
Tuning Strain Stiffening of Protein Hydrogels by Charge Modification




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
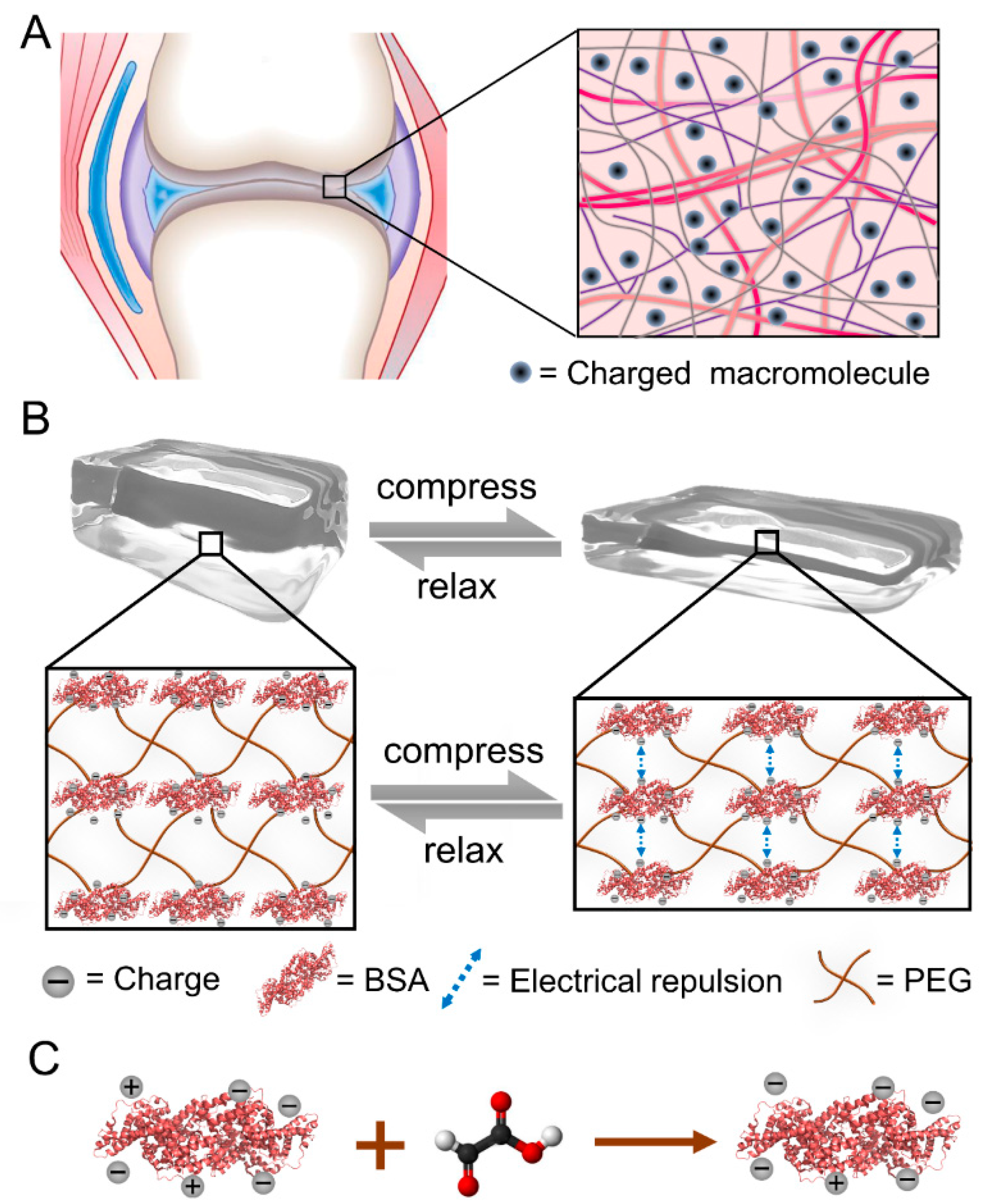
2.1. Design of Tuning the Strain-Stiffening of Protein Hydrogels by Protein Surface Charge Modification
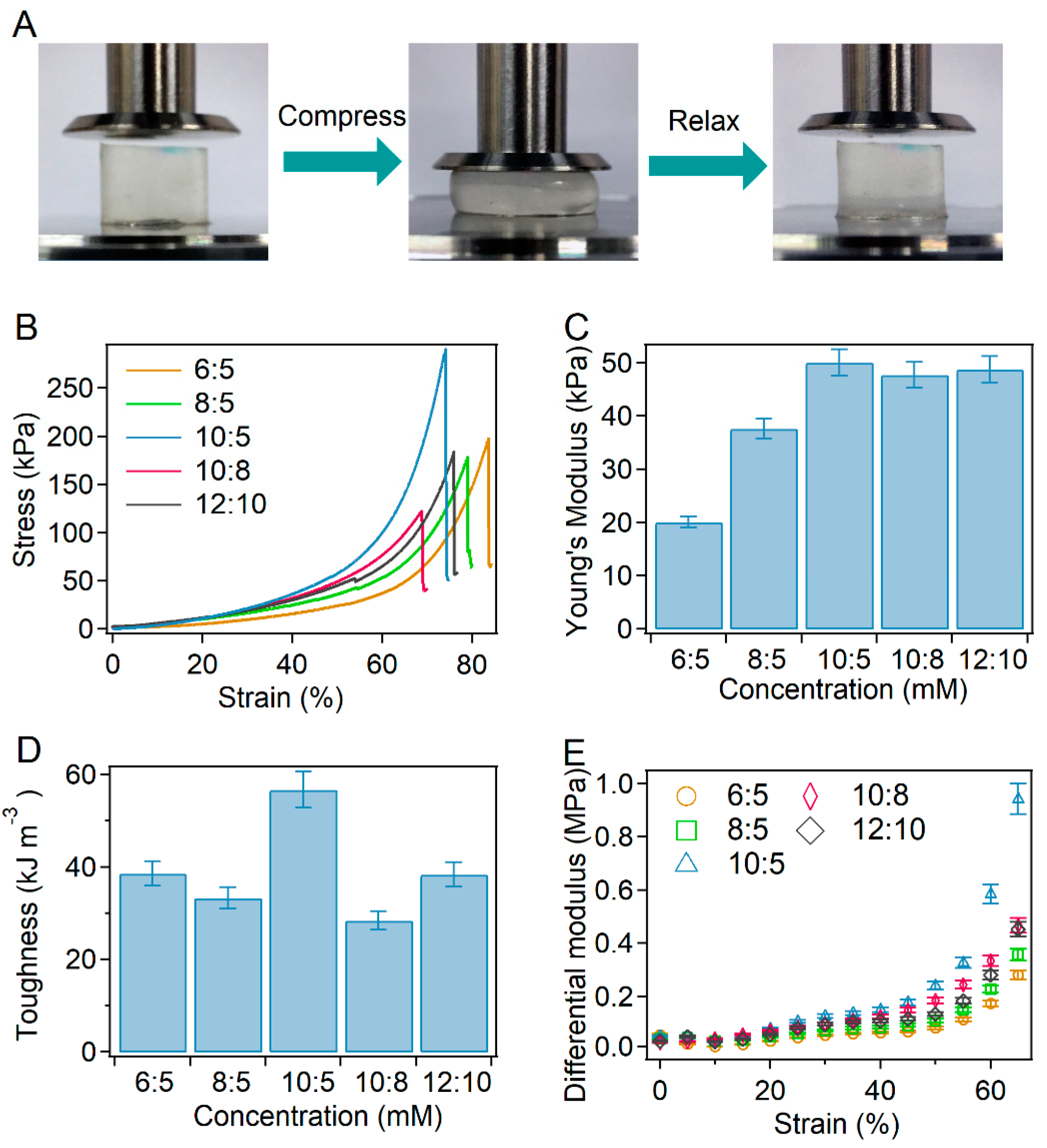
2.2. Mechanical Properties of BSA-PEG Hydrogels
2.3. Charge Modification and Bulk Properties of the BSA-PEG Hydrogels
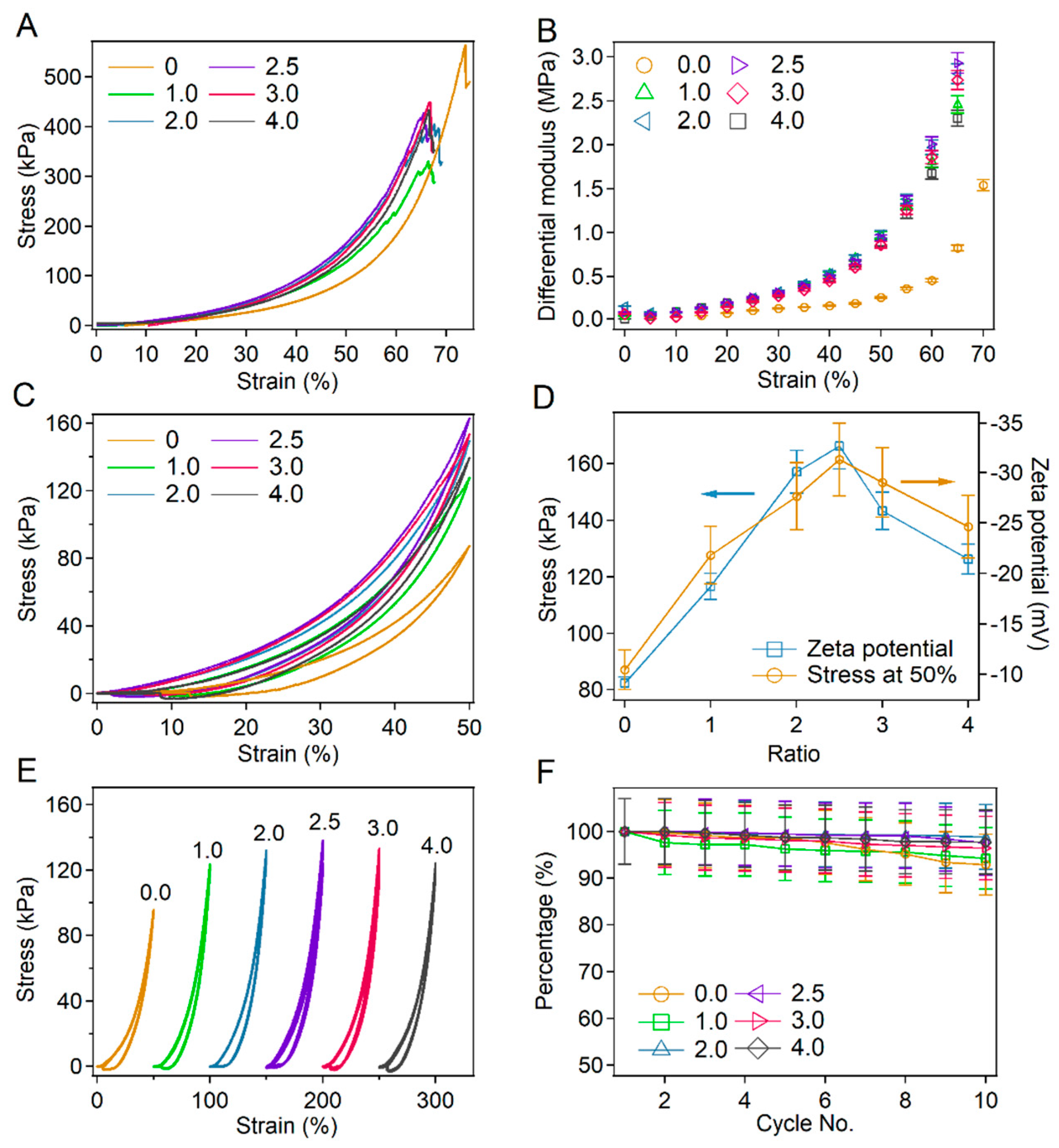
2.4. Tuning Strain-Stiffening of BSA-PEG Hydrogels by Charge Modification
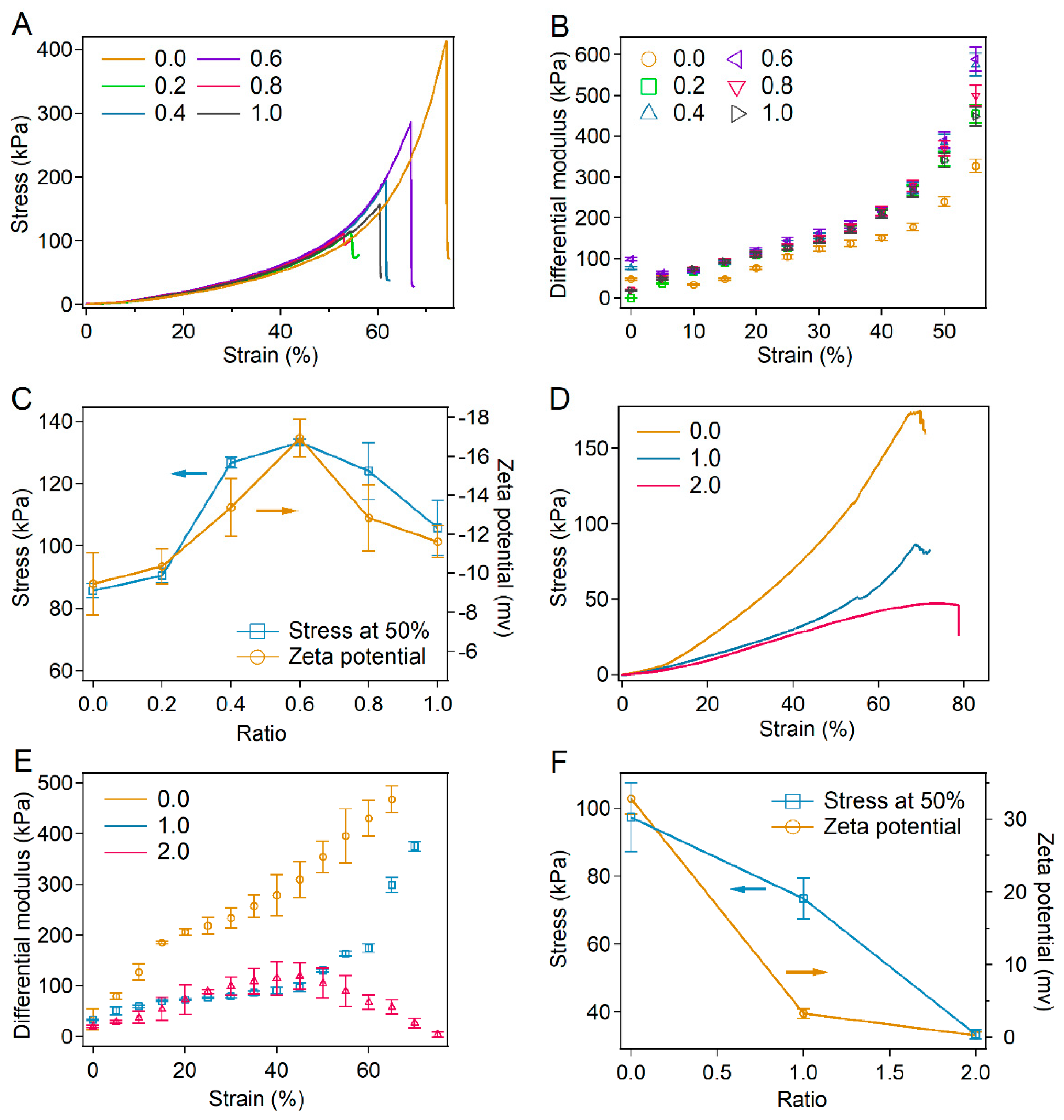
2.5. Universality of Tuning Strain-Stiffening of Hydrogels by Surface Charge Modification of Proteins
2.6. Biocompatibility of the Modified BSA-PEG Hydrogels
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Xu, Z.; Lovrak, M.; le Sage, V.A.A.; Zhang, K.; Guo, X.; Eelkema, R.; Mendes, E.; van Esch, J.H. Biomimetic Strain-Stiffening Self-Assembled Hydrogels. Angew. Chem. Int. Ed. 2020, 59, 4830–4834. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D. Continuum biomechanics of soft biological tissues. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 2003, 459, 3–46. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Gocheva, V.; Hammink, R.; Zouani, O.F.; Rowan, A.E. Stress-stiffening-mediated stem-cell commitment switch in soft responsive hydrogels. Nat. Mater. 2016, 15, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Shin, J.H.; MacKintosh, F.C.; Mahadevan, L.; Matsudaira, P.; Weitz, D.A. Elastic behavior of cross-linked and bundled actin networks. Science 2004, 304, 1301–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurniawan, N.A.; Wong, L.H.; Rajagopalan, R. Early stiffening and softening of collagen: Interplay of deformation mechanisms in biopolymer networks. Biomacromolecules 2012, 13, 691–698. [Google Scholar] [CrossRef]
- Licup, A.J.; Munster, S.; Sharma, A.; Sheinman, M.; Jawerth, L.M.; Fabry, B.; Weitz, D.A.; MacKintosh, F.C. Stress controls the mechanics of collagen networks. Proc. Natl. Acad. Sci. USA 2015, 112, 9573–9578. [Google Scholar] [CrossRef] [Green Version]
- Piechocka, I.K.; Jansen, K.A.; Broedersz, C.P.; Kurniawan, N.A.; MacKintosh, F.C.; Koenderink, G.H. Multi-scale strain-stiffening of semiflexible bundle networks. Soft Matter 2016, 12, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Wen, Q.; Janmey, P.A.; Tang, J.X.; Conti, E.; MacKintosh, F.C. Nonlinear Elasticity of Stiff Filament Networks: Strain Stiffening, Negative Normal Stress, and Filament Alignment in Fibrin Gels. J. Phys. Chem. B 2009, 113, 3799–3805. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.C.; Yao, N.Y.; Broedersz, C.P.; Herrmann, H.; MacKintosh, F.C.; Weitz, D.A. Origins of Elasticity in Intermediate Filament Networks. Phys. Rev. Lett. 2010, 104, 058101. [Google Scholar] [CrossRef]
- Bertula, K.; Martikainen, L.; Munne, P.; Hietala, S.; Klefstrom, J.; Ikkala, O.; Nonappa. Strain-Stiffening of Agarose Gels. ACS Macro Lett. 2019, 8, 670–675. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Castano Romera, M.; Lafleur, R.P.M.; Guibert, C.; Voets, I.K.; Storm, C.; Sijbesma, R.P. Strain Stiffening Hydrogels through Self-Assembly and Covalent Fixation of Semi-Flexible Fibers. Angew. Chem. Int. Ed. 2017, 56, 8771–8775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.X.; Xue, B.; Li, Y.; Qin, M.; Wu, J.Y.; Lu, K.; Wu, J.H.; Cao, Y.; Jiang, Q.; Wang, W. Polymer-Supramolecular Polymer Double-Network Hydrogel. Adv. Funct. Mater. 2016, 26, 9044–9052. [Google Scholar] [CrossRef]
- Li, L.; Zhang, K.J.; Wang, T.K.; Wang, P.; Xue, B.; Cao, Y.; Zhu, L.Y.; Jiang, Q. Biofabrication of a biomimetic supramolecular-polymer double network hydrogel for cartilage regeneration. Mater. Des. 2020, 189, 108492. [Google Scholar] [CrossRef]
- Hashemnejad, S.M.; Kundu, S. Strain stiffening and negative normal stress in alginate hydrogels. J. Polym. Sci. Pol. Phys. 2016, 54, 1767–1775. [Google Scholar] [CrossRef]
- McAllister, J.W.; Lott, J.R.; Schmidt, P.W.; Sammler, R.L.; Bates, F.S.; Lodge, T.P. Linear and Nonlinear Rheological Behavior of Fibrillar Methylcellulose Hydrogels. ACS Macro Lett. 2015, 4, 538–542. [Google Scholar] [CrossRef]
- Schuster, E.; Lundin, L.; Williams, M.A.K. Investigating the Relationship between Network Mechanics and Single-Chain Extension Using Biomimetic Polysaccharide Gels. Macromolecules 2012, 45, 4863–4869. [Google Scholar] [CrossRef]
- Ruskowitz, E.R.; DeForest, C.A. Photoresponsive biomaterials for targeted drug delivery and 4D cell culture. Nat. Rev. Mater. 2018, 3, 17087. [Google Scholar] [CrossRef]
- Wenger, M.P.E.; Bozec, L.; Horton, M.A.; Mesquida, P. Mechanical Properties of Collagen Fibrils. Biophy. J. 2007, 93, 1255–1263. [Google Scholar] [CrossRef] [Green Version]
- Li, H.B.; Cao, Y. Protein Mechanics: From Single Molecules to Functional Biomaterials. Accounts Chem. Res. 2010, 43, 1331–1341. [Google Scholar] [CrossRef]
- Yang, Y.J.; Holmberg, A.L.; Olsen, B.D. Artificially Engineered Protein Polymers. Annu. Rev. Chem. Biomol. 2017, 8, 549–575. [Google Scholar] [CrossRef]
- Vepari, C.; Kaplan, D.L. Silk as a biomaterial. Prog. Polym. Sci. 2007, 32, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xue, B.; Cao, Y. 100th Anniversary of Macromolecular Science Viewpoint: Synthetic Protein Hydrogels. ACS Macro Lett. 2020, 9, 512–524. [Google Scholar] [CrossRef]
- Li, J.Y.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Raines, R.T. Collagen-based biomaterials for wound healing. Biopolymers 2014, 101, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Whelan, D.; Caplice, N.M.; Clover, A.J. Fibrin as a delivery system in wound healing tissue engineering applications. J. Control Release 2014, 196, 1–8. [Google Scholar] [CrossRef]
- Lei, H.; Dong, L.; Li, Y.; Zhang, J.; Chen, H.; Wu, J.; Zhang, Y.; Fan, Q.; Xue, B.; Qin, M.; et al. Stretchable hydrogels with low hysteresis and anti-fatigue fracture based on polyprotein cross-linkers. Nat. Commun. 2020, 11, 4032. [Google Scholar] [CrossRef]
- Wu, J.; Li, P.; Dong, C.; Jiang, H.; Bin, X.; Gao, X.; Qin, M.; Wang, W.; Bin, C.; Cao, Y. Rationally designed synthetic protein hydrogels with predictable mechanical properties. Nat. Commun. 2018, 9, 620. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.-B.; Su, D.-H.; Ding, S.-L.; Zhang, Q.-C.; Li, Z.-F.; Chen, F.-C.; Ding, W.; Zhang, S.-T.; Dong, J. Salt-Assisted Toughening of Protein Hydrogel with Controlled Degradation for Bone Regeneration. Adv. Funct. Mater. 2019, 29, 1901314. [Google Scholar] [CrossRef]
- Kim, C.S.; Yang, Y.J.; Bahn, S.Y.; Cha, H.J. A bioinspired dual-crosslinked tough silk protein hydrogel as a protective biocatalytic matrix for carbon sequestration. NPG Asia Mater. 2017, 9, e391. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Mehlich, A.; Koga, N.; Huang, J.; Koga, R.; Gao, X.; Hu, C.; Jin, C.; Rief, M.; Kast, J.; et al. Forced protein unfolding leads to highly elastic and tough protein hydrogels. Nat. Commun. 2013, 4, 2974. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.A. Joint contact stress: A reasonable surrogate for biological processes? Iowa Orthop. J. 2005, 25, 82–94. [Google Scholar] [PubMed]
- Lee, S.H.; Miller, J.S.; Moon, J.J.; West, J.L. Proteolytically degradable hydrogels with a fluorogenic substrate for studies of cellular proteolytic activity and migration. Biotechnol. Progr. 2005, 21, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Buschle-Diller, G.; Wu, Y. Thermoresponsive hydrogels from BSA esterified with low molecular weight PEG. J. Appl. Polym. Sci. 2014, 131, 40946. [Google Scholar] [CrossRef]
- Demers, N.; Agostinelli, E.; Averill-Bates, D.A.; Fortier, G. Immobilization of native and poly(ethylene glycol)-treated (‘PEGylated’) bovine serum amine oxidase into a biocompatible hydrogel. Biotechnol. Appl. Biochem. 2001, 33, 201–207. [Google Scholar] [CrossRef]
- Gayet, J.C.; Fortier, G. High water content BSA-PEG hydrogel for controlled release device: Evaluation of the drug release properties. J. Control. Release 1996, 38, 177–184. [Google Scholar] [CrossRef]
- JeanFrancois, J.; Fortier, G. Immobilization of L-asparaginase into a biocompatible poly(ethylene glycol)-albumin hydrogel.1. Preparation and in vitro characterization. Biotechnol. Appl. Biochem. 1996, 23, 221–226. [Google Scholar]
- Ma, X.; Sun, X.; Hargrove, D.; Chen, J.; Song, D.; Dong, Q.; Lu, X.; Fan, T.-H.; Fu, Y.; Lei, Y. A Biocompatible and Biodegradable Protein Hydrogel with Green and Red Autofluorescence: Preparation, Characterization and In Vivo Biodegradation Tracking and Modeling. Sci. Rep. 2016, 6, 19370. [Google Scholar] [CrossRef]
- Ong, J.; Zhao, J.; Justin, A.W.; Markaki, A.E. Albumin-based hydrogels for regenerative engineering and cell transplantation. Biotechnol. Bioeng. 2019, 116, 3457–3468. [Google Scholar] [CrossRef]
- Tang, Z.; He, H.; Zhu, L.; Liu, Z.; Yang, J.; Qin, G.; Wu, J.; Tang, Y.; Zhang, D.; Chen, Q.; et al. A General Protein Unfolding-Chemical Coupling Strategy for Pure Protein Hydrogels with Mechanically Strong and Multifunctional Properties. Adv. Sci. 2021, 9, 2102557. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, J.; Guo, Y.; Li, Y.; Wang, J.; Wang, W.; Cao, Y.; Xue, B. Tuning Strain Stiffening of Protein Hydrogels by Charge Modification. Int. J. Mol. Sci. 2022, 23, 3032. https://doi.org/10.3390/ijms23063032
Gu J, Guo Y, Li Y, Wang J, Wang W, Cao Y, Xue B. Tuning Strain Stiffening of Protein Hydrogels by Charge Modification. International Journal of Molecular Sciences. 2022; 23(6):3032. https://doi.org/10.3390/ijms23063032
Chicago/Turabian StyleGu, Jie, Yu Guo, Yiran Li, Juan Wang, Wei Wang, Yi Cao, and Bin Xue. 2022. "Tuning Strain Stiffening of Protein Hydrogels by Charge Modification" International Journal of Molecular Sciences 23, no. 6: 3032. https://doi.org/10.3390/ijms23063032
APA StyleGu, J., Guo, Y., Li, Y., Wang, J., Wang, W., Cao, Y., & Xue, B. (2022). Tuning Strain Stiffening of Protein Hydrogels by Charge Modification. International Journal of Molecular Sciences, 23(6), 3032. https://doi.org/10.3390/ijms23063032