical for the reprogramming of myelin and non-myelin (Remak) SCs into repair cells following injury. SC reprogramming is impaired in some diseases or in ageing due to a slow/weak upregulation of some crucial transcription factors, such as Jun or Stat3 [2,3,39,41,42].
Due to their significant role in nerve regeneration, repair SCs and their activation, transcriptome and proteome have been of increasing interest, and have recently also been studied in humans [43,44].
3.1. The Role of HDACs during Schwann Cell Development
Several epigenetic modifications involved in the regulation of neural crest development have been described. Regarding HDACs in the SC lineage, it has been shown that class I and class II HDACs have an important role in SC development. In mouse models, the use of conditional knockouts (Dhh-Cre, Table 1) has shown that Hdac1 and Hdac2 depletion results in massive SC loss, probably through a significant reduction in Sox10 expression [45]. Hdac1/2 deletion in neural crest cells, using the Wnt1-cre mouse, shows that HDAC1/2 activity is necessary for neural crest cell differentiation into SC precursors and satellite glial cells [45]. In this study, authors show that HDAC1/2 bind and induce the Pax3 promoter. This Pax3 induction is required to maintain SOX10 levels in developing SCs and satellite glia cells. Also, HDAC1, HDAC2, SOX10, and PAX3 are recruited to the Mpz promoter to induce its activation and the expression of MPZ [45]. In addition, SOX10 independently activates the fabp7 promoter and, therefore, regulates FAPB7 protein expression, which is another early marker of the SC lineage [28,45].

Table 1.
Conditional or tamoxifen inducible knockout mice used to study the role of HDACs in Schwann cells (SCs). MGI ID number from the Mouse Genome Informatics (http://www.informatics.jax.org/) (Accessed on 26 February 2022).
3.2. The Role of HDACs in Myelination
SCs wrap around axons several times and insulate them to allow saltatory conduction of action potentials. The process of myelination begins after birth in peripheral nerves, when large diameter axons are already sorted in a 1:1 ratio with myelinating SCs and start to be ensheathed by individual SCs spaced along their length. To produce myelin, SCs upregulate myelin genes, and genes encoding the enzymes that synthesize lipids to form the myelin sheath, as well as several proteins that compact and stabilize myelin (Mpz, Mbp, Cnp and others). This genetic program is determined by several transcription factors, such as SOX10, OCT6, and KROX20, and involves downregulation of negative regulators of myelination, such as JUN, NOTCH, SOX2, or ID2 [34]. It has also been demonstrated that a rise in cAMP induced by the G protein-coupled receptor 126 (Gpr126) controls SC differentiation and the myelination process [50]. Gomis–Coloma et al. (2018) show that Class II HDACs have an important role in SC differentiation and myelination. Cytoplasmic HDAC4 responds to cAMP signaling by shuttling into the nucleus. Once in the nucleus, HDAC4 represses the Jun promoter by binding it in a complex with NCOR1 and HDAC3 [4]. In this study, it was also shown that HDAC4 and HDAC5 have a compensatory contribution in the activation of the myelin transcriptional program and myelination process in vivo. Meanwhile, Hdac4/5 double knockout mice showed elevated Jun expression and delayed axonal myelination, consistent with findings using cultured SCs [4]. Moreover, HDAC5 regulates the repression of Maf during myelination. Maf has been implicated in myelination, as its ablation in myelinating Schwann cells causes hypomyelination. Specifically, HDAC5 is phosphorylated by nuclear CaM-kinases in response to NRG1 signaling and is excluded from the nucleus, increasing Maf activation and induction of cholesterol biosynthetic genes [51].
HDAC1/2 also have critical functions during the myelination process. Hdacs1/2 double cKO mutants are characterized by a radial sorting delay, the absence of myelin, and large amounts of SC apoptosis [15]. In this study, the authors identify specific primary functions for HDAC1 and HDAC2: while HDAC1 maintains SC survival in early postnatal SCs by preventing a precocious increase of active beta-catenin levels, HDAC2 acts together with SOX10 to activate the transcription of Sox10, Krox20, and Mpz and, thereby, induces the myelination program [15]. In a similar study, Chen et al. (2011) show that the absence of HDAC1/2 prevents developmental myelination and leads to low Sox10 expression in SCs, consistent with the study published by Jacob, et al. (2011) [15,52].
HDAC1 and HDAC2 have also been implicated in myelin maintenance in adult nerves. In Hdac1/2 inducible conditional knockouts using inducible Mpz-CreERT2, motor and sensory functions are impaired, and there is a decrease in Mpz expression. These decreased levels of MPZ cause impaired nodal and paranodal structures with lower levels of NFacs186, NFasc155, and Caspr [46]. In addition, authors show that in this inducible conditional knockout (with Plp1-CreERT), demyelination in vitro in SC co-cultures with dorsal root ganglion (DRG) neurons, is decreased when exogenous MPZ is expressed in SC [46].
Two studies have identified HDAC3 as an inhibitor of developmental myelination, allowing the shift to a homeostatic myelination program which is responsible for myelin maintenance in adults nerves. The loss of this stability is associated with neuropathology [5,49]. HDAC3 inhibition during development promotes Krox20 expression, resulting in hypermyelination. Additionally, HDAC3 antagonizes Neuregulin–PI3K–AKT signaling in myelination. In Hdac3 conditional knockouts, under Cnp-cre (Hdac3 cKO) or tamoxifen inducible Plp1-CreERT (Hdac3 iKO), there is an increase in promyelinating transcription factors and myelin gene expression, causing hypermyelinated axons during development and in mature nerves [5]. In Hdac3 conditional mutant mice, under Mpz-cre, it has been shown that HDAC3 is a regulator of myelin gene expression, and HDAC3 loss of function in SC results in hypermyelination in adult nerves [49]. Further studies are, however, needed to elucidate the role of HDAC3 in development and myelin maintenance in SCs.
In addition, other studies show that HDAC1, HDAC2, and HDAC3 form a complex together with Schwann cell factor 1/positive regulatory domain protein 4 (SC1/PRDM4) [53]. This complex represses Cyclin E transcription through the binding of SC1/PRDM4 to the Cyclin E promoter and causes cell proliferation arrest. SC1 is a p75NTR-interacting zinc finger protein [53]. The role of this complex has, however, not yet been studied in vivo.
HDAC1 and HDAC2 have also been implicated in myelination as part of the NuRD complex. Along with SWI/SNF-like ATPase subunit (CHD3 or CHD4, chromodomain helicase DNA-binding protein), they can reposition nucleosomes. A SC-specific deletion of Chd4 caused a transient delay in myelination, and demyelination in mature nerves [54]. The Nab1/2 and Chd4 knockouts, however, did not show as profound of a loss of SOX10 and KROX20 transcription factors as the Hdac1/2 double knockout did. While the NuRD complex was originally thought to be primarily repressive, either CHD4 or the associated NuRD complex can also activate transcription. Accordingly, the CHD4 conditional knockout nerves showed deficient repression of Krox20 target genes that are normally repressed during SC development, but also reduced activation of some major myelin genes [54].
Histones are not the only target of HDACs, as they also regulate transcription factors like NF-κB. HDAC1 and HDAC2 modulate the acetylation state of NF-κB, a process that is critical to switch on the myelination program in SCs. HDAC1/2 mutants, using Dhh-cre mice, and Hdac1 and Hdac2 floxed mice, present heavily acetylated NF-κB-p65, blocking the expression of positive regulators of myelination such as Sox10, Oct6, and Krox-20 (Egr2), and inducing the expression of differentiation inhibitors, such as Sox2, Sox11, Jagged1, Jun (c-Jun), Hes1, Hes5, Id2, and Id4 [52]. HDAC1 also directly interacts with SOX10, and this appears to be required for gene activation and maintenance of Mpz expression even in mature SCs [15,46].
While there are no in vivo studies in the PNS involving HDAC11, the unique class IV HDAC, there are some developmental studies in oligodendrocytes, the myelinating glial cells in the CNS. Specifically, Hdac11 KO (null KO) mice show reduced Mbp and Plp expression due to a reduction of acetylation levels in histone H3 at residues lysine 9 and lysine 14 (H3K9/K14ac) [55].
3.3. Hdac Expression during Development and Myelination
Recent studies using RNA sequencing (RNA-seq) and single cell RNA-seq granted the opportunity to review gene expression in peripheral nerves at different time points in different cell types. The Sciatic Nerve Atlas (SNAT) webpage from the University of Zurich allows simple and quick checks of gene expression in the SC lineage [56]. The description of gene expression in SNAT during nerve development (from E13.5 to P60), has become an important tool for the identification of important genes and the discrimination of cell type specific expression (Figure 2).
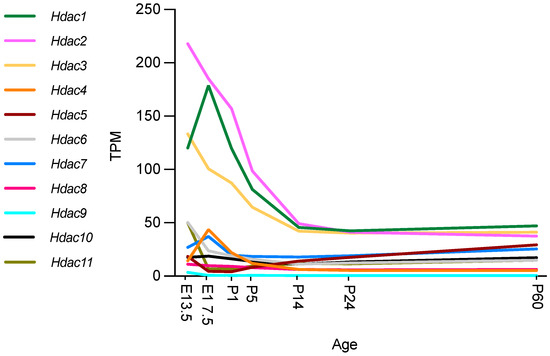
Figure 2.
Hdac expression during mouse nerve development. Expression in transcripts per million (TPM). Graphs ggenerated based on RNA-seq data from Gerber et al., 2021, GEO database accession number GSE137870 [56]. Authors used four independent samples for bulk RNA sequencing at every time point, two from male and two from female mice. At E13.5 and E17.5, sciatic nerves were pooled from two embryos of the same sex per sample. At all postnatal time points examined (P1, P5, P14, P24, and P60), sciatic nerves from one mouse were used for each independent sample [56].
All HDAC subtypes are expressed in nerves. Class I Hdacs, which include Hdac1, Hdac2 and Hdac3, are mostly expressed during development and at P60. Hdac6 and Hdac11 are predominantly expressed during embryonic development (E13.5) after which expression decreases significantly. Similarly, Hdac4 is mainly expressed during embryonic development, although expression is significantly upregulated between E13.5 and E17.5 and then again significantly downregulated by P1. Gerber et al. further describe in their single cell sequencing data (see Figure 3) that SCs are not the main cell source for Hdac4, Hdac6, Hdac7, and Hdac11 gene expression levels during development, with fibroblast-related cells and endothelial cells contributing. Expression levels of these Hdacs are, overall, not as high as Hdac1, Hdac2 and Hdac3, which are highly expressed during embryonic development and then reach significantly lower levels by P14, that are maintained into adulthood. Hdac5 gene expression is significantly upregulated during early embryonic development (E13.5), and then drops before birth (E17.5). After birth, Hdac5 and Hdac7 are both gradually, but significantly, upregulated until P60. SCs are, however, also not the main cell source contributing to the expression of these HDAC subtypes, with especially pericytes/endothelial cells contributing (Figure 3) [56].
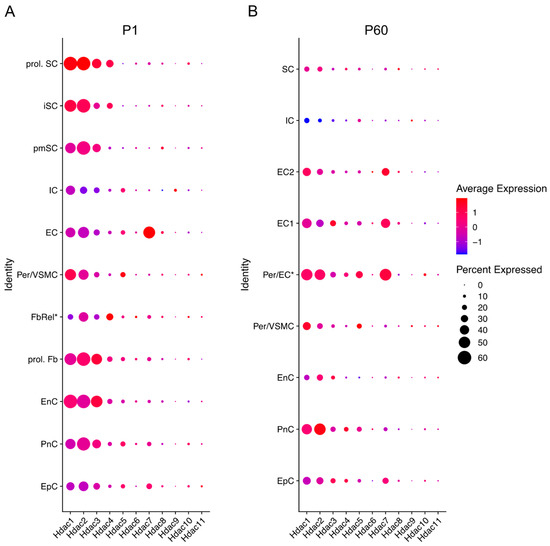
Figure 3.
Hdac expression across different cells in the sciatic nerve at P1 (early developmental stage) and P60 (mouse adulthood). Data based on 10× genomics single cell RNA sequencing data from Gerber et al. (2021). Graphs generated based on RNA-seq data from Gerber et al., 2021 (GEO database accession number GSE137870). [56]. Three independent samples were included for each time point, processed as three independent 10× Genomics runs. Expression is detailed in proliferating (prol. SC), immature (iSC), and pro-myelinating (pmSC) Schwann cells; immune cells (IC), endothelial cells (EC), pericytes, and vascular smooth muscle cells (per/VMSC), pericytes/endothelial cells (Per/EC*), endothelial cells 1 and 2 (EC1&2), proliferating fibroblast-like cells (prol. Fb), fibroblast-related cells (FbRel*), endoneurial cells (EnC, also known as endoneurial fibroblasts or fibroblast-like cells), perineurial cells (PnC), and epineurial cells (EpC). Dot colour represents average expression level in cells expressing the Hdac of interest. Dot size represents percentage of cells expressing the Hdac of interest. (* indicates tentative label suggested by Gerber et al., 2021). Futher single cell RNA-seq gene expression can be checked in the Sciatic Nerves Atlas: https://snat.ethz.ch/index.html (Accessed on 26 February 2022).
Hdac8 and Hdac10 show low expression levels overall, while Hdac9 is undetectable in nerves after birth. These Hdac subtypes are mainly expressed in epineurial, perineurial, and endoneurial cells (Figure 3) [56].
3.4. The Role of HDACs in Repair Schwann Cells and Remyelination
To understand the role of HDACs in nerve regeneration and repair SC reprograming, the use of HDAC inhibitors and genetically modified organisms, predominantly mutant mice, is essential. Kim et al. (2019) used a reversible HDAC class I and II inhibitor, Trichostatin A (TSA), to study the role of HDACs in nerve regeneration. TSA is an antimicrobial drug extracted from Streptomyces hygroscopicus, which acts as a noncompetitive enzyme inhibitor. It effectively suppresses SC reprograming after nerve injury, and inhibits myelin and axon clearance, as well as the reprogramming and proliferation of SCs [57]. The application of TSA to regulate HDAC levels in SCs could consequently be an effective way to delay peripheral neurodegeneration (Table 2).
Table 2.
HDACs inhibitor or activator used in in vivo Schwann cell (SC) or peripheral nervous system (PNS) studies. (Note: Mocetinostat is also an HDAC3 and HDAC11 inhibitor. Theophylline is also an HDAC1 enhancer).
The class I HDAC inhibitor valproic acid (VPA), which is extensively used as an antiepileptic drug, has been shown to improve nerve regeneration as well. It selectively induces proteasomal degradation of HDAC2. In rats, when VPA conduits (silicon tubes coated with VPA) are used following sciatic nerve injury, regeneration and functional recovery are improved [58,59]. A recent study using bioabsorbable conduits (hydroxyapatite/poly d-l-lactic acid, PDLLA) with sustained release of VPA in injured rats has shown improved peripheral nerve regeneration that is comparable to autografts. These autografts are regularly used to repair peripheral nerve injuries and have better results than empty conduits, but they have the disadvantages of limited availability and donor site morbidity [60]. These studies using TSA and VPA are of particular interest, as they show that inhibition, degradation, or knockout of class I HDACs does not necessarily have the same effect on regeneration. This can be explained by contextual differences in animal models, different mechanisms of action of inhibitors used, subcellular localization and post-translational modifications of HDACs, or different substrates or diverse binding proteins [61].
In vivo studies, using conditional knockout mice of HDACs class I and II, support HDACs playing a major role in repair SC regulation and nerve regeneration. Brügger et al. (2017) describe how HDAC1 and HDAC2 are strongly upregulated in SC after nerve injury and show that, in Hdac1/2 dKO (using Mpz-CreERT2), remyelination is impaired after injury in the adult [47]. They identified that SUMOylated HDAC2 is responsible for the early injury response in SCs. Specifically, Sox10, in coordination with HDAC2, recruits other chromatin-remodeling enzymes (demethylases), such as JMJD2C and KDM3A, to activate Oct6 and Krox20 genes (sox10 targets) after nerve injury [47].
Another study with class I HDACs shows that a class I HDAC knockout mediated reduction in Oct6 levels results in faster conversion into repair SCs, increased myelin clearance, and improved axonal regrowth after injury [62]. To stimulate early remyelination in wildtype C57BL/6 mice, Brügger et al. (2017) used a short treatment with Mocetinostat, a HDAC1/2 inhibitor, which increased regeneration and remyelination after injury [47]. They also documented that by stimulating eEF1A1 deacetylation, myelination is stimulated. Acetylated eEF1A1 translocates to the nucleus where it binds to Sox10 and transports it out of the nucleus. By stimulating eEF1A1 deacetylation, Sox10 remains inside the nucleus where it targets promyelinating genes increasing the remyelination process in PNS and CNS [48].
Conditional or inducible knockout of Hdac3 enhances SC remyelination and myelin thickness after injury [5]. These observations suggest that HDAC3 inhibition accelerates SC differentiation, enhances remyelination, and improves functional recovery in repair SCs. Inhibitors such as sodium phenylbutyrate (PBA) decrease inflammation induced by SC through the inhibition of HDAC3 activity and regulation of protein expression via the NFκB-p65 pathway [63]. When PBA is used in injured mice, axonal regeneration is stimulated, as well as remyelination. When HDAC3 protein levels decrease, nuclear translocation of NFκB-p65 is induced, reducing the levels of pro-inflammatory cytokine expression (such as TNFα, IL-1β, and IL-6) in SCs. These findings support the idea that HDACs play a role in the transcriptional regulation of pro-inflammatory cytokines [63] and suggest a potential application for HDAC3 inhibition in improving peripheral nerve regeneration.
Recent work carried out by Velasco–Aviles et al. (2022) focusing on class II HDACs has shown that they also play an important role in repair SC biology and remyelination, in particular Hdac4, Hdac5, and Hdac7, which are expressed in SCs [6]. In a SC-specific triple cKO of these HDACs, using Mpz-Cre mice, they saw that remyelination is impaired, but repair SC activation is accelerated due to increased myelin clearance in mutant mice after injury. The same effect can be observed in development in the Hdac4/5 dKO or the Hdac4/5/7 triple knockout (tKO). Hdac4/5/7 tKO mice show elevated JUN levels after nerve injury, and this JUN upregulation has been shown to delay remyelination in nerve regeneration, the same delay seen in Jun_OE overexpressing mice [64]. In injured nerves of single cKO for Hdac5 and Hdac7, Velasco et al. (2022), did not find a significant effect on remyelination or in repair SC activation, and in Hdac4 single cKO there was only a slight delay in remyelination, but not in repair SC activation [6].
To date, no nerve regeneration studies have been performed with Hdac11 knockouts (class IV HDAC) or a specific class IV drug such as elevenostat (JB3-22) [26,65].
Further to the role of HDACs in SCs, levels of histone acetylation in axons have vital roles after injury and in repair (reviewed in [66,67,68,69]). In sensory neurons, injury produces a calcium wave that induces the PKCγ-dependent nuclear export of HDAC5, and consequently axon regeneration is accelerated [70]. HDAC5 is also transported to the growth cone in injured axons, which modulates growth-cone dynamics to sustain axon regeneration [70]. In addition, it has been described that calcium-dependent activation of PP4/2 signaling controls the axonal regenerative ability via HDAC3 inhibition [71]. Furthermore, HDAC6 inhibition increases growth cone size in sensory axons and increases axonal acetylated Miro1, which prevents chondroitin sulfate proteoglycans-dependent decreases in mitochondrial transport and sensory axon growth [72].
A further factor determining the effect of HDACs during myelination and remyelinations is the dependence of myelin on high amounts of fatty acids for its assembly and maintenance. SCs are consequently especially vulnerable to lipid synthesis dysregulation. Acetyl-CoA (acetyl coenzyme A) is a crucial metabolite for this, as it is at the crossroads of lipid metabolism and energy generation. It is the substrate of HATs and HDACs, and, therefore, inhibition of HDAC activity, both genetically or pharmacologically, could impact on acetyl-CoA metabolism. Specifically, it could create a metabolic deficit of Acetyl-CoA in SCs that could affect fatty acid synthesis, impacting myelination. While regulation of SC fatty acid synthesis has been described in several papers, with studies focusing on the synthesis of the fatty acids precursor, acetyl-CoA, there are no studies which use Hdac mutant mice or pharmacological inhibitors that consider Acetyl-CoA levels [73,74,75,76,77]. Myelination or remyelination impairment in some HDAC knockouts could, however, be caused by Acetyl-CoA dysregulation in the SC and it would, consequently be interesting to explore this in future studies using Hdac or Hat mutant mice.
3.5. HDACs in Ageing or Disease
The PNS, too often the ignored younger cousin of the CNS, is a wonderfully intricate system composed of neurons, glia, immune, and connective cells, all operating in concert to provide some functions in common with the brain, and some, such as the capacity to regenerate, that are unique. Given this complexity, it is not surprising that many age-associated disorders of the PNS are well described and yet sorely lacking in mechanistic insight. For example, the incidence of peripheral neuropathies increases rapidly with advancing age, with some estimates suggesting that nearly one third of the population over the age of 65 suffers from some form of neuropathy [78]. The consequences of peripheral neuropathies are disabling, commonly manifesting as sensations of burning, tingling, or numbness in the extremities. Furthermore, the accompanying loss of balance can contribute to traumatic falls in the elderly [79,80,81].
In the aged injured PNS, the interaction between SC and regenerative axons takes longer, and myelin clearance, macrophage recruitment, and the amount of trophic factors secreted by repair SC and target organs decreases. Recently, five separate studies focussing on the ageing PNS in murine systems came to a similar, surprising conclusion: aged SC or macrophage dysfunction, and not neuronal dysfunction, ultimately restrains the re-growth of axons after nerve injury [41,82,83,84,85,86,87]. In addition, it has been demonstrated that age-associated or long-term denervated stumps in injured mice show defective SCs with impaired nerve regeneration after injury [87,88,89]. This deterioration of nerves distal to the injury site has been modelled and documented in rodents and is considered a major reason for regeneration failure in humans [84,85,87,90]. The mechanisms that control the phenotypic instability of repair cells are, therefore, attractive therapeutic targets, as well as of great biological interest, yet so far unexplored. Aged SC have a reduced capacity to be reprogramed into repair SC. Downregulation of myelin genes is impaired, and repair associated gene upregulation is also decreased. There is a poor upregulation of Jun in aged SC after injury [41]. When this diminished Jun upregulation is restored in SC, regeneration in aged nerves is improved [87]. Recently, it has been described that the transcription factors JUN and STAT3 play a fundamental role in the long-term maintenance of repair or ageing SCs [87,88]. In summary, aged repair SCs have a reduced ability to induce Jun activation, a decreased exogenous myelin debris ingestion, and a low cytokine expression after nerve injury [42,84]. It has also been shown that repair SC length diminishes in long-term denervated nerve stumps [91].
During the last 20 years, it has become increasingly clear that epigenetics, including DNA methylation, histone modifications, non-coding RNAs, and microRNA, plays a crucial role in the ageing process [92]. Histone modifications, including methylation and acetylation, have been intimately linked to lifespan regulation [93]. These modifications are reversible, and their reversal could consequently be used as a tool to improve ageing dependent degeneration and increase lifespan. The possibility for lifespan regulation is best characterized in this regard, mainly due to the advent of HDAC inhibitors from the cancer biology field [94]. In yeast, C. elegans and Drosophila Melanogaster it has been shown that class II HDAC inhibition or deletion extends lifespan through trehalose metabolism (a conserved storage carbohydrate) [95]. The function of Class II HDACs in ageing is relatively unknown and the existing data is sometimes contradictory. For example, in a Huntington’s disease model, the reduction of HDAC4 is shown to facilitate clearance of pathogenic huntingtin protein that aggregates and enhances neuron degeneration [96]. In contrast, another study reported HDAC4 overexpression delaying cellular senescence via SUMOylation of the class III HDAC SIRT1, whereas HDAC4 knockdown leads to premature senescence in human fibroblasts [97]. Class I HDACs such as HDAC1 may also play an important role in ageing. In the ageing brain, in a model of Alzheimer’s disease, HDAC1 deficiency causes impaired OGG1-initated 8-oxoguanine activity, an enzyme that is responsible for repairing oxidative DNA damage in the brain [98]. Additionally, the role of HDAC11 has been investigated in multiple sclerosis, schizophrenia, learning, and memory [99,100,101].
Although the role of HDACs in regeneration in the aged PNS has been poorly studied, it has been broadly studied in cancer, neurodegeneration, cardiometabolic diseases, liver dysfunction, sarcopenia, inflammation, and arthritis, as well as in diseases characterized by premature ageing in preclinical mouse studies. For some of these ageing-related diseases (such as liver dysfunction or sarcopenia), evidence for HDAC inhibitors as an effective treatment is only now emerging. There is substantial evidence for a role of HDACs in many physiological processes and, therefore, potential therapeutic applications are being investigated in a range of ongoing clinical trials in various fields, including neurodegeneration and regeneration [102].
3.6. Hdac Levels after Injury and Ageing
Using RNA-seq data from a nerve injury study [87], it is possible to gain insight into the expression of Hdac genes after nerve injury in young and aged mice, and in chronic injury in young mice (long-term nerve injury without axonal regeneration). While all Hdacs are expressed to some extent after injury, the expression levels of some of them are low (Figure 4).
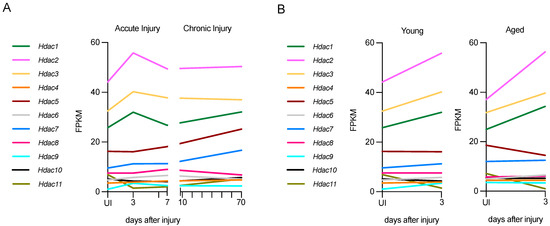
Figure 4.
Hdac expression in acute and chronic injury in young nerves or acute injury in aged nerves. (A) Hdac expression in young nerves, both in acute (up to 7 days) and chronic injury (up to 70 days). (B) Hdac expression in acute injury (3 days) in young and aged nerves. Expression in fragments per kilobase of exon per million mapped fragments (FPKM. Graphs generated based on RNA-seq data from Wagstaff et al., 2021 (ArrayExpress ID E-MTAB-9640) [87].
In acutely injured young nerves (P60), class I HDACs, Hdac1, Hdac2, and Hdac3, are the most highly expressed after injury. At 3 days post-injury, both Hdac1 and Hdac2 are up-regulated significantly, and at 7 days these HDACs are down-regulated to levels similar to the uninjured nerve (Figure 4A). The class II HDACs Hdac9 and Hdac10 are also up-regulated significantly, but expression is lower than that of class I HDACs. Hdac11 is down-regulated significantly after injury.
In chronic lesions, where distal stumps have no axonal contact for a long period of time, some Hdacs are persistently upregulated for up to 10 weeks. In particular, Hdac5 and Hdac7, which are not upregulated in acute injury, show slight but not significant upregulation, and Hdac11 is upregulated significantly. By contrast, Hdac6 and Hdac8 are significantly downregulated in chronically injured nerves (Figure 4A). It is remarkable that in chronic injuries, several Hdacs are expressed differently 70 days after injury than in acute injuries. This indicates that a precise modulation of Hdac expression during acute and chronic injury is important, and it should be considered in chronic neurodegenerative contexts from a therapeutic perspective. It would also be interesting to check Hdac levels and the effect of different inhibitors in this context.
In aged nerves (12-month-old mice), the up-regulation of Hdacs 3 days after injury is very similar to that in young, injured nerves, and levels are not significantly different between young and old mice 3 days after injury. Specifically, the upregulation of the class I HDACs, Hdac1 and Hdac2, and the downregulation of the class IV HDAC, Hdac11, is significant in aged mice as well. Interestingly, while the upregulation of Hdac9 and 10 is significant after acute injury in young mice, it is not in old mice (Figure 4B). Overall, all Hdacs have a similar pattern of regulation in young and aged mice, and similar expression levels in uninjured nerves, with only Hdac9 levels being significantly elevated in uninjured older mice.
4. HDAC Genetic Compensation in Repair Schwann Cells
Genetic compensation, or genetic buffering, is a process by which a cell, tissue, or organism with a pathogenic mutation does not develop the expected adverse phenotype due to gene duplications or compensatory actions of another gene or genes, which functionally compensate for the loss-of-function genotypes, restoring normal function of the mutated gene and maintaining a normal physiology [103]. Almost 90 years ago, the first genetic compensation was reported in Drosophila Melanogaster, and further compensations have since then been reported in other organisms, such as plants (Arabidopsis thaliana), yeasts (Saccharomyces sp.), zebrafish (Danio rerio), mice (Mus musculus), and humans (Homo sapiens) [104].
Genetic compensation can occur at the transcriptional level. Thus, the observation that some mutations can trigger the transcriptional modulation of other genes, a process also named transcriptional adaptation, has provided a novel explanation for the contradictory phenotypes observed in knockdown versus knockout models and has also increased awareness of the use of these technologies [105]. Different studies have predicted that redundancies are evolutionarily unstable and only have a transitory lifetime. Despite this, there are many examples of transcriptional adaptations or functional redundancies that have been conserved during evolution [104,105,106].
Gene dosage fluctuations (noise) are a well-known phenomenon that has been studied from bacteria to mammalian cells and that could have a significant influence on the fitness of an organism [107]. It has been hypothesized that gene redundancy could have been selected to overcome the negative effects of gene expression noise [106]. Therefore, an adverse effect of a possibly reduced expression of a noisy gene which is fundamental for a certain biological process (such as differentiation) can theoretically be mitigated by the expression of a paralogous gene, a redundant gene controlled by a different promoter [105].
Several studies have reported that genetic compensation of HDACs occurs in different tissues. In adult skeletal muscle, for instance, four class IIa Hdacs alleles (Hdac4, Hdac5, Hdac7, and Hdac9) need to be deleted to observe an increase in slow-fiber gene expression. These class IIa HDACs normally repress the transcription factor family myocyte enhancing factor 2 (MEF2), regulating slow fiber caliber [108].
In brain development, genetic compensation of HDACs also occurs. The combined deletion of Hdac1 and Hdac2 results in severely impaired brain development and embryonic lethality. This can be prevented by a single allele of either Hdac1 or Hdac2, suggesting that both HDACs have the capacity to compensate and rescue normal cellular function required in the brain [109]. Additionally, a similar compensatory effect between Hdac4 and Hdac5 has recently been reported in learning and memory [110].
Genetic compensation is also important during peripheral nerve development. It has been shown that Hdac1 and Hdac2 function redundantly during SC development [15,52]. Indeed, ablation of Hdac1/2 in SCs leads to impaired myelination during development as well as impaired remyelination after lesion. Despite this, no phenotype was found in single Hdac1 or Hdac2 mutants [47]. In the double KO mice, Sox10 levels were normal in the repair SCs, but Krox20 upregulation was strongly reduced during remyelination. In addition, SUMOylated HDAC2, JMJD2C, and KDM3A collaborate in repair SCs to sequentially de-repress Oct6 and Krox20, two SOX10 target genes that are critical for SC development and repair SC reprogramming after nerve injury [47].
In Class IIa HDACs, genetic compensation has recently been reported as well. Velasco-Aviles et al. (2022) describe genetic redundancy in repair SCs (as well as in development, see Section 2.2) using cKO in Mpz-Cre mice [6]. In regeneration and during remyelination, single cKO of Hdac4, Hdac5, or Hdac7 does not have any effect on remyelination. When both Hdac4 and Hdac5 were depleted in Hdac4/5 dKO mice, both myelination during development and remyelination after injury were delayed. Interestingly, in these mice, the transcription factor JUN induces the compensatory overexpression of Hdac7, which can partially compensate for the absence of Hdac4 and Hdac5. Indeed, when all three genes were removed from SCs (Hdac4/5/7 tKO) myelin development and remyelination after injury was impaired for a significantly longer period of time. Strikingly, Hdac9, the fourth class IIa HDAC, which is not normally expressed in nerves, is highly upregulated in tKO nerves, which could partially compensate for the absence of other class IIa HDACs. Hdac9 de novo expression is induced by the expression of MEF2D, which binds to the Hdac9 promoter activating its expression. In addition, in this tKO mouse, the authors show that myelin clearance after injury is accelerated [6].
HDACs genetic redundancy, however, does not always happen or does not necessarily have a compensatory effect to prevent the appearance of a pathologic phenotype. For instance, Hdac3 depletion in the liver leads to increased hepatocellular damage and disrupts metabolic homeostasis; however, in these mice, Hdac1 or Hdac2 do not show compensatory upregulation [111].
Several previous studies investigating class I or class II HDACs do not consider that one HDAC gene depletion could be upregulating another paralogue HDAC gene to compensate for this deletion. Most studies using HDAC inhibitors do not take into account the putative compensatory overexpression of another HDAC when drawing their conclusions [112]. In any future studies where HDAC activity is blocked genetically or pharmacologically, the process of compensation should always be considered.
5. HDACs Therapies: Approved, Trials and Future Directions
Over the last three decades, at least 30 HDAC inhibitors (HDACi) have been extensively studied in clinical trials, several of which have already received regulatory approval. They have been shown to be effective in a broad range of diseases, especially in cancer treatment, where they have been mainly applied to treat hematological malignancies. HDACi have also been shown to be potential treatments for metabolic diseases such as diabetes, neurodegenerative diseases such as Multiple sclerosis, viral infections such as HIV, and peripheral neuropathies such as Charcot–Marie–Tooth disease (CMT) [113,114].
Scientific publications and clinical trials around this topic are rapidly increasing. To date, at least 700 clinical trials are registered and more than 200 are currently recruiting participants. From 2011 to 2020 at least 2000 HDACi papers were published per year, with patents representing 6–11% of the total publications [113]. The global market for HDAC research has been predicted to grow by 32% annually [114].
Five of these agents with deacetylase mechanisms of action have already been approved by regulatory agencies: Vorinostat, Belinostat, Romidepsin, Tucidinostat, and Panobinostat. Vorinostat, a hydroxamic acid derivate (SAHA, suberoylanilide hydroxamic acid) was the first HDACi to be approved by the FDA in 2006 for the treatment of cutaneous T cell lymphoma (CTCL). Romidepsin was approved in 2009 for CTCL as well, Belinostat was approved in 2014 for the treatment of peripheral T cell lymphoma (PTCL), and Parabinostat received FDA approval in 2015 for Multiple Myeloma treatment. Tucidinostat, a non-hydroxamic benzamide class HDAC was approved by China’s National Medical Products Administration for PTCL in 2014 and for postmenopausal advanced breast cancer in 2019 [114].
The molecular targets for all approved inhibitors are classical HDACs (class I, II, and IV), as dysfunctional expression of classical HDACs is associated with tumorigenesis and a higher tumor burden. Class I HDACs and class III HDACs have tumor suppressor functions: conditional knockout mice for Hdac1 and Hdac2 develop lymphoid malignancies [115], as well as non-hematopoietic tumors.
The FDA-approved HDACis, originally approved for the treatment of hematopoietic cancers, are currently also being studied for non-hematological malignancies. HDACis are also being investigated in a range of other diseases due to their well-documented roles in human pathology. These include viral silencing, and HDACi have been widely tested in clinical trials for their ability to reverse HIV latency. They have also been investigated as a potential therapeutic avenue to treat diabetes as HDACs contribute to glucose homeostasis in mammals [116,117,118,119,120]. Immune-mediated diseases such as chronic obstructive pulmonary disease, rheumatoid arthritis, and inflammatory bowel disease are further examples of diseases where HDACi treatment is being investigated [121,122].
Importantly, it has been well-documented in animal models that HDACs play a significant role in the neuropathology of the CNS, particularly in conditions such as Multiple Sclerosis, Amyotrophic Lateral Sclerosis (ALS), Parkinson’s disease, Alzheimer’s disease (AD), Huntington’s disease, frontotemporal dementia, in psychiatric conditions such as Schizophrenia or mood disorders such as depression or anxiety [69,123].
In the last decade, growing evidence has also shown that HDACs have a significant impact on the development of PNS disease. HDAC6 has been implicated in peripheral neuropathy, neuropathic pain, in CMT, and in chemotherapy-induced peripheral neuropathy. The proposed mechanism for pathogenesis is mitochondrial dysfunction, altered mitochondrial transport, and microtubule instability [124]. The study of HDACis in diabetic neuropathy is yielding very promising results [112,125]. Ricolinostat, an oral, selective HDAC6i is being investigated in a phase 2 clinical trial for diabetic neuropathic pain (see Table 3) [126].
Table 3.
Examples of HDACs inhibitors in clinical trials of diseases of CNS and PNS (from https://clinicaltrials.gov/) (Accessed on 18 February 2022).
HDACs also play an important role in the adult nervous system and in degeneration. HDAC2 plays a role in synaptic plasticity, dendritic spine density, and memory formation [127]. HDAC2 dysregulation is also associated with neuronal degeneration during AD progression [128]. HDAC6 inhibitors have been shown to be of potential benefit in neurodegeneration in ALS, as they can restore histone acetylation levels and consequently slow down disease progression in a FUS (fused in sarcoma) mouse model of ALS [129]. HDAC6 is also a potential target for peripheral neurodegenerative neuropathy, as it has been demonstrated that increases in deacetylated α-TUBULIN and dysregulated axonal transport could be a common pathogenic mechanism of various genetic forms of CMT [130].
Two selective HDAC6 inhibitors, ACY-1215 (Ricolinostat), and ACY-241 (Citarinostat) are currently being studied in clinical trials for hematologic and non-hematologic cancers [130]. Interestingly, ACY-1215 has also been shown to restore nerve damage and reduce pain, numbness, and muscle weakness resulting from chemotherapy and CMT [126,131,132]. A Phase II trial of ACY-1215 for the treatment of diabetic neuropathic pain is currently in the recruitment phase.
The currently approved treatments are all pan HDAC inhibitors, and this lack of specificity is responsible for common side effects such as fatigue, cytopenia, nausea, and, in some cases, cardiotoxicity. Future research needs to be more focused on selective inhibition of HDAC subtypes and future efforts should focus on describing new molecules with increased tissue specificity.
Author Contributions
Conceived the idea and wrote the paper: N.P., F.M., S.V.F., C.M., H.C. and J.A.G.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been funded by grants from the Ministerio de Economía y Competitividad (BFU2016-75864R and PID2019-109762RB-I00), ISABIAL (UGP18-257 and UGP-2019-128) to H. Cabedo and Conselleria Educació Generalitat Valenciana (PROMETEO 2018/114) to H. Cabedo. Medical Research Council (UK) studentship (2251399) to C. Mutschler.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hopf, A.; Schaefer, D.J.; Kalbermatten, D.F.; Guzman, R.; Madduri, S. Schwann Cell-Like Cells: Origin and Usability for Repair and Regeneration of the Peripheral and Central Nervous System. Cells 2020, 9, 1990. [Google Scholar] [CrossRef] [PubMed]
- Arthur-Farraj, P.J.; Latouche, M.; Wilton, D.K.; Quintes, S.; Chabrol, E.; Banerjee, A.; Woodhoo, A.; Jenkins, B.; Rahman, M.; Turmaine, M.; et al. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 2012, 75, 633–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, K.R.; Mirsky, R. Schwann Cells in Nerve Repair and Regeneration. In Peripheral Nerve Tissue Engineering and Regeneration; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 1–17. [Google Scholar]
- Gomis-Coloma, C.; Velasco-Aviles, S.; Gomez-Sanchez, J.A.; Casillas-Bajo, A.; Backs, J.; Cabedo, H. Class IIa histone deacetylases link cAMP signaling to the myelin transcriptional program of Schwann cells. J. Cell Biol. 2018, 217, 1249–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Zhang, L.; Queme, L.F.; Liu, X.; Lu, A.; Waclaw, R.R.; Dong, X.; Zhou, W.; Kidd, G.; Yoon, S.O.; et al. A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat. Med. 2018, 24, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Aviles, S.; Patel, N.; Casillas-Bajo, A.; Frutos-Rincón, L.; Velasco-Serna, E.; Gallar, J.; Arthur-Farraj, P.; Gomez-Sanchez, J.A.; Cabedo, H. A genetic compensatory mechanism regulated by Jun and Mef2d modulates the expression of distinct class IIa Hdacs to ensure peripheral nerve myelination and repair. eLife 2022, 11, e72917. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janzen, W.P.; Wigle, T.J.; Jin, J.; Frye, S.V. Epigenetics: Tools and technologies. Drug Discov. Today Technol. 2010, 7, e59–e65. [Google Scholar] [CrossRef] [Green Version]
- Bertos, N.R.; Wang, A.H.; Yang, X.-J. Class II histone deacetylases: Structure, function, and regulation. Biochem. Cell Biol. 2001, 79, 243–252. [Google Scholar] [CrossRef]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta Gene Regul. Mech. 2010, 1799, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Li, H. Structure-based inhibitor discovery of class i histone deacetylases (HDACS). Int. J. Mol. Sci. 2020, 21, 8828. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Histone Deacetylase in Neuropathology, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 104, ISBN 9780128246221. [Google Scholar]
- Montgomery, R.L.; Davis, C.A.; Potthoff, M.J.; Haberland, M.; Fielitz, J.; Qi, X.; Hill, J.A.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007, 21, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Christen, C.N.; Pereira, J.A.; Somandin, C.; Baggiolini, A.; Lötscher, P.; Özçelik, M.; Tricaud, N.; Meijer, D.; Yamaguchi, T.; et al. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat. Neurosci. 2011, 14, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 1004. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürger, M.; Chory, J. Structural and chemical biology of deacetylases for carbohydrates, proteins, small molecules and histones. Commun. Biol. 2018, 1, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ververis, K.; Karagiannis, T.C. Overview of the Classical Histone Deacetylase Enzymes and Histone Deacetylase Inhibitors. ISRN Cell Biol. 2012, 2012, 130360. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Parra, M. Class IIa HDACs—New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Zheng, B.; Wang, J.; Zhao, G.; Yao, Z.; Niu, Z.; He, W. Histone deacetylase 10, a potential epigenetic target for therapy. Biosci. Rep. 2021, 41, 20210462. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Kontek, R. Sirtuins in DNA repair. Postepy Biochem. 2020, 66, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Wu, F.; Jin, Y.M.; Chang, W.Q.; Xu, T.M. HDAC11: A rising star in epigenetics. Biomed. Pharmacother. 2020, 131, 110607. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef]
- Santosa, K.B.; Keane, A.M.; Jablonka-Shariff, A.; Vannucci, B.; Snyder-Warwick, A.K. Clinical relevance of terminal Schwann cells: An overlooked component of the neuromuscular junction. J. Neurosci. Res. 2018, 96, 1125–1135. [Google Scholar] [CrossRef]
- Abdo, H.; Calvo-Enrique, L.; Lopez, J.M.; Song, J.; Zhang, M.-D.; Usoskin, D.; El Manira, A.; Adameyko, I.; Hjerling-Leffler, J.; Ernfors, P. Specialized cutaneous Schwann cells initiate pain sensation. Science 2019, 365, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Michailov, G.V.; Sereda, M.W.; Brinkmann, B.G.; Fischer, T.M.; Haug, B.; Birchmeier, C.; Role, L.; Lai, C.; Schwab, M.H.; Nave, K.-A. Axonal neuregulin-1 regulates myelin sheath thickness. Science 2004, 304, 700–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, K.R.; Arthur-Farraj, P. Repair Schwann cell update: Adaptive reprogramming, EMT, and stemness in regenerating nerves. Glia 2019, 67, 421–437. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R.; Arthur-Farraj, P. The Role of Cell Plasticity in Tissue Repair: Adaptive Cellular Reprogramming. Dev. Cell 2015, 34, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessen, K.R.; Mirsky, R. The success and failure of the schwann cell response to nerve injury. Front. Cell. Neurosci. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Hirata, K.; Kawabuchi, M. Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc. Res. Technol. 2002, 57, 541–547. [Google Scholar] [CrossRef]
- Lutz, A.B.; Chung, W.-S.; Sloan, S.A.; Carson, G.A.; Zhou, L.; Lovelett, E.; Posada, S.; Zuchero, J.B.; Barres, B.A.; Freeman, M.; et al. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. USA 2017, 114, E8072–E8080. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Lee, H.J.; Yoo, Y.H.; Kim, J.K.; Park, H.T. Autophagic myelin destruction by schwann cells during wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [Green Version]
- De Felipe, C.; Hunt, S.P. The differential control of c-jun expression in regenerating sensory neurons and their associated glial cells. J. Neurosci. 1994, 14, 2911. [Google Scholar] [CrossRef]
- Painter, M.W.; Lutz, A.B.; Cheng, Y.-C.; Latremoliere, A.; Duong, K.; Miller, C.M.; Posada, S.; Cobos, E.J.; Zhang, A.X.; Wagers, A.J.; et al. Diminished Schwann cell repair responses underlie age-associated impaired axonal regeneration. Neuron 2014, 83, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Painter, M.W. Aging Schwann cells: Mechanisms, implications, future directions. Curr. Opin. Neurobiol. 2017, 47, 203–208. [Google Scholar] [CrossRef]
- Weiss, T.; Taschner-Mandl, S.; Bileck, A.; Slany, A.; Kromp, F.; Rifatbegovic, F.; Frech, C.; Windhager, R.; Kitzinger, H.; Tzou, C.H.; et al. Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 2016, 64, 2133–2153. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.B.; Laranjeira, S.G.; Eriksson, T.M.; Jessen, K.R.; Mirsky, R.; Quick, T.J.; Phillips, J.B. Characterising cellular and molecular features of human peripheral nerve degeneration. Acta Neuropathol. Commun. 2020, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Lotscher, P.; Engler, S.; Baggiolini, A.; Varum Tavares, S.; Brugger, V.; John, N.; Buchmann-Moller, S.; Snider, P.L.; Conway, S.J.; et al. HDAC1 and HDAC2 Control the Specification of Neural Crest Cells into Peripheral Glia. J. Neurosci. 2014, 34, 6112–6122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brügger, V.; Engler, S.; Pereira, J.A.; Ruff, S.; Horn, M.; Welzl, H.; Münger, E.; Vaquié, A.; Sidiropoulos, P.N.M.; Egger, B.; et al. HDAC1/2-Dependent P0 Expression Maintains Paranodal and Nodal Integrity Independently of Myelin Stability through Interactions with Neurofascins. PLoS Biol. 2015, 13, e1002258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brügger, V.; Duman, M.; Bochud, M.; Münger, E.; Heller, M.; Ruff, S.; Jacob, C. Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nat. Commun. 2017, 8, 14272. [Google Scholar] [CrossRef] [PubMed]
- Duman, M.; Vaquié, A.; Nocera, G.; Heller, M.; Stumpe, M.; Siva Sankar, D.; Dengjel, J.; Meijer, D.; Yamaguchi, T.; Matthias, P.; et al. EEF1A1 deacetylation enables transcriptional activation of remyelination. Nat. Commun. 2020, 11, 3420. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L.H.; Cattin, A.L.; Fontana, X.; Harford-Wright, E.; Burden, J.J.; White, I.J.; Smith, J.G.; Napoli, I.; Quereda, V.; Policarpi, C.; et al. HDAC3 Regulates the Transition to the Homeostatic Myelinating Schwann Cell State. Cell Rep. 2018, 25, 2755–2765.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, S.C.; Luo, R.; Liebscher, I.; Giera, S.; Jeong, S.J.; Mogha, A.; Ghidinelli, M.; Feltri, M.L.; Schöneberg, T.; Piao, X.; et al. The Adhesion GPCR GPR126 Has Distinct, Domain-Dependent Functions in Schwann Cell Development Mediated by Interaction with Laminin-211. Neuron 2015, 85, 755–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Wende, H.; Walcher, J.; Küehnemund, J.; Cheret, C.; Kempa, S.; McShane, E.; Selbach, M.; Lewin, G.R.; Birchmeier, C. Maf links Neuregulin1 signaling to cholesterol synthesis in myelinating Schwann cells. Genes Dev. 2018, 32, 645–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, H.; Yoon, S.O.; Xu, X.; Hottiger, M.O.; Svaren, J.; Nave, K.A.; Kim, H.A.; Olson, E.N.; Lu, Q.R. HDAC-mediated deacetylation of NF-κB is critical for Schwann cell myelination. Nat. Neurosci. 2011, 14, 437–441. [Google Scholar] [CrossRef] [Green Version]
- Chittka, A.; Arevalo, J.C.; Rodriguez-Guzman, M.; Pérez, P.; Chao, M.V.; Sendtner, M. The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. J. Cell Biol. 2004, 164, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, H.; Kohnken, R.; Svaren, J. The nucleosome remodeling and deacetylase chromatin remodeling (NuRD) complex is required for peripheral nerve myelination. J. Neurosci. 2012, 32, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hu, Q.; D’Ercole, A.J.; Ye, P. Histone Deacetylase 11 Regulates Oligodendrocyte-Specific Gene Expression and Cell Development in OL-1 Oligodendroglia Cells. Glia 2009, 57, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, D.; Pereira, J.A.; Gerber, J.; Tan, G.; Dimitrieva, S.; Yángüez, E.; Suter, U. Transcriptional profiling of mouse peripheral nerves to the single-cell level to build a sciatic nerve ATlas (SNAT). eLife 2021, 10, e58591. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, C.; Jung, J.; Yeo, S.G. The histone deacetylase class I, II inhibitor trichostatin A delays peripheral neurodegeneration. J. Mol. Histol. 2019, 50, 167–178. [Google Scholar] [CrossRef]
- Wu, F.; Xing, D.; Peng, Z.; Rao, T. Enhanced rat sciatic nerve regeneration through silicon tubes implanted with valproic acid. J. Reconstr. Microsurg. 2008, 24, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.; Wu, F.; Xing, D.; Peng, Z.; Ren, D.; Feng, W.; Chen, Y.; Zhao, Z.; Wang, H.; Wang, J.; et al. Effects of Valproic Acid on Axonal Regeneration and Recovery of Motor Function after Peripheral Nerve Injury in the Rat. Arch. Bone Jt. Surg. 2014, 2, 17. [Google Scholar]
- Wu, F.; Jiao, C.; Yang, Y.; Liu, F.; Sun, Z. Nerve conduit based on HAP/PDLLA/PRGD for peripheral nerve regeneration with sustained release of valproic acid. Cell Biol. Int. 2021, 45, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A.; D’Mello, S.R. Complex neuroprotective and neurotoxic effects of histone deacetylases. J. Neurochem. 2018, 145, 96–110. [Google Scholar] [CrossRef] [Green Version]
- Duman, M.; Martinez-Moreno, M.; Jacob, C.; Tapinos, N. Functions of histone modifications and histone modifiers in Schwann cells. Glia 2020, 68, 1584–1595. [Google Scholar] [CrossRef] [Green Version]
- Yadav, A.; Huang, T.-C.; Chen, S.-H.; Ramasamy, T.S.; Hsueh, Y.-Y.; Lin, S.-P.; Lu, F.-I.; Liu, Y.-H.; Wu, C.-C. Sodium phenylbutyrate inhibits Schwann cell inflammation via HDAC and NFκB to promote axonal regeneration and remyelination. J. Neuroinflamm. 2021, 18, 238. [Google Scholar] [CrossRef] [PubMed]
- Fazal, S.V.; Gomez-Sanchez, J.A.; Wagstaff, L.J.; Musner, N.; Otto, G.; Janz, M.; Mirsky, R.; Jessen, K.R. Graded elevation of c-Jun in Schwann cells in vivo: Gene dosage determines effects on development, re-myelination, tumorigenesis and hypomyelination. J. Neurosci. 2017, 37, 12297–12313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, L.; Zhang, S.; Huang, R.; Li, Z. HDAC11 promotes meiotic apparatus assembly during mouse oocyte maturation via decreasing H4K16 and α-tubulin acetylation. Cell Cycle 2020, 19, 354–362. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.E.; Cho, Y. Epigenetic Regulation of Axon Regeneration after Neural Injury. Mol. Cells 2017, 40, 10–16. [Google Scholar] [CrossRef]
- Wahane, S.; Halawani, D.; Zhou, X.; Zou, H. Epigenetic regulation of axon regeneration and glial activation in injury responses. Front. Genet. 2019, 10, 640. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 2012, 31, 3063–3078. [Google Scholar] [CrossRef] [Green Version]
- Hervera, A.; Zhou, L.; Palmisano, I.; McLachlan, E.; Kong, G.; Hutson, T.H.; Danzi, M.C.; Lemmon, V.P.; Bixby, J.L.; Matamoros-Angles, A.; et al. PP4-dependent HDAC3 dephosphorylation discriminates between axonal regeneration and regenerative failure. EMBO J. 2019, 38, e101032. [Google Scholar] [CrossRef]
- Kalinski, A.L.; Kar, A.N.; Craver, J.; Tosolini, A.P.; Sleigh, J.N.; Lee, S.J.; Hawthorne, A.; Brito-Vargas, P.; Miller-Randolph, S.; Passino, R.; et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J. Cell Biol. 2019, 218, 1871–1890. [Google Scholar] [CrossRef] [Green Version]
- Montani, L.; Pereira, J.A.; Norrmén, C.; Pohl, H.B.F.; Tinelli, E.; Trötzmüller, M.; Figlia, G.; Dimas, P.; von Niederhäusern, B.; Schwager, R.; et al. De novo fatty acid synthesis by Schwann cells is essential for peripheral nervous system myelination. J. Cell Biol. 2018, 217, 1353–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della-Flora Nunes, G.; Mueller, L.; Silvestri, N.; Patel, M.S.; Wrabetz, L.; Feltri, M.L.; Poitelon, Y. Acetyl-CoA production from pyruvate is not necessary for preservation of myelin. Glia 2017, 65, 1626–1639. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Golden, J.P.; Baloh, R.H.; Schmidt, R.E.; Hunter, D.A.; Milbrandt, J. Schwann Cell Mitochondrial Metabolism Supports Long-Term Axonal Survival and Peripheral Nerve Function. J. Neurosci. 2011, 31, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Sasaki, Y.; Kim, S.; Strickland, A.; Workman, C.S.; Yang, K.; Gross, R.W.; Milbrandt, J. Aberrant schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 2013, 77, 886–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, M.K.; Passero, J.V.; Rawat, A.; Ament, X.H.; Yang, F.; Vidensky, S.; Collins, S.L.; Horton, M.R.; Hoke, A.; Rutter, G.A.; et al. Macrophage monocarboxylate transporter 1 promotes peripheral nerve regeneration after injury in mice. J. Clin. Investig. 2021, 13, e141964. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.K.; Ashton-Miller, J.A. Peripheral neuropathy. Postgrad. Med. 1996, 99, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Navarro, X.; Geuna, S.; Grothe, C.; Haastert-Talini, K. Introduction: Thematic Papers Issue on Peripheral Nerve Regeneration and Repair. Anat. Rec. 2018, 301, 1614–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwer, B.A.; de Greef, B.T.A.; Hoeijmakers, J.G.J.; Geerts, M.; van Kleef, M.; Merkies, I.S.J.; Faber, C.G. Neuropathic Pain due to Small Fiber Neuropathy in Aging: Current Management and Future Prospects. Drugs Aging 2015, 32, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Verdú, E.; Ceballos, D.; Vilches, J.J.; Navarro, X. Influence of aging on peripheral nerve function and regeneration. J. Peripher. Nerv. Syst. 2008, 5, 191–208. [Google Scholar] [CrossRef]
- Kang, H.; Lichtman, J.W. Motor axon regeneration and muscle reinnervation in young adult and aged animals. J. Neurosci. 2013, 33, 19480–19491. [Google Scholar] [CrossRef]
- Yuan, X.; Klein, D.; Kerscher, S.; West, B.L.; Weis, J.; Katona, I.; Martini, R. Macrophage Depletion Ameliorates Peripheral Neuropathy in Aging Mice. J. Neurosci. 2018, 38, 4610–4620. [Google Scholar] [CrossRef] [Green Version]
- Scheib, J.; Höke, A. Advances in peripheral nerve regeneration. Nat. Rev. Neurol. 2013, 9, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Gordon, T. Peripheral Nerve Regeneration and Muscle Reinnervation. Int. J. Mol. Sci. 2020, 21, 8652. [Google Scholar] [CrossRef] [PubMed]
- Stratton, J.A.; Eaton, S.; Rosin, N.L.; Jawad, S.; Holmes, A.; Yoon, G.; Midha, R.; Biernaskie, J. Macrophages and Associated Ligands in the Aged Injured Nerve: A Defective Dynamic That Contributes to Reduced Axonal Regrowth. Front. Aging Neurosci. 2020, 12, 174. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, L.J.; Gomez-Sanchez, J.A.; Fazal, S.V.; Otto, G.W.; Kilpatrick, A.M.; Michael, K.; Wong, L.Y.N.; Ma, K.H.; Turmaine, M.; Svaren, J.; et al. Failures of nerve regeneration caused by aging or chronic denervation are rescued by restoring schwann cell c-jun. Elife 2021, 10, e62232. [Google Scholar] [CrossRef]
- Benito, C.; Davis, C.M.; Gomez-Sanchez, J.A.; Turmaine, M.; Meijer, D.; Poli, V.; Mirsky, R.; Jessen, K.R. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. J. Neurosci. 2017, 37, 4225–4269. [Google Scholar] [CrossRef]
- Ronchi, G.; Cillino, M.; Gambarotta, G.; Fornasari, B.E.; Raimondo, S.; Pugliese, P.; Tos, P.; Cordova, A.; Moschella, F.; Geuna, S. Irreversible changes occurring in long-term denervated Schwann cells affect delayed nerve repair. J. Neurosurg. 2017, 127, 843–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, R.; de Winter, F.; Arkenaar, C.; Tannemaat, M.R.; Verhaagen, J. Enhanced regeneration and reinnervation following timed GDNF gene therapy in a cervical ventral root avulsion. Exp. Neurol. 2019, 321, 113037. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, J.A.; Pilch, K.S.; van der Lans, M.; Fazal, S.V.; Benito, C.; Wagstaff, L.J.; Mirsky, R.; Jessen, K.R. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J. Neurosci. 2017, 37, 9086–9099. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Maleszewska, M.; Mawer, J.S.P.; Tessarz, P. Histone Modifications in Ageing and Lifespan Regulation. Curr. Mol. Biol. Rep. 2016, 2, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Cao, X.; Sun, L.; Zhu, J.Y.; Wasko, B.M.; Liu, W.; Crutcher, E.; Liu, H.; Jo, M.C.; Qin, L.; et al. Inactivating histone deacetylase HDA promotes longevity by mobilizing trehalose metabolism. Nat. Commun. 2021, 12, 1981. [Google Scholar] [CrossRef]
- Mielcarek, M.; Landles, C.; Weiss, A.; Bradaia, A.; Seredenina, T.; Inuabasi, L.; Osborne, G.F.; Wadel, K.; Touller, C.; Butler, R.; et al. HDAC4 Reduction: A Novel Therapeutic Strategy to Target Cytoplasmic Huntingtin and Ameliorate Neurodegeneration. PLOS Biol. 2013, 11, e1001717. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Quezada, M.; Levine, P.; Han, Y.; McDaniel, K.; Zhou, T.; Lin, E.; Glaser, S.; Meng, F.; Francis, H.; et al. Functional Role of Cellular Senescence in Biliary Injury. Am. J. Pathol. 2015, 185, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, P.C.; Patnaik, D.; Watson, L.A.; Gao, F.; Pan, L.; Wang, J.; Adaikkan, C.; Penney, J.; Cam, H.P.; Huang, W.C.; et al. HDAC1 modulates OGG1-initiated oxidative DNA damage repair in the aging brain and Alzheimer’s disease. Nat. Commun. 2020, 11, 2484. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Telles, E.; Karl, M.; Cheng, F.; Luetteke, N.; Sotomayor, E.M.; Miller, R.H.; Seto, E. Loss of HDAC11 ameliorates clinical symptoms in a multiple sclerosis mouse model. Life Sci. Alliance 2018, 1, e201800039. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Sagarkar, S.; Balasubramanian, N.; Mishra, S.; Choudhary, A.G.; Kokare, D.M.; Sakharkar, A.J. Repeated mild traumatic brain injury causes persistent changes in histone deacetylase function in hippocampus: Implications in learning and memory deficits in rats. Brain Res. 2019, 1711, 183–192. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, R.L.; Daniels, E.G.; Molenaars, M.; Houtkooper, R.H.; Janssens, G.E. From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 2019, 11, e9854. [Google Scholar] [CrossRef]
- Buglo, E.; Sarmiento, E.; Martuscelli, N.B.; Sant, D.W.; Danzi, M.C.; Abrams, A.J.; Dallman, J.E.; Züchner, S. Genetic compensation in a stable slc25a46 mutant zebrafish: A case for using F0 CRISPR mutagenesis to study phenotypes caused by inherited disease. PLoS ONE 2020, 15, e0230566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J. Gene redundancy and gene compensation: An updated view. J. Genet. Genom. 2019, 46, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Jakutis, G.; Stainier, D.Y. Genotype–Phenotype Relationships in the Context of Transcriptional Adaptation and Genetic Robustness. Annu. Rev. Genet. 2021, 55, 71–91. [Google Scholar] [CrossRef] [PubMed]
- Kafri, R.; Levy, M.; Pilpel, Y. The regulatory utilization of genetics redundancy through responsive backup circuits. Proc. Natl. Acad. Sci. USA 2006, 103, 11653–11658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raser, J.M. Noise in Gene Expression. Science 2010, 2010, 2010–2014. [Google Scholar]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation andMEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagelkruys, A.; Lagger, S.; Krahmer, J.; Leopoldi, A.; Artaker, M.; Pusch, O.; Zezula, J.; Weissmann, S.; Xie, Y.; Schöfer, C.; et al. A single allele of Hdac2 but not Hdac1 is sufficient for normal mouse brain development in the absence of its paralog. Development 2014, 141, 604–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Huang, M.; Bushong, E.; Phan, S.; Uytiepo, M.; Beutter, E.; Boemer, D.; Tsui, K.; Ellisman, M.; Maximov, A. Class IIa HDACs regulate learning and memory through dynamic experience-dependent repression of transcription. Nat. Commun. 2019, 10, 3469. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Chyla, B.J.; Amann, J.M.; Bhaskara, S.; Huppert, S.S.; Hiebert, S.W. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008, 27, 1017–1028. [Google Scholar] [CrossRef] [Green Version]
- An, J.; Zhang, X.; Jia, K.; Zhang, C.; Zhu, L.; Cheng, M.; Li, F.; Zhao, S.; Hao, J. Trichostatin A increases BDNF protein expression by improving XBP-1s/ATF6/GRP78 axis in Schwann cells of diabetic peripheral neuropathy. Biomed. Pharmacother. 2021, 133, 111062. [Google Scholar] [CrossRef]
- He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.Y. Medicinal chemistry updates of novel HDACs inhibitors (2020 to present). Eur. J. Med. Chem. 2022, 227, 113946. [Google Scholar] [CrossRef] [PubMed]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef] [PubMed]
- Heideman, M.R.; Wilting, R.H.; Yanover, E.; Velds, A.; De Jong, J.; Kerkhoven, R.M.; Jacobs, H.; Wessels, L.F.; Dannenberg, J.H. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013, 121, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Beliakova-Bethell, N.; Mukim, A.; White, C.H.; Deshmukh, S.; Abewe, H.; Richman, D.D.; Spina, C.A. Histone deacetylase inhibitors induce complex host responses that contribute to differential potencies of these compounds in HIV reactivation. J. Biol. Chem. 2019, 294, 5576–5589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific Control of Pancreatic Endocrine β- and δ-Cell Mass by Class IIa Histone Deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Lundh, M.; Galbo, T.; Poulsen, S.S.; Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 2015, 17, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Vasquez, D.S.; Ravnskjaer, K.; Denechaud, P.D.; Yu, R.T.; Alvarez, J.G.; Downes, M.; Evans, R.M.; Montminy, M.; Shaw, R.J. Class IIa Histone Deacetylases Are Hormone-Activated Regulators of FOXO and Mammalian Glucose Homeostasis. Cell 2011, 145, 607–621. [Google Scholar] [CrossRef] [Green Version]
- Daneshpajooh, M.; Bacos, K.; Bysani, M.; Bagge, A.; Ottosson Laakso, E.; Vikman, P.; Eliasson, L.; Mulder, H.; Ling, C. HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia 2017, 60, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Hamminger, P.; Rica, R.; Ellmeier, W. Histone deacetylases as targets in autoimmune and autoinflammatory diseases. Adv. Immunol. 2020, 147, 1–59. [Google Scholar] [CrossRef]
- Bahari-Javan, S.; Sananbenesi, F.; Fischer, A. Histone-acetylation: A link between Alzheimer’s disease and post-traumatic stress disorder? Front. Neurosci. 2014, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, K.; Barton, M.C. HDAC6: A Key Link Between Mitochondria and Development of Peripheral Neuropathy. Front. Mol. Neurosci. 2021, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, M.; Novakovic, I.; Nikolic, D.; Maksic, J.M.; Brankovic, S.; Petronic, I.; Cirovic, D.; Ducic, S.; Grajic, M.; Bogicevic, D. Genetic and Epigenomic Modifiers of Diabetic Neuropathy. Int. J. Mol. Sci. 2021, 22, 4887. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Trinh, R.T.; Mahant, I.D.; Peng, B.; Matthias, P.; Heijnen, C.J.; Kavelaars, A. Cell-specific role of histone deacetylase 6 in chemotherapy-induced mechanical allodynia and loss of intraepidermal nerve fibers. Pain 2019, 160, 2877–2890. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Mahady, L.; Nadeem, M.; Malek-Ahmadi, M.; Chen, K.; Perez, S.E.; Mufson, E.J. HDAC2 dysregulation in the nucleus basalis of Meynert during the progression of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2019, 45, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Rossaert, E.; Pollari, E.; Jaspers, T.; Van Helleputte, L.; Jarpe, M.; Van Damme, P.; De Bock, K.; Moisse, M.; Van Den Bosch, L. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kozikowski, A.P. A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014–2019). Expert Opin. Ther. Pat. 2019, 30, 121–136. [Google Scholar] [CrossRef]
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; Van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef]
- Benoy, V.; Vanden Berghe, P.; Jarpe, M.; Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot–Marie–Tooth Disease. Neurotherapeutics 2017, 14, 417–428. [Google Scholar] [CrossRef] [Green Version]
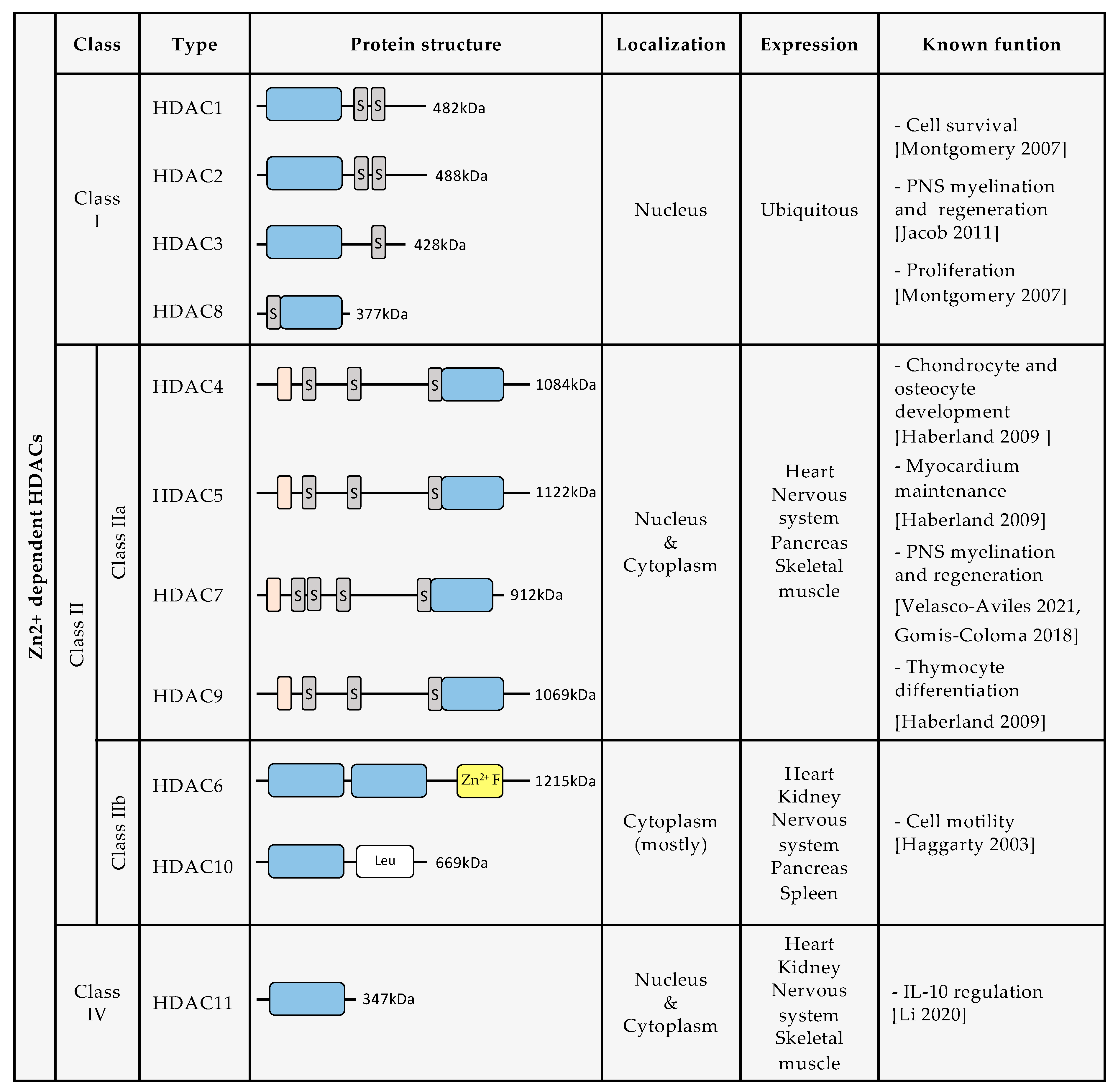
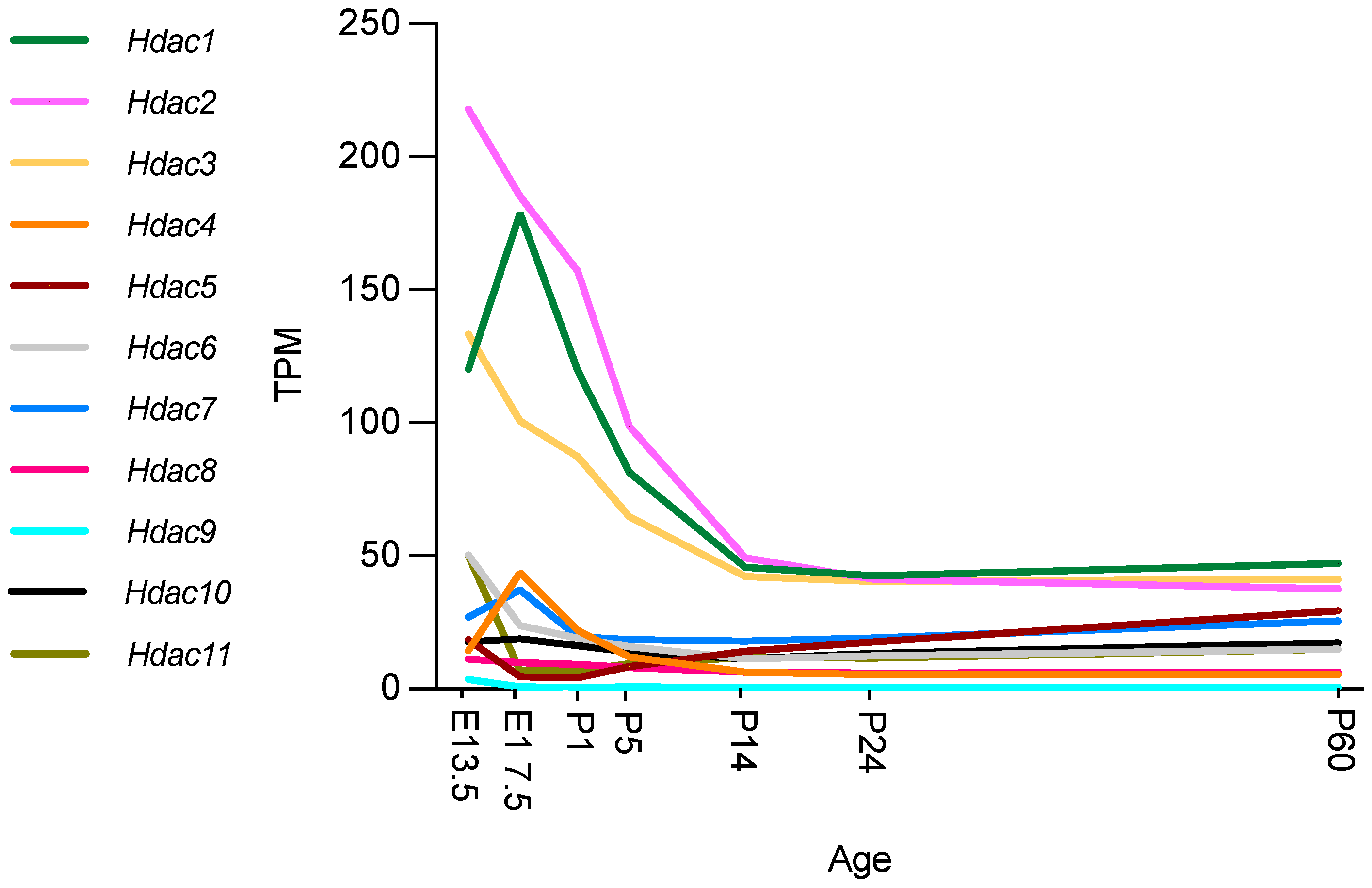
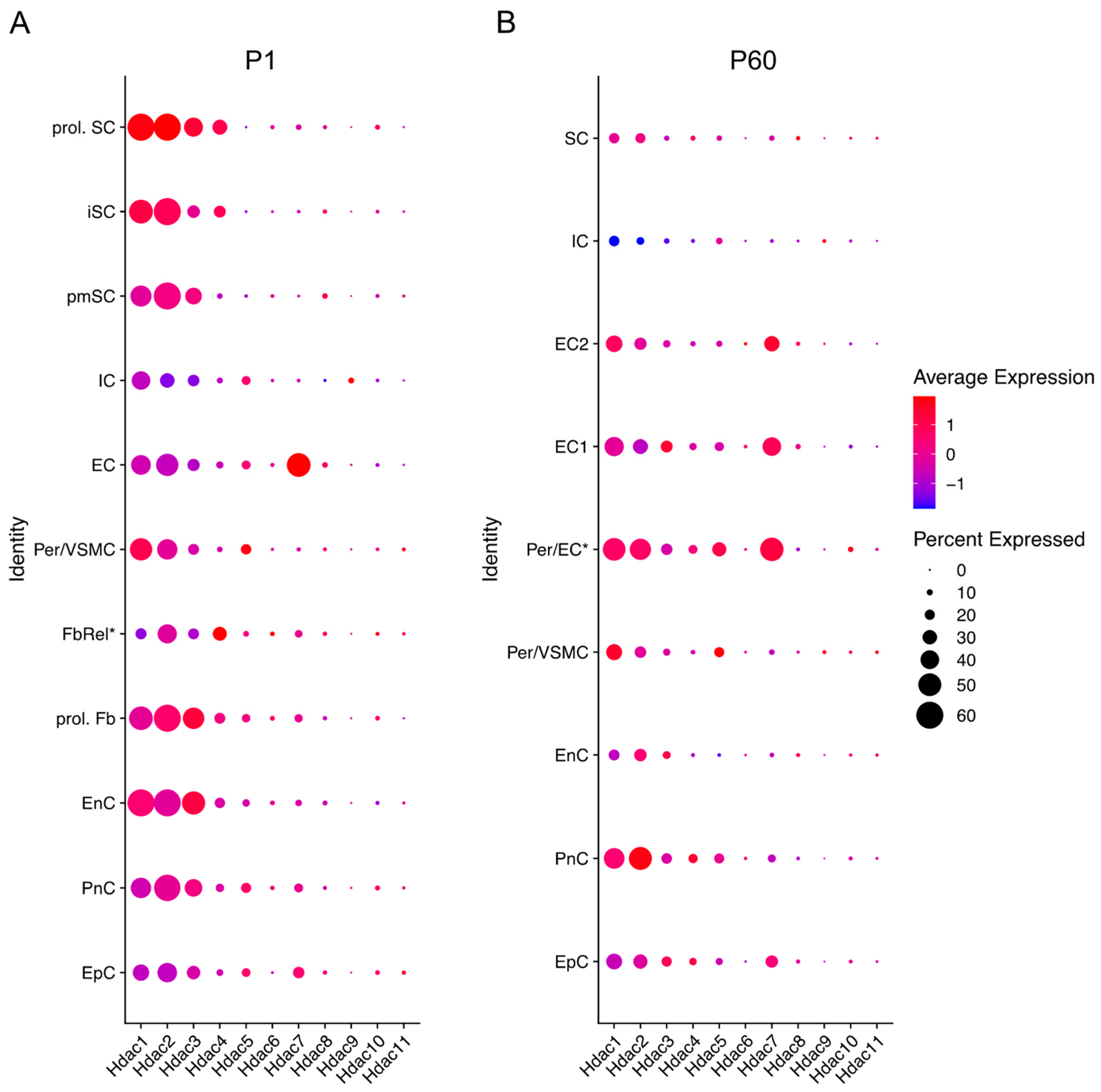
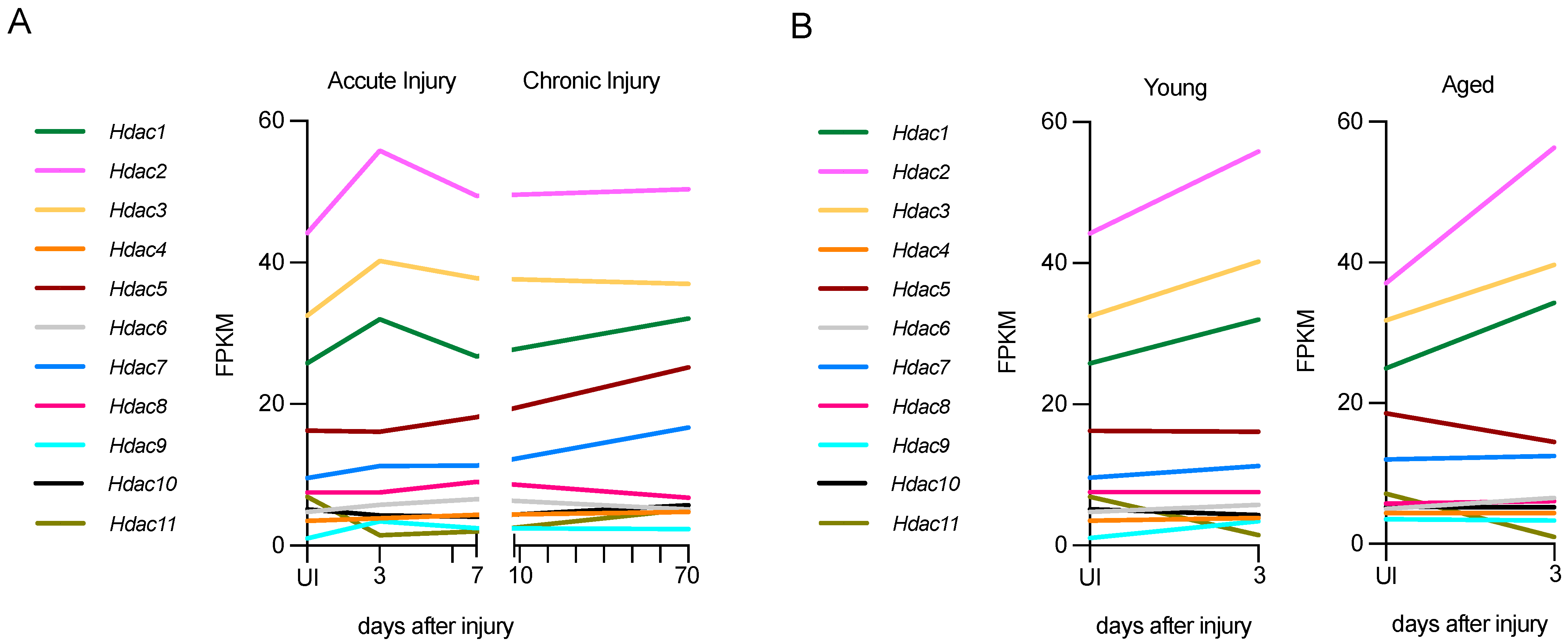
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).