
A Comparative Analysis of Weizmannia coagulans Genomes Unravels the Genetic Potential for Biotechnological Applications

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Genomic Features of W. coagulans Strains
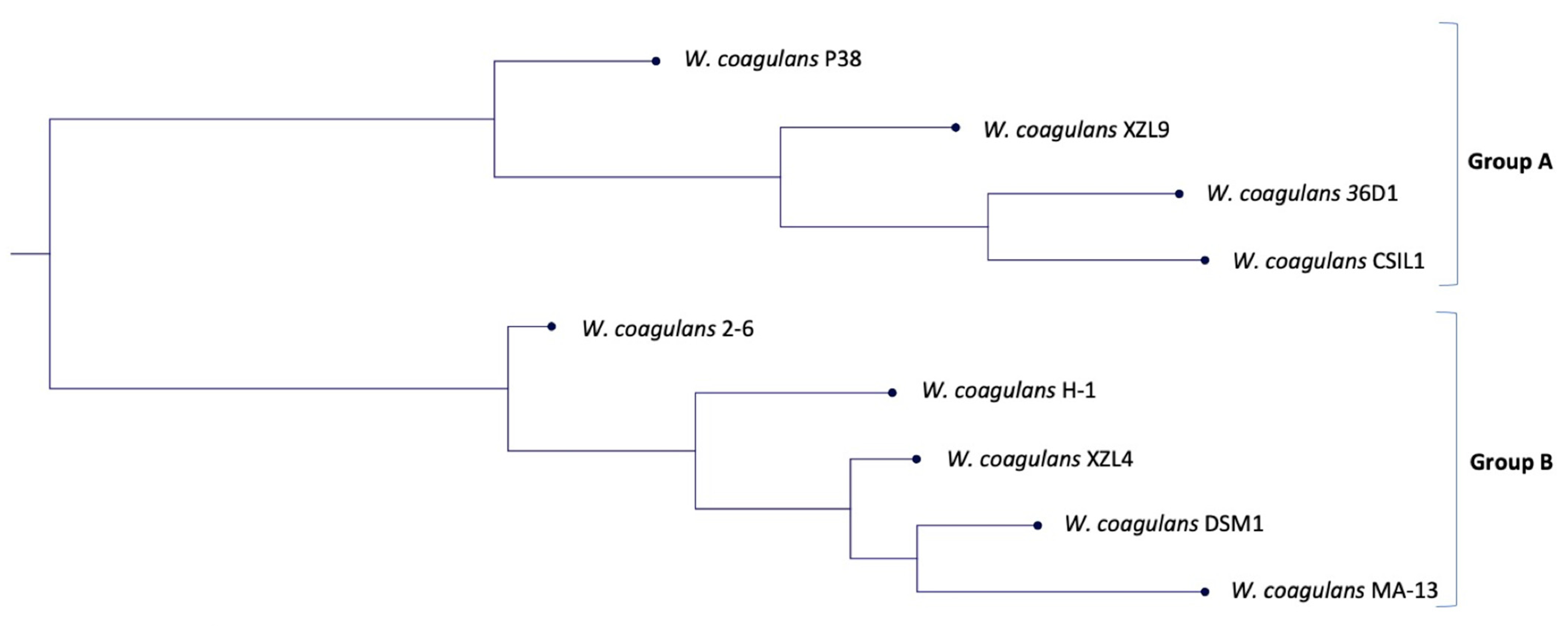
2.2. Phylogenetic and Comparative Genomic Analysis
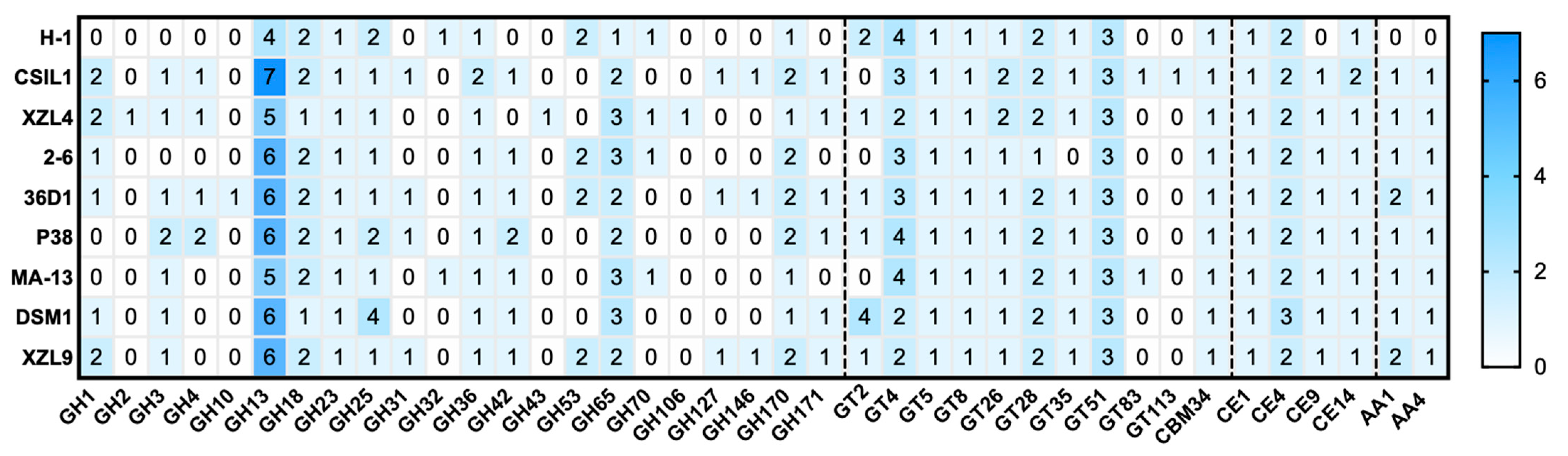
2.3. CAZome of W. coagulans
2.4. Protein Secretion Systems
2.5. Resistance Mechanisms to Environmental Challenges
2.5.1. Toxin–Antitoxin Systems
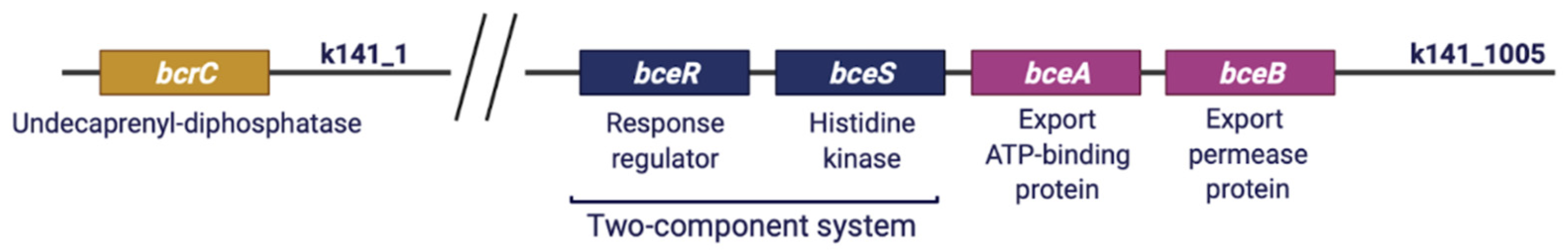
2.5.2. Genetic Determinants of Resistance to Bacitracin
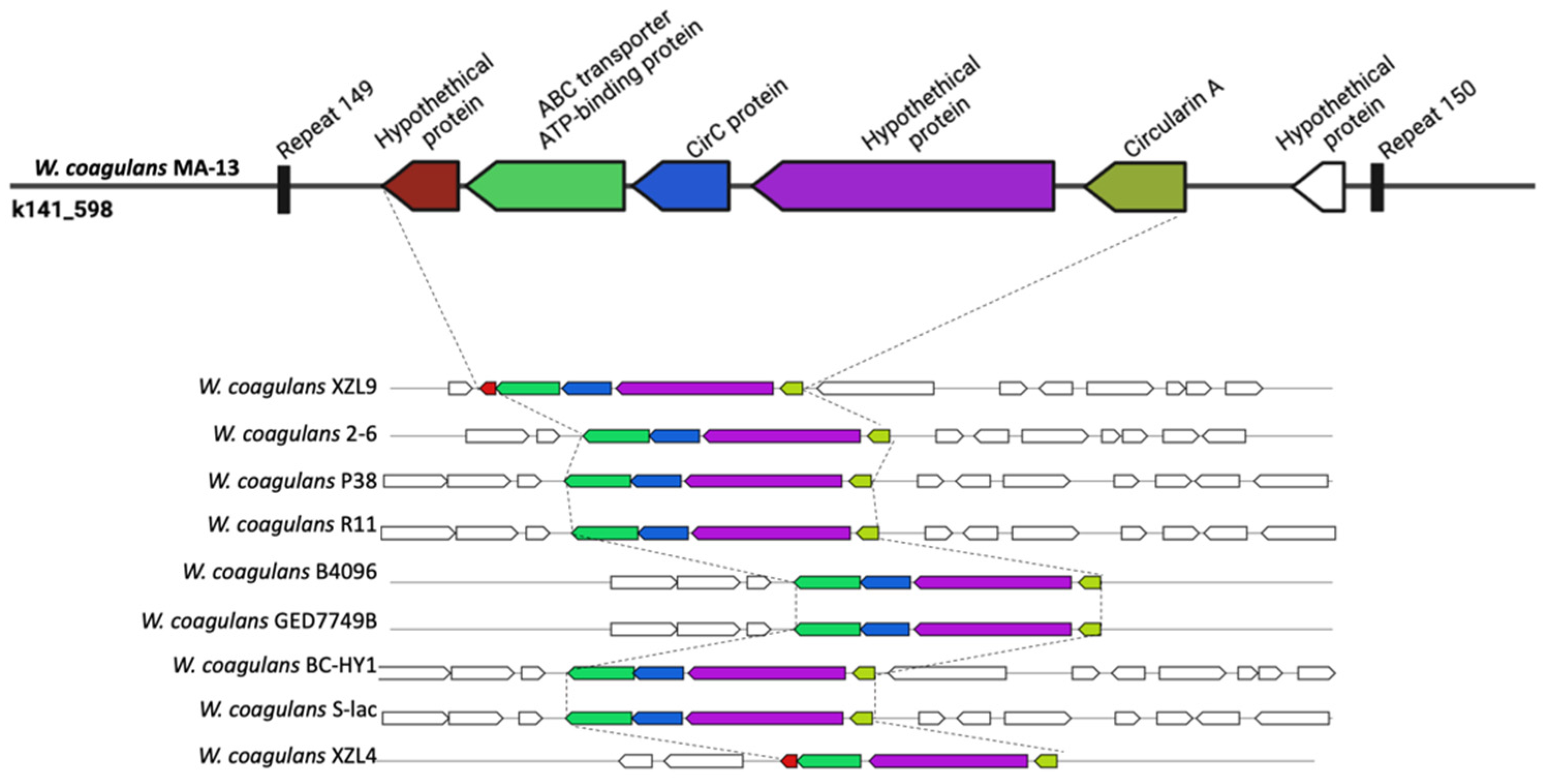
2.5.3. Secondary Metabolites: Bacteriocins
2.6. Innate and Adaptive Immunity
2.6.1. Innate Immunity
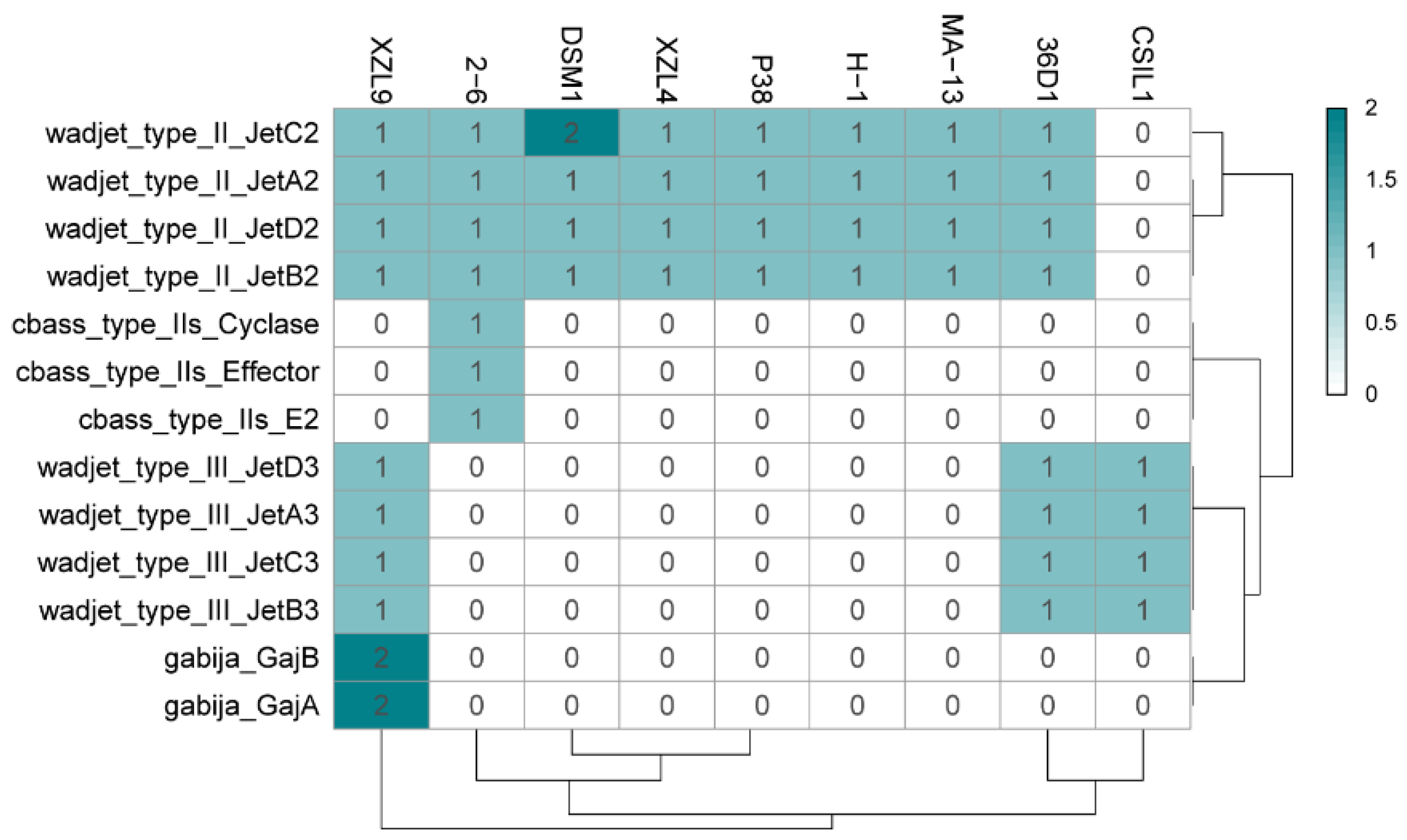
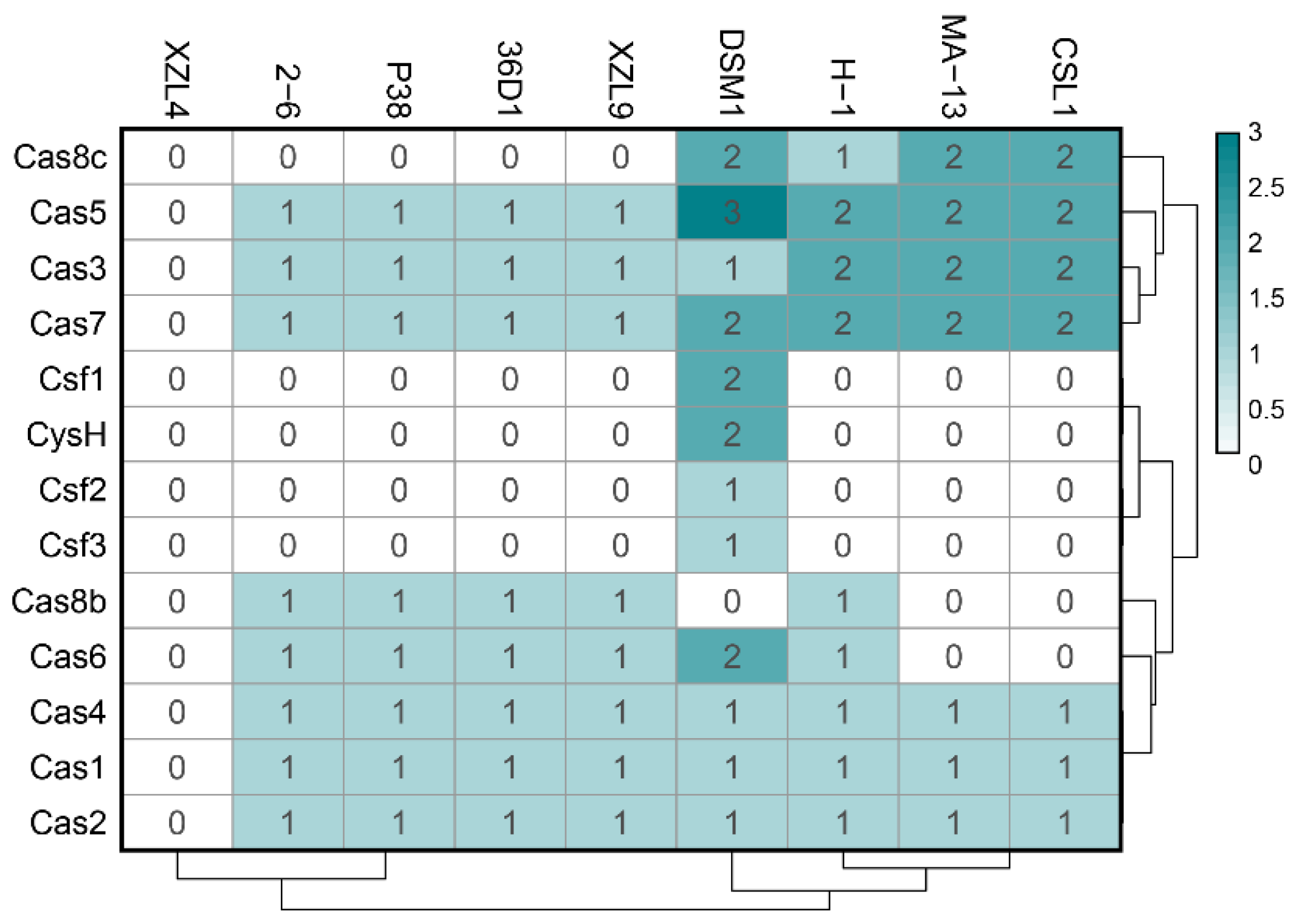
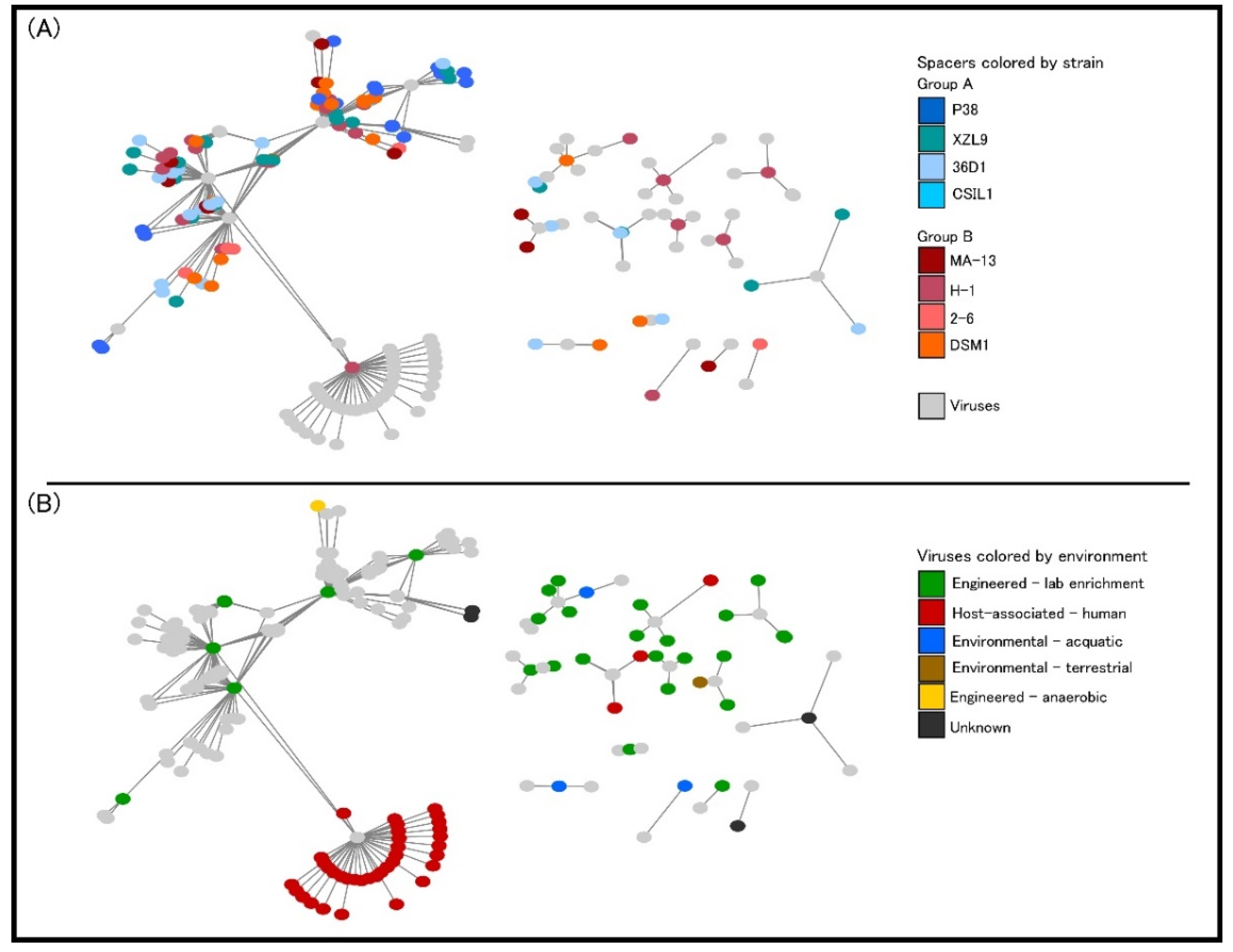
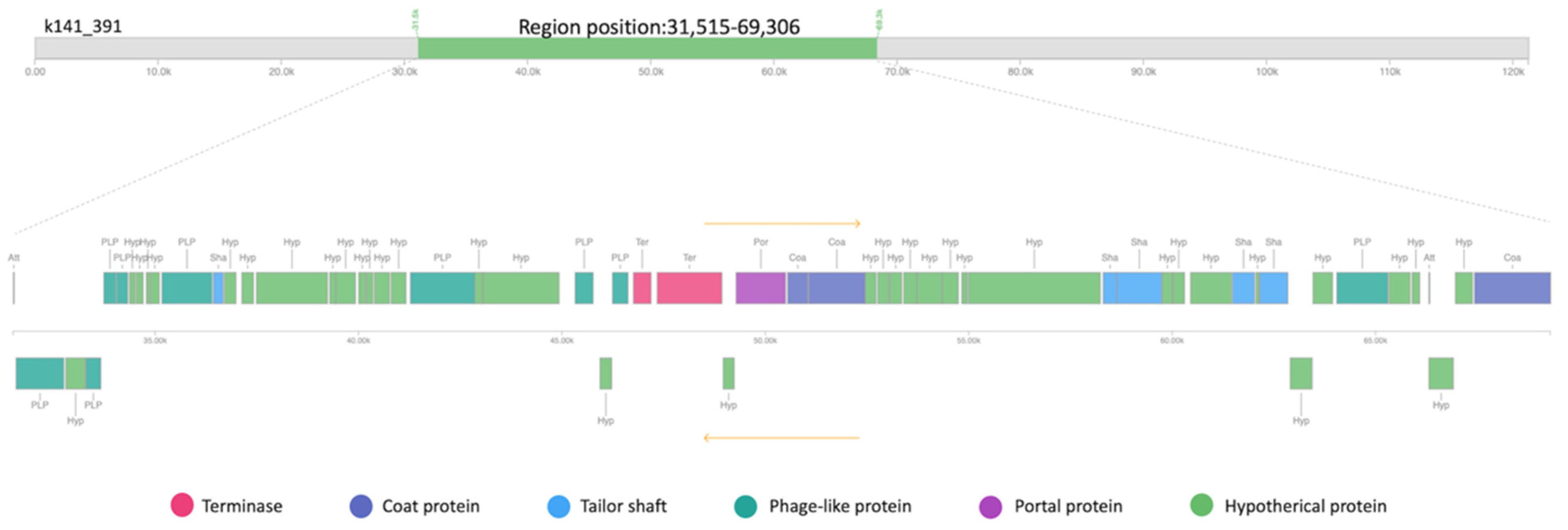
2.6.2. Adaptive Immunity: CRISPR-Cas System and Mobile Elements
3. Materials and Methods
3.1. Genome Assembly and Annotation
3.2. Construction of Whole-Genomes Sequence-Based Phylogenetic Tree and Comparative Analysis of Orthologous Genes
3.3. Analysis of Resistance Mechanisms to Environmental Challenges
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aulitto, M.; Tom, L.M.; Ceja-Navarro, J.A.; Simmons, B.A.; Singer, S.W. Whole-Genome Sequence of Brevibacillus borstelensis SDM, Isolated from a Sorghum-Adapted Microbial Community. Microbiol. Resour. Announc. 2020, 9, e01046-20. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, L.J.; Martín, C. Pretreatment of lignocellulose: Formation of inhibitory by-products and strategies for minimizing their effects. Bioresour. Technol. 2016, 199, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofvendahl, K.; Hahn-Hägerdal, B. Factors affecting the fermentative lactic acid production from renewable resources. Enzym. Microb. Technol. 2000, 26, 87–107. [Google Scholar] [CrossRef]
- Su, F.; Xu, P. Genomic analysis of thermophilic Bacillus coagulans strains: Efficient producers for platform bio-chemicals. Sci. Rep. 2014, 4, 3926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.S.; Patel, S.; Saini, N.; Chen, S. Robust demarcation of 17 distinct bacillus species clades, proposed as novel bacillaceae genera, by phylogenomics and comparative genomic analyses: Description of Robertmurraya kyonggiensis sp. nov. and proposal for an emended genus bacillus limiting it o. Int. J. Syst. Evol. Microbiol. 2020, 70, 5753–5798. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of prokaryotic names with standing in nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Aulitto, M.; Fusco, S.; Bartolucci, S.; Franzén, C.J.; Contursi, P. Bacillus coagulans MA-13: A promising thermophilic and cellulolytic strain for the production of lactic acid from lignocellulosic hydrolysate. Biotechnol. Biofuels 2017, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Aulitto, M.; Fusco, S.; Nickel, D.B.; Bartolucci, S.; Contursi, P.; Franzén, C.J. Seed culture pre-adaptation of Bacillus coagulans MA-13 improves lactic acid production in simultaneous saccharification and fermentation. Biotechnol. Biofuels 2019, 12, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Cai, C.; Chen, H.; Jiang, T.; Zheng, Z. Efficient non-sterilized fermentation of biomass-derived Xylose to lactic acid by a thermotolerant Bacillus coagulans NL01. Appl. Biochem. Biotechnol. 2012, 168, 2387–2397. [Google Scholar] [CrossRef]
- Ou, M.S.; Ingram, L.O.; Shanmugam, K.T. L(+)-Lactic acid production from non-food carbohydrates by thermotolerant Bacillus coagulans. J. Ind. Microbiol. Biotechnol. 2011, 38, 599–605. [Google Scholar] [CrossRef]
- Ye, L.; Zhou, X.; Bin Hudari, M.S.; Li, Z.; Wu, J.C. Highly efficient production of l-lactic acid from xylose by newly isolated Bacillus coagulans C106. Bioresour. Technol. 2013, 132, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Konuray, G.; Erginkaya, Z. Potential use of Bacillus coagulans in the food industry. Foods 2018, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Kapse, N.G.; Engineer, A.S.; Gowdaman, V.; Wagh, S.; Dhakephalkar, P.K. Functional annotation of the genome unravels probiotic potential of Bacillus coagulans HS243. Genomics 2019, 111, 921–929. [Google Scholar] [CrossRef]
- Cao, J.; Yu, Z.; Liu, W.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Probiotic characteristics of Bacillus coagulans and associated implications for human health and diseases. J. Funct. Foods 2020, 64, 103643. [Google Scholar] [CrossRef]
- Aulitto, M.; Strazzulli, A.; Sansone, F.; Cozzolino, F.; Monti, M.; Moracci, M.; Fiorentino, G.; Limauro, D.; Bartolucci, S.; Contursi, P. Prebiotic properties of Bacillus coagulans MA-13: Production of galactoside hydrolyzing enzymes and characterization of the transglycosylation properties of a GH42 β-galactosidase. Microb. Cell Fact. 2021, 20, 71. [Google Scholar] [CrossRef] [PubMed]
- Le Marrec, C.; Hyronimus, B.; Bressollier, P.; Verneuil, B.; Urdaci, M.C. Biochemical and genetic characterization of coagulin, a new antilisterial bacteriocin in the pediocin family of bacteriocins, produced by Bacillus coagulans I4. Appl. Environ. Microbiol. 2000, 66, 5213–5220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellegaard, K.M.; Engel, P. Beyond 16S rRNA community profiling: Intra-species diversity in the gut microbiota. Front. Microbiol. 2016, 7, 1475. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Yu, X.; Ouyang, J.; Ma, X. Complete Genome Sequence of Bacillus coagulans BC01, a Promising Human Probiotic Strain Isolated from Thick Broad Bean Sauce. Microbiol. Resour. Announc. 2021, 10, e00392-21. [Google Scholar] [CrossRef]
- Rhee, M.S.; Moritz, B.E.; Xie, G.; Glavina del Rio, T.; Dalin, E.; Tice, H.; Bruce, D.; Goodwin, L.; Chertkov, O.; Brettin, T.; et al. Complete genome sequence of a thermotolerant sporogenic lactic acid bacterium, Bacillus coagulans strain 36D1. Stand. Genom. Sci. 2011, 5, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Su, F.; Tao, F.; Tang, H.; Xu, P. Genome sequence of the thermophile Bacillus coagulans hammer, the type strain of the species. J. Bacteriol. 2012, 194, 6294–6295. [Google Scholar] [CrossRef] [Green Version]
- Nema, V. The Role and Future Possibilities of Next-Generation Sequencing in Studying Microbial Diversity. Microb. Divers. Genom. Era 2019, 611–630. [Google Scholar] [CrossRef]
- Rodríguez-Beltrán, J.; DelaFuente, J.; León-Sampedro, R.; MacLean, R.C.; San Millán, Á. Beyond horizontal gene transfer: The role of plasmids in bacterial evolution. Nat. Rev. Microbiol. 2021, 19, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Aulitto, M.; Fusco, S.; Franzén, C.J.; Strazzulli, A.; Moracci, M.; Bartolucci, S.; Contursi, P. Draft genome sequence of Bacillus coagulans ma-13, a thermophilic lactic acid producer from lignocellulose. Microbiol. Resour. Announc. 2019, 8, e00341-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Tettelin, H.; Medini, D. The Pangenome: Diversity, Dynamics and Evolution of Genomes; Springer Nature: Cham, Switzerland, 2020; ISBN 9783030382810. [Google Scholar]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Mao, X.; Yang, J.; Chen, X.; Mao, F.; Xu, Y. DbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012, 40, W445–W451. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, H.; Wu, P.; Entwistle, S.; Li, X.; Yohe, T.; Yi, H.; Yang, Z.; Yin, Y. DbCAN-seq: A database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res. 2018, 46, D516–D521. [Google Scholar] [CrossRef] [PubMed]
- Abdul Manas, N.H.; Md Illias, R.; Mahadi, N.M. Strategy in manipulating transglycosylation activity of glycosyl hydrolase for oligosaccharide production. Crit. Rev. Biotechnol. 2018, 38, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Bang, W.Y.; Ban, O.H.; Lee, B.S.; Oh, S.; Park, C.; Park, M.K.; Jung, S.K.; Yang, J.; Jung, Y.H. Genomic-, phenotypic-, and toxicity-based safety assessment and probiotic potency of Bacillus coagulans IDCC 1201 isolated from green malt. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab026. [Google Scholar] [CrossRef] [PubMed]
- Aulitto, M.; Fusco, F.A.; Fiorentino, G.; Bartolucci, S.; Contursi, P.; Limauro, D. A thermophilic enzymatic cocktail for galactomannans degradation. Enzym. Microb. Technol. 2018, 111, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Aulitto, M.; Fusco, S.; Limauro, D.; Fiorentino, G.; Bartolucci, S.; Contursi, P. Galactomannan degradation by thermophilic enzymes: A hot topic for biotechnological applications. World J. Microbiol. Biotechnol. 2019, 35, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulitto, M.; Fusco, S.; Fiorentino, G.; Limauro, D.; Pedone, E.; Bartolucci, S.; Contursi, P. Thermus thermophilus as source of thermozymes for biotechnological applications: Homologous expression and biochemical characterization of an α-galactosidase. Microb. Cell Fact. 2017, 16, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ing, N.; Deng, K.; Chen, Y.; Aulitto, M.; Gin, J.W.; Pham, T.L.M.; Petzold, C.J.; Singer, S.W.; Bowen, B.; Sale, K.L.; et al. A multiplexed nanostructure-initiator mass spectrometry (NIMS) assay for simultaneously detecting glycosyl hydrolase and lignin modifying enzyme activities. Sci. Rep. 2021, 11, 11803. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Henrissat, B. Glycoside hydrolases and glycosyltransferases: Families and functional modules. Curr. Opin. Struct. Biol. 2001, 11, 593–600. [Google Scholar] [CrossRef]
- Xavier, J.R.; Ramana, K.V.; Sharma, R.K. β-galactosidase: Biotechnological applications in food processing. J. Food Biochem. 2018, 42, e12564. [Google Scholar] [CrossRef]
- Coutinho, P.M.; Deleury, E.; Davies, G.J.; Henrissat, B. An evolving hierarchical family classification for glycosyltransferases. J. Mol. Biol. 2003, 328, 307–317. [Google Scholar] [CrossRef]
- Ulvskov, P.; Paiva, D.S.; Domozych, D.; Harholt, J. Classification, Naming and Evolutionary History of Glycosyltransferases from Sequenced Green and Red Algal Genomes. PLoS ONE 2013, 8, e76511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaivits, E.; Siaperas, R.; Agrafiotis, A.; Ouazzani, J.; Magoulas, A.; Gioti, A.; Topakas, E. Functional and transcriptomic investigation of laccase activity in the presence of PCB29 identifies two novel enzymes and the multicopper oxidase repertoire of a marine-derived fungus. Sci. Total Environ. 2021, 775, 145818. [Google Scholar] [CrossRef] [PubMed]
- Gygli, G.; de Vries, R.P.; van Berkel, W.J.H. On the origin of vanillyl alcohol oxidases. Fungal Genet. Biol. 2018, 116, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Chang, S.H.; Sun, H.L.; Chang, Y.C.; Hsin, I.L.; Lue, K.H.; Ko, J.L. IFN-γ induction on carbohydrate binding module of fungal immunomodulatory protein in human peripheral mononuclear cells. J. Agric. Food Chem. 2012, 60, 4914–4922. [Google Scholar] [CrossRef] [PubMed]
- Goosens, V.J.; Monteferrante, C.G.; Van Dijl, J.M. The Tat system of Gram-positive bacteria. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 1698–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentschev, I.; Dietrich, G.; Goebel, W. The E. coli α-hemolysin secretion system and its use in vaccine development. Trends Microbiol. 2002, 10, 39–45. [Google Scholar] [CrossRef]
- Rivera-Calzada, A.; Famelis, N.; Llorca, O.; Geibel, S. Type VII secretion systems: Structure, functions and transport models. Nat. Rev. Microbiol. 2021, 19, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Peñil-Celis, A.; Garcillán-Barcia, M.P. Crosstalk Between Type VI Secretion System and Mobile Genetic Elements. Front. Mol. Biosci. 2019, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Kędzierska, B.; Potrykus, K. Minigene as a novel regulatory element in toxin-antitoxin systems. Int. J. Mol. Sci. 2021, 22, 3389. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Wood, T.K. A Primary Physiological Role of Toxin/Antitoxin Systems Is Phage Inhibition. Front. Microbiol. 2020, 11, 1895. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L. Toxin–antitoxin systems: Why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Bukowski, M.; Hyz, K.; Janczak, M.; Hydzik, M.; Dubin, G.; Wladyka, B. Identification of novel mazEF/pemIK family toxin-antitoxin loci and their distribution in the Staphylococcus genus. Sci. Rep. 2017, 7, 13462. [Google Scholar] [CrossRef] [Green Version]
- Loh, J.M.S.; Soh, K.Y.; Proft, T. Generation of Bioluminescent Group A Streptococcus for Biophotonic Imaging. In Methods in Molecular Biology; Proft, T., Loh, J.M.S., Eds.; Springer: New York, NY, USA, 2020; Volume 2136, pp. 71–77. ISBN 978-1-0716-0467-0. [Google Scholar]
- Davis, J.J.; Boisvert, S.; Brettin, T.; Kenyon, R.W.; Mao, C.; Olson, R.; Overbeek, R.; Santerre, J.; Shukla, M.; Wattam, A.R.; et al. Antimicrobial Resistance Prediction in PATRIC and RAST. Sci. Rep. 2016, 6, 27930. [Google Scholar] [CrossRef] [PubMed]
- Dintner, S.; Heermann, R.; Fang, C.; Jung, K.; Gebhard, S. A sensory complex consisting of an ATP-binding cassette transporter and a two-component regulatory system controls bacitracin resistance in Bacillus subtilis. J. Biol. Chem. 2014, 289, 27899–27910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohki, R.; Giyanto, T.K.; Masuyama, W.; Moriya, S.; Kobayashi, K.; Ogasawara, N. The BceRS two-component regulatory system induces expression of the bacitracin transporter, BceAB, in Bacillus subtilis. Mol. Microbiol. 2003, 49, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Harel, Y.M.; Bailone, A.; Bibi, E. Resistance to bacitracin as modulated by an Escherichia coli homologue of the bacitracin ABC transporter BcrC subunit from Bacillus licheniformis. J. Bacteriol. 1999, 181, 6176–6178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, R.; El Ghachi, M.; Mengin-Lecreulx, D.; Chippaux, M.; Denizot, F. BcrC from Bacillus subtilis acts as an undecaprenyl pyrophosphate phosphatase in bacitracin resistance. J. Biol. Chem. 2005, 280, 28852–28857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demain, A.L.; Fang, A. The natural functions of secondary metabolites. Adv. Biochem. Eng. Biotechnol. 2000, 69, 1–39. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Meirelles, L.A.; Newman, D.K. From the soil to the clinic: The impact of microbial secondary metabolites on antibiotic tolerance and resistance. Nat. Rev. Microbiol. 2022, 20, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Kemperman, R.; Jonker, M.; Nauta, A.; Kuipers, O.P.; Kok, J. Functional Analysis of the Gene Cluster Involved in Production of the Bacteriocin Circularin A by Clostridium beijerinckii ATCC 25752. Appl. Environ. Microbiol. 2003, 69, 5839–5848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of anti-phage defense systems in the microbialpan-genome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Altae-Tran, H.; Böhning, F.; Makarova, K.S.; Segel, M.; Schmid-Burgk, J.L.; Koob, J.; Wolf, Y.I.; Koonin, E.V.; Zhang, F. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 2020, 369, 1077–1084. [Google Scholar] [CrossRef]
- Panas, M.W.; Jain, P.; Yang, H.; Mitra, S.; Biswas, D.; Wattam, A.R.; Letvin, N.L.; Jacobs, W.R. Noncanonical SMC protein in Mycobacterium smegmatis restricts maintenance of Mycobacterium fortuitum plasmids. Proc. Natl. Acad. Sci. USA 2014, 111, 13264–13271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millman, A.; Melamed, S.; Amitai, G.; Sorek, R. Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat. Microbiol. 2020, 5, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Huang, F.; Wu, H.; Lu, X.; Yan, Y.; Yu, B.; Wang, X.; Zhu, B. A nucleotide-sensing endonuclease from the Gabija bacterial defense system. Nucleic Acids Res. 2021, 49, 5216–5229. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, A.D.; Gilmore, M.S. CRISPR-Cas: To take up DNA or not—That is the question. Cell Host Microbe 2012, 12, 125–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newsom, S.; Parameshwaran, H.P.; Martin, L.; Rajan, R. The CRISPR-Cas Mechanism for Adaptive Immunity and Alternate Bacterial Functions Fuels Diverse Biotechnologies. Front. Cell. Infect. Microbiol. 2021, 10, 898. [Google Scholar] [CrossRef] [PubMed]
- Almendros, C.; Guzmán, N.M.; García-Martínez, J.; Mojica, F.J.M. Anti-cas spacers in orphan CRISPR4 arrays prevent uptake of active CRISPR–Cas I-F systems. Nat. Microbiol. 2016, 1, 16081. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.A.; Utkina, I.; Wolf, Y.I.; Makarova, K.S.; Severinov, K.V.; Koonin, E.V. CRISPR Arrays Away from cas Genes. Cris. J. 2020, 3, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Páez-Espino, D.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Reddy, T.B.K.; Nayfach, S.; Schulz, F.; Call, L.; et al. IMG/VR v3: An integrated ecological and evolutionary framework for interrogating genomes of uncultivated viruses. Nucleic Acids Res. 2021, 49, D764–D775. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, A. Genomic islands mediate environmental adaptation and the spread of antibiotic resistance in multiresistant Enterococci—Evidence from genomic sequences. BMC Microbiol. 2021, 21, 55. [Google Scholar] [CrossRef] [PubMed]
- Owen, S.V.; Wenner, N.; Dulberger, C.L.; Rodwell, E.V.; Bowers-Barnard, A.; Quinones-Olvera, N.; Rigden, D.J.; Rubin, E.J.; Garner, E.C.; Baym, M.; et al. Prophage-encoded phage defence proteins with cognate self-immunity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Aulitto, M.; Gallo, G.; Puopolo, R.; Mormone, A.; Limauro, D.; Contursi, P.; Piochi, M.; Bartolucci, S.; Fiorentino, G. Genomic Insight of Alicyclobacillus mali FL18 Isolated from an Arsenic-Rich Hot Spring. Front. Microbiol. 2021, 12, 669. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.; Mahillon, J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, present and future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardeau, M.; Lehtinen, M.J.; Forssten, S.D.; Nurminen, P. Importance of the gastrointestinal life cycle of Bacillus for probiotic functionality. J. Food Sci. Technol. 2017, 54, 2570. [Google Scholar] [CrossRef] [PubMed]
- Ben Amor, M.G.; Siala, M.; Zayani, M.; Grosset, N.; Smaoui, S.; Messadi-Akrout, F.; Baron, F.; Jan, S.; Gautier, M.; Gdoura, R. Isolation, identification, prevalence, and genetic diversity of Bacillus cereus group bacteria from different foodstuffs in Tunisia. Front. Microbiol. 2018, 9, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, P.; Yu, S.; Wang, J.; Guo, H.; Zhang, Y.; Liao, X.; Zhang, J.; Wu, S.; Gu, Q.; Xue, L.; et al. Bacillus cereus Isolated from vegetables in China: Incidence, genetic diversity, virulence genes, and antimicrobial resistance. Front. Microbiol. 2019, 10, 948. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikheenko, A.; Valin, G.; Prjibelski, A.; Saveliev, V.; Gurevich, A. Icarus: Visualizer for de novo assembly evaluation. Bioinformatics 2016, 32, 3321–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Arkin, A.; Stevens, R.; Cottingham, R.; Maslov, S.; Perez, F. KBase: The Department of Energy systems biology knowledgebase. Nat. Biotechnol. 2015, 36, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selengut, J.D.; Haft, D.H.; Davidsen, T.; Ganapathy, A.; Gwinn-Giglio, M.; Nelson, W.C.; Richter, A.R.; White, O. TIGRFAMs and Genome Properties: Tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 2007, 35, D260–D264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and classification of antiviral defence systems in bacteria and archaea with PADLOC reveals new system types. Nucleic Acids Res. 2021, 49, 10868–10878. [Google Scholar] [CrossRef] [PubMed]
- Russel, J.; Pinilla-Redondo, R.; Mayo-Muñoz, D.; Shah, S.A.; Sørensen, S.J. CRISPRCasTyper: Automated Identification, Annotation, and Classification of CRISPR-Cas Loci. CRISPR J. 2020, 3, 462–469. Available online: https://home.liebertpub.com/crispr (accessed on 15 September 2021). [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Fusco, S.; Aulitto, M.; Iacobucci, I.; Crocamo, G.; Pucci, P.; Bartolucci, S.; Monti, M.; Contursi, P. The interaction between the F55 virus-encoded transcription regulator and the RadA host recombinase reveals a common strategy in Archaea and Bacteria to sense the UV-induced damage to the host DNA. Biochim. Biophys. Acta-Gene Regul. Mech. 2020, 1863, 194493. [Google Scholar] [CrossRef] [PubMed]
- Beeby, M.; O’Connor, B.D.; Ryttersgaard, C.; Boutz, D.R.; Perry, L.J.; Yeates, T.O. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol. 2005, 3, 1549–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, P.; Mamo, G.; Karlsson, E.N. Potential and utilization of thermophiles and thermostable enzymes in biorefining. Microb. Cell Fact. 2007, 6, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atalah, J.; Cáceres-Moreno, P.; Espina, G.; Blamey, J.M. Thermophiles and the applications of their enzymes as new biocatalysts. Bioresour. Technol. 2019, 280, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Notomista, E.; Falanga, A.; Fusco, S.; Pirone, L.; Zanfardino, A.; Galdiero, S.; Varcamonti, M.; Pedone, E.; Contursi, P. The identification of a novel Sulfolobus islandicus CAMP-like peptide points to archaeal microorganisms as cell factories for the production of antimicrobial molecules. Microb. Cell Fact. 2015, 14, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ae, P.C.; Pisani, F.M.; Grigoriev, A.; Raffaele, A.E.; Simonetta, C.; Ae, B.; Rossi, M.; Contursi, P.; Pisani, F.M.; Grigoriev, A.; et al. Identification and autonomous replication capability of a chromosomal replication origin from the archaeon Sulfolobus solfataricus. Extremophiles 2004, 8, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Prato, S.; Vitale, R.M.; Contursi, P.; Lipps, G.; Saviano, M.; Rossi, M.; Bartolucci, S. Molecular modeling and functional characterization of the monomeric primase-polymerase domain from the Sulfolobus solfataricus plasmid pIT3. FEBS J. 2008, 275, 4389–4402. [Google Scholar] [CrossRef] [PubMed]
- Contursi, P.; Cannio, R.; Prato, S.; She, Q.; Rossi, M.; Bartolucci, S. Transcriptional analysis of the genetic element pSSVx: Differential and temporal regulation of gene expression reveals correlation between transcription and replication. J. Bacteriol. 2007, 189, 6339–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contursi, P.; Fusco, S.; Limauro, D.; Fiorentino, G. Host and viral transcriptional regulators in Sulfolobus: An overview. Extremophiles 2013, 17, 881–895. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Query | Reference | ANI Estimate (%) |
|---|---|---|
| MA-13 | CSL1 | 94.68 |
| MA-13 | 36D1 | 94.85 |
| MA-13 | XZL9 | 95.04 |
| MA-13 | P38 | 95.15 |
| MA-13 | H-1 | 97.89 |
| MA-13 | 2–6 | 97.98 |
| MA-13 | XZL4 | 98.22 |
| MA-13 | DSM1 | 98.43 |
| Strain | Total No. of Genes | No. of CAZymes | % CAZymes |
|---|---|---|---|
| MA-13 | 2689 | 40 | 1.49 |
| DSM1 | 2766 | 45 | 1.62 |
| 36D1 | 3128 | 48 | 1.53 |
| XZL9 | 3112 | 46 | 1.47 |
| P38 | 3097 | 46 | 1.48 |
| 2–6 | 2743 | 39 | 1.42 |
| H-1 | 2536 | 36 | 1.42 |
| XZL4 | 2593 | 43 | 1.65 |
| CSIL1 | 3165 | 50 | 1.56 |
| Accession Number | W. coagulans Strain | Function |
|---|---|---|
| WP_035181994.1 | 2–6 | 6 kDa early secretory antigenic target ESAT-6 (EsxA) |
| WP_041818879.1 | 2–6 | Putative secretion accessory protein EsaA/YueB @ Bacteriophage SPP1 receptor |
| WP_013858856.1 | 2–6 | FtsK/SpoIIIE family protein, putative EssC/YukB component of Type VII secretion system |
| WP_035181994.1 | 36D1 | 6 kDa early secretory antigenic target ESAT-6 (EsxA) |
| WP_014096028.1 | 36D1 | secretion protein HlyD |
| WP_029141776.1 | DSM_1 | Putative secretion system component EssB/YukC |
| MBF8418457.1 | MA-13 | type VII secretion protein EssA |
| MBF8418459.1 | MA-13 | type VII secretion protein EssB |
| WP_026685044.1 | CSIL1 | secretion protein HlyD |
| WP_052123334.1 | P38 | FtsK/SpoIIIE family protein, putative EssC/YukB component of Type VII secretion system |
| WP_035190278.1 | P38 | FtsK/SpoIIIE family protein, putative EssC/YukB component of Type VII secretion system |
| WP_035190280.1 | P38 | Putative secretion system component EssB/YukC |
| WP_035190310.1 | P38 | Putative secretion accessory protein EsaA/YueB @ Bacteriophage SPP1 receptor |
| WP_035190312.1 | P38 | 6 kDa early secretory antigenic target ESAT-6 (EsxA) |
| Strain | Contig | Prediction | Start | End | Consensus_repeat | Orientation | Cas_associated | N_repeats |
|---|---|---|---|---|---|---|---|---|
| 2–6 | 2–6_NC_015634.1 | I-B | 2,499,794 | 2,503,038 | GTTGAACTTTAACATTGGATGTATTTAAAT | R | yes | 50 |
| I-B | 2,497,017 | 2,497,373 | GTTTCAATTCCTCATAGGTAAAATACAAAC | R | yes | 6 | ||
| 36D1 | 36D1_NC_016023.1 | Unknown | 320,637 | 321,284 | TTTTGAAGCCGTCAAAAGGACAAAA | F | orphan | 13 |
| I-B | 1,096,065 | 1,097,943 | GTTAGTATTTTACCTATGAGGAATTGAAAC | R | orphan | 29 | ||
| I-B | 2,117,433 | 2,121,795 | GTTTGTATTTTACCTATGAGGAATTGAAAC | F | yes | 66 | ||
| I-B | 2,123,872 | 2,124,703 | GTTTGTATTTTACCTATGAGGAATTGAAAC | F | yes | 13 | ||
| I-B | 2,126,172 | 2,127,007 | GTTTGTATTTTACCTATGAGGAATTGAAAC | F | yes | 13 | ||
| CSIL1 | CSIL1_NZ_AXVW01000068.1_scaffold00063.63_C | I-C | 13,264 | 13,753 | GTCACACTCCTTGCGAGTGTGTGGATTGAAAT | F | yes | 8 |
| CSIL1_NZ_AXVW01000098.1_scaffold00091.91_C | I-C | 7 | 236 | GTCGCTCCCTACATGGGGGCGTGGATTGAAATC | F | orphan | 4 | |
| CSIL1_NZ_KI519465.1_scaffold00026.26 | I-B | 19,196 | 19,356 | GTTAGTATTTTACCTATGAGGAATTGAAA | F | orphan | 3 | |
| DSM1 | DSM1_NZ_CP009709.1 | I-C | 1,536,669 | 1,541,901 | GTCGCTCCCTACGTGGGGGCGTGGATTGAAAT | R | yes | 80 |
| I-B | 2,584,186 | 2,585,866 | GTTAGTATTTTACCTATGAGGAATTGAAAC | F | orphan | 26 | ||
| I-C | 2,785,593 | 2,786,680 | GTCACACTCCTCGTGAGTGTGTGGATTGAAAT | F | yes | 17 | ||
| H-1 | H-1_NZ_ANAQ01000049.1_000049 | I-C | 9436 | 9599 | GTCACACTCCTCGTGAGTGTGTGAATTGAAAT | F | yes | 3 |
| H-1_NZ_ANAQ01000116.1_000116 | I-C | 43 | 204 | GTCACACTCCTCGTGAGTGTGTGAATTGAAAT | F | orphan | 3 | |
| H-1_NZ_ANAQ01000125.1_000125 | I-C | 3689 | 3983 | GTCACACTCCTCGTGAGTGTGTGAATTGAAAT | R | orphan | 5 | |
| H-1_NZ_ANAQ01000203.1_000203 | I-B | 74 | 2139 | GTTGAACTTTAACATTGGATGTATTTAAAT | F | orphan | 32 | |
| H-1_NZ_ANAQ01000268.1_000268 | I-B | 17,520 | 20,714 | GTTGAACTTTAACATTGGATGTATTTAAAT | F | yes | 49 | |
| H-1_NZ_ANAQ01000293.1_000293 | I-B | 112 | 339 | GTTGAACTTTAACATTGGATGTATTTAAATT | R | orphan | 4 | |
| MA-13 | MA-13_NZ_SMSP02000017.1_69 | I-C | 32 | 1577 | GTCGCTCCCTACATGGGGGCGTGGATTGAAAT | F | orphan | 24 |
| MA-13_NZ_SMSP02000061.1_151 | I-C | 8671 | 10,021 | GTCGCTCCCTACATGGGGGCGTGGATTGAAAT | R | yes | 21 | |
| MA-13_NZ_SMSP02000062.1_309 | I-C | 52,270 | 52,630 | GTCGCTCCCTACATGGGGGCGTGGATTGAAAT | F | yes | 6 | |
| P38 | P38_NZ_JSVI01000006.1_6 | I-B | 99,580 | 101,051 | GTTGAACTTTAACATTGGATGTATTTAAAT | F | yes | 23 |
| P38_NZ_JSVI01000107.1_111 | I-B | 446 | 1784 | GTTGAACTTTAACATTGGATGTATTTAAAT | R | orphan | 21 | |
| XZL9 | XZL9_NZ_ANAP01000038.1_000038 | I-B | 122 | 953 | GTTTGTATTTTACCTATGAGGAATTGAAAC | R | yes | 13 |
| XZL9_NZ_ANAP01000157.1_000160 | I-B | 97 | 1197 | GTTTGTATTTTACCTATGAGGAATTGAAAC | F | orphan | 17 | |
| XZL9_NZ_ANAP01000220.1_000226 | I-B | 68 | 3018 | GTTTGTATTTTACCTATGAGGAATTGAAAC | R | orphan | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aulitto, M.; Martinez-Alvarez, L.; Fiorentino, G.; Limauro, D.; Peng, X.; Contursi, P. A Comparative Analysis of Weizmannia coagulans Genomes Unravels the Genetic Potential for Biotechnological Applications. Int. J. Mol. Sci. 2022, 23, 3135. https://doi.org/10.3390/ijms23063135
Aulitto M, Martinez-Alvarez L, Fiorentino G, Limauro D, Peng X, Contursi P. A Comparative Analysis of Weizmannia coagulans Genomes Unravels the Genetic Potential for Biotechnological Applications. International Journal of Molecular Sciences. 2022; 23(6):3135. https://doi.org/10.3390/ijms23063135
Chicago/Turabian StyleAulitto, Martina, Laura Martinez-Alvarez, Gabriella Fiorentino, Danila Limauro, Xu Peng, and Patrizia Contursi. 2022. "A Comparative Analysis of Weizmannia coagulans Genomes Unravels the Genetic Potential for Biotechnological Applications" International Journal of Molecular Sciences 23, no. 6: 3135. https://doi.org/10.3390/ijms23063135
APA StyleAulitto, M., Martinez-Alvarez, L., Fiorentino, G., Limauro, D., Peng, X., & Contursi, P. (2022). A Comparative Analysis of Weizmannia coagulans Genomes Unravels the Genetic Potential for Biotechnological Applications. International Journal of Molecular Sciences, 23(6), 3135. https://doi.org/10.3390/ijms23063135