
Repositioning the Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) on the TRAIL to the Development of Diabetes Mellitus: An Update of Experimental and Clinical Evidence


Abstract
:1. Introduction
2. Literature Search and Review Criteria
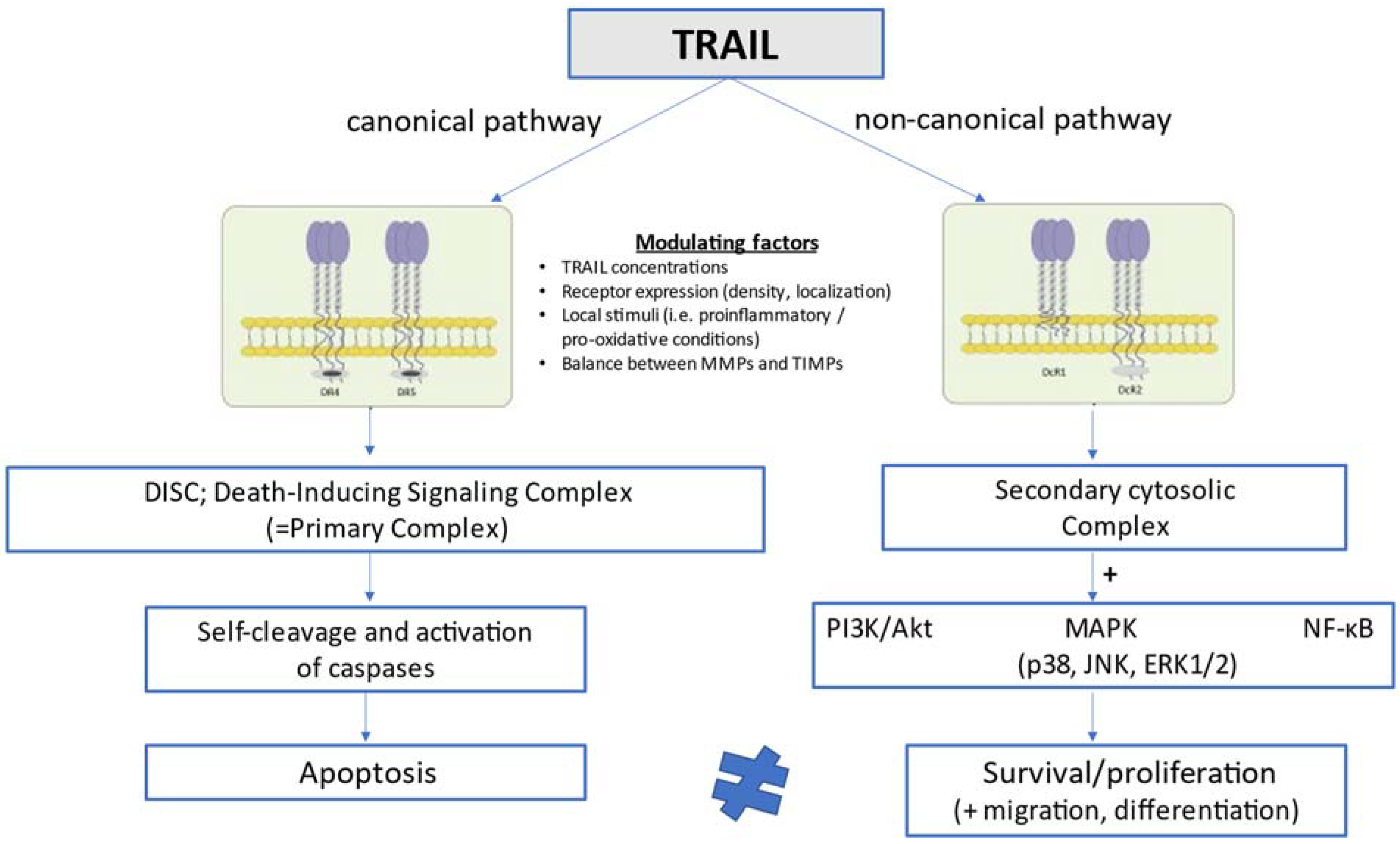
3. Brief Overview of TRAIL Biology and Signaling Pathways
4. The Role of TRAIL in T1DM
5. The Role of TRAIL in T2DM
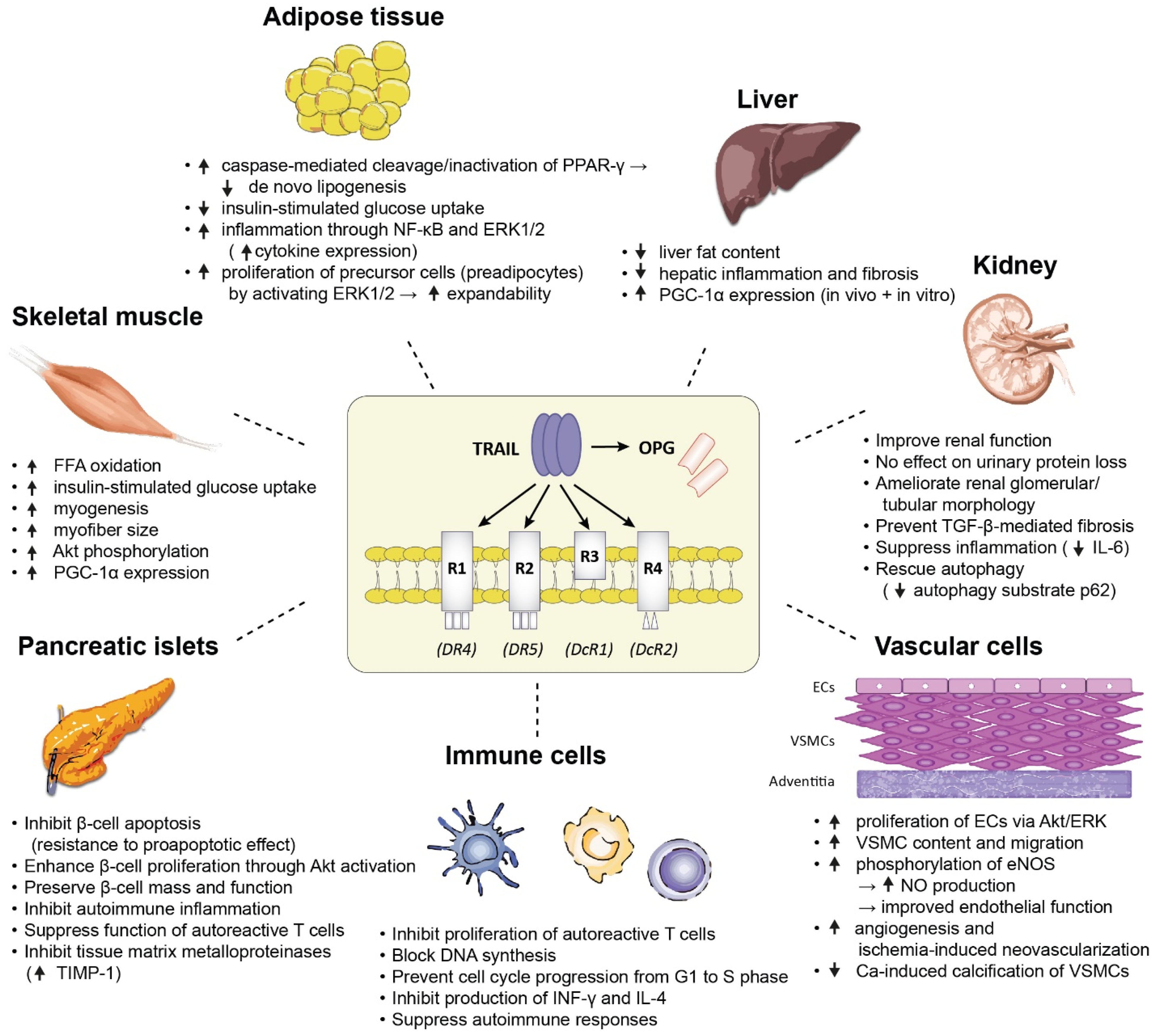
6. Proposed Mechanisms Underlying the Protective Role of TRAIL in T1DM and T2DM
7. The role of TRAIL in Diabetes-Related Complications
7.1. Atherosclerotic Cardiovascular Disease (ACVD)
7.2. Microvascular Complications
8. Summary and Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The Molecular Architecture of the TNF Superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased Susceptibility to Tumor Initiation and Metastasis in TNF-Related Apoptosis-Inducing Ligand-Deficient Mice. J. Immunol. Baltim. Md. 2002, 168, 1356–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical Role for Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Immune Surveillance against Tumor Development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossin, A.; Miloro, G.; Hueber, A.O. TRAIL and FasL Functions in Cancer and Autoimmune Diseases: Towards an Increasing Complexity. Cancers 2019, 11, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and Characterization of a New Member of the TNF Family That Induces Apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazi, A.; Herbst, R.S. To Kill a Tumor Cell: The Potential of Proapoptotic Receptor Agonists. J. Clin. Investig. 2008, 118, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Secchiero, P.; Zerbinati, C.; Rimondi, E.; Corallini, F.; Milani, D.; Grill, V.; Forti, G.; Capitani, S.; Zauli, G. TRAIL Promotes the Survival, Migration and Proliferation of Vascular Smooth Muscle Cells. Cell. Mol. Life Sci. 2004, 61, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Carnevale, E.; Milani, D.; Pandolfi, A.; Zella, D.; Zauli, G. TRAIL Promotes the Survival and Proliferation of Primary Human Vascular Endothelial Cells by Activating the Akt and ERK Pathways. Circulation 2003, 107, 2250–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Wang, Q.; Xu, J.; Xu, X.; Padilla, M.T.; Ren, G.; Gou, X.; Lin, Y. Attenuation of TNFSF10/TRAIL-Induced Apoptosis by an Autophagic Survival Pathway Involving TRAF2- And RIPK1/RIP1-Mediated MAPK8/JNK Activation. Autophagy 2012, 8, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Milani, D.; Zauli, G.; Rimondi, E.; Celeghini, C.; Marmiroli, S.; Narducci, P.; Capitani, S.; Secchiero, P. Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand Sequentially Activates pro-Survival and pro-Apoptotic Pathways in SK-N-MC Neuronal Cells. J. Neurochem. 2003, 86, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Song, J.H.; Tse, M.C.L.; Bellail, A.; Phuphanich, S.; Khuri, F.; Kneteman, N.M.; Hao, C. Lipid Rafts and Nonrafts Mediate Tumor Necrosis Factor Related Apoptosis-Inducing Ligand Induced Apoptotic and Nonapoptotic Signals in Non Small Cell Lung Carcinoma Cells. Cancer Res. 2007, 67, 6946–6955. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Norcio, A.; Toffoli, B.; Zauli, G.; Secchiero, P. Potential Role of TRAIL in the Management of Autoimmune Diabetes Mellitus. Curr. Pharm. Des. 2012, 18, 5759–5765. [Google Scholar] [CrossRef]
- Harith, H.H.; Morris, M.J.; Kavurma, M.M. On the TRAIL of Obesity and Diabetes. Trends Endocrinol. Metab. 2013, 24, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Bossi, F.; Bernardi, S.; Zauli, G.; Secchiero, P.; Fabris, B. TRAIL Modulates the Immune System and Protects against the Development of Diabetes. J. Immunol. Res. 2015, 2015, 680749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamhamedi-Cherradi, S.E.; Zheng, S.; Tisch, R.M.; Chen, Y.H. Critical Roles of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in Type 1 Diabetes. Diabetes 2003, 52, 2274–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartland, S.P.; Harith, H.H.; Genner, S.W.; Dang, L.; Cogger, V.C.; Vellozzi, M.; di Bartolo, B.A.; Thomas, S.R.; Adams, L.A.; Kavurma, M.M. Non-Alcoholic Fatty Liver Disease, Vascular Inflammation and Insulin Resistance Are Exacerbated by TRAIL Deletion in Mice. Sci. Rep. 2017, 7, 1898. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, B.A.; Chan, J.; Bennett, M.R.; Cartland, S.; Bao, S.; Tuch, B.E.; Kavurma, M.M. TNF-Related Apoptosis-Inducing Ligand (TRAIL) Protects against Diabetes and Atherosclerosis in Apoe/Mice. Diabetologia 2011, 54, 3157–3167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zauli, G.; Toffoli, B.; di Iasio, M.G.; Celeghini, C.; Fabris, B.; Secchiero, P. Treatment with Recombinant Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Alleviates the Severity of Streptozotocin-Induced Diabetes. Diabetes 2010, 59, 1261–1265. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, S.; Zauli, G.; Tikellis, C.; Candido, R.; Fabris, B.; Secchiero, P.; Cooper, M.E.; Thomas, M.C. TNF-Related Apoptosis-Inducing Ligand Significantly Attenuates Metabolic Abnormalities in High-Fat-Fed Mice Reducing Adiposity and Systemic Inflammation. Clin. Sci. 2012, 123, 547–555. [Google Scholar] [CrossRef]
- Bernardi, S.; Toffoli, B.; Tisato, V.; Bossi, F.; Biffi, S.; Lorenzon, A.; Zauli, G.; Secchiero, P.; Fabris, B. TRAIL Reduces Impaired Glucose Tolerance and NAFLD in the High-Fat Diet Fed Mouse. Clin. Sci. 2018, 132, 69–83. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Park, E.J.; Joe, Y.; Seo, E.; Park, M.K.; Seo, S.Y.; Chung, H.Y.; Yoo, Y.H.; Kim, D.K.; Lee, H.J. Systemic Delivery of TNF-Related Apoptosis-Inducing Ligand (TRAIL) Elevates Levels of Tissue Inhibitor of Metalloproteinase-1 (TIMP-1) and Prevents Type 1 Diabetes in Nonobese Diabetic Mice. Endocrinology 2010, 151, 5638–5646. [Google Scholar] [CrossRef] [Green Version]
- Dirice, E.; Sanlioglu, A.D.; Kahraman, S.; Ozturk, S.; Balci, M.K.; Omer, A.; Griffith, T.S.; Sanlioglu, S. Adenovirus-Mediated TRAIL Gene (Ad5hTRAIL) Delivery into Pancreatic Islets Prolongs Normoglycemia in Streptozotocin-Induced Diabetic Rats. Hum. Gene Ther. 2009, 20, 1177–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornese, G.; Iafusco, D.; Monasta, L.; Agnoletto, C.; Tisato, V.; Ventura, A.; Zauli, G.; Secchiero, P. The Levels of Circulating TRAIL at the Onset of Type 1 Diabetes Are Markedly Decreased in Patients with Ketoacidosis and with the Highest Insulin Requirement. Acta Diabetol. 2014, 51, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Bisgin, A.; Yalcin, A.D.; Gorczynski, R.M. Circulating Soluble Tumor Necrosis Factor Related Apoptosis Inducing-Ligand (TRAIL) Is Decreased in Type-2 Newly Diagnosed, Non-Drug Using Diabetic Patients. Diabetes Res. Clin. Pract. 2012, 96, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Corallini, F.; Ceconi, C.; Parrinello, G.; Volpato, S.; Ferrari, R.; Zauli, G. Potential Prognostic Significance of Decreased Serum Levels of TRAIL after Acute Myocardial Infarction. PLoS ONE 2009, 4, e4442. [Google Scholar] [CrossRef] [PubMed]
- Michowitz, Y.; Goldstein, E.; Roth, A.; Afek, A.; Abashidze, A.; ben Gal, Y.; Keren, G.; George, J. The Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) in Atherosclerosis. J. Am. Coll. Cardiol. 2005, 45, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Arik, H.O.; Yalcin, A.D.; Gumuslu, S.; Genç, G.E.; Turan, A.; Sanlioglu, A.D. Association of Circulating STRAIL and High-Sensitivity CRP with Type 2 Diabetic Nephropathy and Foot Ulcers. Med. Sci. Monit. 2013, 19, 712–715. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.W.; Liang, W.; Yao, X.M.; Zhang, L.; Zhu, L.J.; Yan, C.; Jin, Y.L.; Yao, Y.S. Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand Expression in Patients with Diabetic Nephropathy. J. Renin-Angiotensin-Aldosterone Syst. 2018, 19, 1470320318785744. [Google Scholar] [CrossRef] [Green Version]
- Abu El-Asrar, A.M.; Ahmad, A.; Alam, K.; Bittoun, E.; Siddiquei, M.M.; Mohammad, G.; Mousa, A.; de Hertogh, G.; Opdenakker, G. Unbalanced Vitreous Levels of Osteoprotegerin, RANKL, RANK, and TRAIL in Proliferative Diabetic Retinopathy. Ocul. Immunol. Inflamm. 2018, 26, 1248–1260. [Google Scholar] [CrossRef]
- Xiang, G.; Zhang, J.; Ling, Y.; Zhao, L. Circulating Level of TRAIL Concentration Is Positively Associated with Endothelial Function and Increased by Diabetic Therapy in the Newly Diagnosed Type 2 Diabetic Patients. Clin. Endocrinol. 2014, 80, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Mi, Q.-S.; Ly, D.; Lamhamedi-Cherradi, S.-E.; Salojin, K.V.; Zhou, L.; Grattan, M.; Meagher, C.; Zucker, P.; Chen, Y.H.; Nagle, J.; et al. Blockade of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Exacerbates Type 1 Diabetes in NOD Mice. Diabetes 2003, 52, 1967–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahraman, S.; Yilmaz, O.; Altunbas, H.A.; Dirice, E.; Sanlioglu, A.D. TRAIL Induces Proliferation in Rodent Pancreatic Beta Cells via AKT Activation. J. Mol. Endocrinol. 2021, 66, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, B.; Tonon, F.; Tisato, V.; Zauli, G.; Secchiero, P.; Fabris, B.; Bernardi, S. TRAIL/DR5 Pathway Promotes AKT Phosphorylation, Skeletal Muscle Differentiation, and Glucose Uptake. Cell Death Dis. 2021, 12, 1089. [Google Scholar] [CrossRef]
- Toffoli, B.; Tonon, F.; Tisato, V.; Michelli, A.; Zauli, G.; Secchiero, P.; Fabris, B.; Bernardi, S. TRAIL Treatment Prevents Renal Morphological Changes and TGF-β-Induced Mesenchymal Transition Associated with Diabetic Nephropathy. Clin. Sci. 2020, 134, 2337–2352. [Google Scholar] [CrossRef]
- Bernardi, S.; Bossi, F.; Toffoli, B.; Fabris, B. Roles and Clinical Applications of OPG and TRAIL as Biomarkers in Cardiovascular Disease. BioMed Res. Int. 2016, 2016, 1752854. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-Induced Apoptosis by a Family of Signaling and Decoy Receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.I.; Gentz, R.; Dixit, V.M. An Antagonist Decoy Receptor and a Death Domain-Containing Receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.S.; Burns, T.F.; McDonald, E.R.; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 Is a DNA Damage-Inducible P53-Regulated Death Receptor Gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and Characterization of TRAIL-R3, a Novel Member of the Emerging TRAIL Receptor Family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Zauli, G.; Melloni, E.; Capitani, S.; Secchiero, P. Role of Full-Length Osteoprotegerin in Tumor Cell Biology. Cell. Mol. Life Sci. 2009, 66, 841–851. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The Novel Receptor TRAIL-R4 Induces NF-ΚB and Protects against TRAIL-Mediated Apoptosis, yet Retains an Incomplete Death Domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular Cloning and Functional Analysis of the Mouse Homologue of the KILLER/DR5 Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Death Receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar] [PubMed]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human Dendritic Cells Mediate Cellular Apoptosis via Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Yamaguchi, N.; Yagita, H.; Okumura, K. Involvement of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in NK Cell-Mediated and IFN-Gamma-Dependent Suppression of Subcutaneous Tumor Growth. Cell. Immunol. 2001, 214, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.M.; Krammer, P.H. Surface Expression of TRAIL/Apo-2 Ligand in Activated Mouse T and B Cells. Eur. J. Immunol. 1998, 28, 1492–1498. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Hiroshi, E.; Okumura, K.; Yagita, H. Type I Interferons (IFNs) Regulate Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Expression on Human T Cells: A Novel Mechanism for the Antitumor Effects of Type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Voltan, R.; Secchiero, P.; Casciano, F.; Milani, D.; Zauli, G.; Tisato, V. Redox Signaling and Oxidative Stress: Cross Talk with TNF-Related Apoptosis Inducing Ligand Activity. Int. J. Biochem. Cell Biol. 2016, 81, 364–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; le Moigne-Muller, G.; van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL Induces Necroptosis Involving RIPK1/RIPK3-Dependent PARP-1 Activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A.E. Non-Canonical Kinase Signaling by the Death Ligand TRAIL in Cancer Cells: Discord in the Death Receptor Family. Cell Death Differ. 2013, 20, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Kavurma, M.M.; Bennett, M.R. Expression, Regulation and Function of Trail in Atherosclerosis. Biochem. Pharmacol. 2008, 75, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Corallini, F.; Ceconi, C.; Ferrari, R.; Zauli, G. Metalloproteinase 2 Cleaves in Vitro Recombinant TRAIL: Potential Implications for the Decreased Serum Levels of TRAIL after Acute Myocardial Infarction. Atherosclerosis 2010, 211, 333–336. [Google Scholar] [CrossRef]
- Tornese, G.; Tisato, V.; Monasta, L.; Vecchi Brumatti, L.; Zauli, G.; Secchiero, P. Serum TRAIL Levels Increase Shortly after Insulin Therapy and Metabolic Stabilization in Children with Type 1 Diabetes Mellitus. Acta Diabetol. 2015, 52, 1003–1006. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Oschilewski, M.; Oschilewski, U.; Schwab, E.; Moumé, C.M.; Greulich, B.; Burkart, V.; Zielasek, J.; Kiesel, U. Analysis of 22 Immunomodulatory Substances for Efficacy in Low-Dose Streptozotocin-Induced Diabetes. Diabetes Res. 1987, 6, 21–27. [Google Scholar]
- Cheung, S.S.C.; Metzger, D.L.; Wang, X.; Huang, J.; Tai, J.; Tingle, A.J.; Ou, D. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand and CD56 Expression in Patients with Type 1 Diabetes Mellitus. Pancreas 2005, 30, 105–114. [Google Scholar] [CrossRef]
- Choi, J.W.; Fujii, T.; Fujii, N. Fas and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Are Closely Linked to the Levels of Glycated and Fetal Hemoglobin in Patients with Diabetes Mellitus. Clin. Lab. 2018, 64, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Keuper, M.; Wernstedt Asterholm, I.; Scherer, P.E.; Westhoff, M.A.; Möller, P.; Debatin, K.M.; Strauss, G.; Wabitsch, M.; Fischer-Posovszky, P. TRAIL (TNF-Related Apoptosis-Inducing Ligand) Regulates Adipocyte Metabolism by Caspase-Mediated Cleavage of PPARgamma. Cell Death Dis. 2013, 4, 474. [Google Scholar] [CrossRef]
- Zoller, V.; Funcke, J.B.; Roos, J.; Dahlhaus, M.; Abd El Hay, M.; Holzmann, K.; Marienfeld, R.; Kietzmann, T.; Debatin, K.M.; Wabitsch, M.; et al. Trail (TNF-Related Apoptosis-Inducing Ligand) Induces an Inflammatory Response in Human Adipocytes. Sci. Rep. 2017, 7, 5691. [Google Scholar] [CrossRef] [Green Version]
- Maixner, N.; Pecht, T.; Haim, Y.; Chalifa-Caspi, V.; Goldstein, N.; Tarnovscki, T.; Liberty, I.F.; Kirshtein, B.; Golan, R.; Berner, O.; et al. A Trail-Tl1a Paracrine Network Involving Adipocytes, Macrophages, and Lymphocytes Induces Adipose Tissue Dysfunction Downstream of E2f1 in Human Obesity. Diabetes 2020, 69, 2310–2323. [Google Scholar] [CrossRef] [PubMed]
- Zoller, V.; Funcke, J.B.; Keuper, M.; Abd El Hay, M.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P. TRAIL (TNF-Related Apoptosis-Inducing Ligand) Inhibits Human Adipocyte Differentiation via Caspase-Mediated Downregulation of Adipogenic Transcription Factors. Cell Death Dis. 2016, 7, 2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idrissova, L.; Malhi, H.; Werneburg, N.W.; Lebrasseur, N.K.; Bronk, S.F.; Fingas, C.; Tchkonia, T.; Pirtskhalava, T.; White, T.A.; Stout, M.B.; et al. TRAIL Receptor Deletion in Mice Suppresses the Inflammation of Nutrient Excess. J. Hepatol. 2015, 62, 1156–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funcke, J.B.; Zoller, V.; Abd El Hay, M.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P. TNF-Related Apoptosis-Inducing Ligand Promotes Human Preadipocyte Proliferation via ERK1/2 Activation. FASEB J. 2015, 29, 3065–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose Tissue Remodeling and Obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Ashley, D.T.; O’Sullivan, E.P.; Davenport, C.; Devlin, N.; Crowley, R.K.; McCaffrey, N.; Moyna, N.M.; Smith, D.; O’Gorman, D.J. Similar to Adiponectin, Serum Levels of Osteoprotegerin Are Associated with Obesity in Healthy Subjects. Metab. Clin. Exp. 2011, 60, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Brombo, G.; Volpato, S.; Secchiero, P.; Passaro, A.; Bosi, C.; Zuliani, G.; Zauli, G. Association of Soluble Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) with Central Adiposity and Low-Density Lipoprotein Cholesterol. PLoS ONE 2013, 8, 58225. [Google Scholar] [CrossRef]
- Choi, J.W.; Song, J.S.; Pai, S.H. Associations of Serum TRAIL Concentrations, Anthropometric Variables, and Serum Lipid Parameters in Healthy Adults. Ann. Clin. Lab. Sci. 2004, 34, 400–404. [Google Scholar]
- Kawano, N.; Mori, K.; Emoto, M.; Lee, E.; Kobayashi, I.; Yamazaki, Y.; Urata, H.; Morioka, T.; Koyama, H.; Shoji, T.; et al. Association of Serum TRAIL Levels with Atherosclerosis in Patients with Type 2 Diabetes Mellitus. Diabetes Res. Clin. Pract. 2011, 91, 316–320. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Waiczies, S.; Ehrlich, S.; Wendling, U.; Seeger, B.; Kamradt, T.; Zipp, F. Death Ligand TRAIL Induces No Apoptosis but Inhibits Activation of Human (Auto)Antigen-Specific T Cells. J. Immunol. 2002, 168, 4881–4888. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Chen, Y.; Göke, R.; Wilmen, A.; Seidel, C.; Göke, A.; Hilliard, B.; Chen, Y. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Is an Inhibitor of Autoimmune Inflammation and Cell Cycle Progression. J. Exp. Med. 2000, 191, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.K.; Williams, O.; Mongkolsapaya, J.; Jin, B.; Xu, X.N.; Walczak, H.; Screaton, G.R. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand in T Cell Development: Sensitivity of Human Thymocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 5158–5163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamhamedi-Cherradi, S.E.; Zheng, S.J.; Maguschak, K.A.; Peschon, J.; Chen, Y.H. Defective Thymocyte Apoptosis and Accelerated Autoimmune Diseases in TRAIL/Mice. Nat. Immunol. 2003, 4, 255–260. [Google Scholar] [CrossRef]
- Dadey, R.E.; Grebinoski, S.; Zhang, Q.; Brunazzi, E.A.; Burton, A.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cell–Derived TRAIL Is Not Required for Peripheral Tolerance. ImmunoHorizons 2021, 5, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Hirata, S.; Fukushima, S.; Matsunaga, Y.; Ito, T.; Uchino, M.; Nishimura, Y.; Senju, S. Dual Effects of TRAIL in Suppression of Autoimmunity: The Inhibition of Th1 Cells and the Promotion of Regulatory T Cells. J. Immunol. 2010, 185, 5259–5267. [Google Scholar] [CrossRef] [PubMed]
- Sanlioglu, A.D.; Griffith, T.S.; Orner, A.; Dirice, E.; Sari, R.; Altunbas, H.A.; Balci, M.K.; Sanlioglu, S. Molecular Mechanisms of Death Ligand-Mediated Immune Modulation: A Gene Therapy Model to Prolong Islet Survival in Type 1 Diabetes. J. Cell. Biochem. 2008, 104, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Falschlehner, C.; Schaefer, U.; Walczak, H. Following TRAIL’s Path in the Immune System. Immunology 2009, 127, 145–154. [Google Scholar] [CrossRef]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin Resistance and Sarcopenia: Mechanistic Links between Common Co-Morbidities. J. Endocrinol. 2016, 229, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, B.; Fabris, B.; Bartelloni, G.; Bossi, F.; Bernardi, S. Dyslipidemia and Diabetes Increase the OPG/TRAIL Ratio in the Cardiovascular System. Mediat. Inflamm. 2016, 2016, 6529728. [Google Scholar] [CrossRef] [Green Version]
- Zauli, G.; Pandolfi, A.; Gonelli, A.; di Pietro, R.; Guarnieri, S.; Ciabattoni, G.; Rana, R.; Vitale, M.; Secchiero, P. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Sequentially Upregulates Nitric Oxide and Prostanoid Production in Primary Human Endothelial Cells. Circ. Res. 2003, 92, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartland, S.P.; Genner, S.W.; Martínez, G.J.; Robertson, S.; Kockx, M.; Lin, R.C.; O’Sullivan, J.F.; Koay, Y.C.; Manuneedhi Cholan, P.; Kebede, M.A.; et al. TRAIL-Expressing Monocyte/Macrophages Are Critical for Reducing Inflammation and Atherosclerosis. iScience 2019, 12, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchiero, P.; Candido, R.; Corallini, F.; Zacchigna, S.; Toffoli, B.; Rimondi, E.; Fabris, B.; Giacca, M.; Zauli, G. Systemic Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Delivery Shows Antiatherosclerotic Activity in Apolipoprotein E-Null Diabetic Mice. Circulation 2006, 114, 1522–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bartolo, B.A.; Cartland, S.P.; Harith, H.H.; Bobryshev, Y.V.; Schoppet, M.; Kavurma, M.M. TRAIL-Deficiency Accelerates Vascular Calcification in Atherosclerosis via Modulation of RANKL. PLoS ONE 2013, 8, 74211. [Google Scholar] [CrossRef] [Green Version]
- Toffoli, B.; Bernardi, S.; Candido, R.; Zacchigna, S.; Fabris, B.; Secchiero, P. TRAIL Shows Potential Cardioprotective Activity. Investig. New Drugs 2012, 30, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.; Giannopoulos, G.; Kossyvakis, C.; Kaoukis, A.; Raisakis, K.; Panagopoulou, V.; Miliou, A.; Theodorakis, A.; Driva, M.; Pyrgakis, V.; et al. Association of Soluble Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand Levels with Coronary Plaque Burden and Composition. Heart 2012, 98, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Volpato, S.; Ferrucci, L.; Secchiero, P.; Corallini, F.; Zuliani, G.; Fellin, R.; Guralnik, J.M.; Bandinelli, S.; Zauli, G. Association of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand with Total and Cardiovascular Mortality in Older Adults. Atherosclerosis 2011, 215, 452–458. [Google Scholar] [CrossRef] [Green Version]
- Mattisson, I.Y.; Björkbacka, H.; Wigren, M.; Edsfeldt, A.; Melander, O.; Fredrikson, G.N.; Bengtsson, E.; Gonçalves, I.; Orho-Melander, M.; Engström, G.; et al. Elevated Markers of Death Receptor-Activated Apoptosis Are Associated with Increased Risk for Development of Diabetes and Cardiovascular Disease. EBioMedicine 2017, 26, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.H.; Park, M.G.; Noh, K.H.; Park, H.R.; Lee, H.W.; Son, S.M.; Park, K.P. Low Serum TNF-Related Apoptosis-Inducing Ligand (TRAIL) Levels Are Associated with Acute Ischemic Stroke Severity. Atherosclerosis 2015, 240, 228–233. [Google Scholar] [CrossRef]
- Moon, A.R.; Park, Y.; Chang, J.H.; Lee, S.S. Inverse Regulation of Serum Osteoprotegerin and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Levels in Patients with Leg Lesional Vascular Calcification: An Observational Study. Medicine 2019, 98, 14489. [Google Scholar] [CrossRef]
- Di Bartolo, B.A.; Cartland, S.P.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.M.; Thai, T.; Yeung, A.W.S.; Thomas, S.R.; et al. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Promotes Angiogenesis and Ischemia-Induced Neovascularization via NADPH Oxidase 4 (NOX4) and Nitric Oxide-Dependent Mechanisms. J. Am. Heart Assoc. 2015, 4, e002527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, S.; Voltan, R.; Rimondi, E.; Melloni, E.; Milani, D.; Cervellati, C.; Gemmati, D.; Celeghini, C.; Secchiero, P.; Zauli, G.; et al. TRAIL, OPG, and TWEAK in Kidney Disease: Biomarkers or Therapeutic Targets? Clin. Sci. 2019, 113, 1145–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorz, C.; Benito-Martin, A.; Boucherot, A.; Ucero, A.C.; Rastaldi, M.P.; Henger, A.; Armelloni, S.; Santamaría, B.; Berthier, C.C.; Kretzler, M.; et al. The Death Ligand TRAIL in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartland, S.P.; Erlich, J.H.; Kavurma, M.M. TRAIL Deficiency Contributes to Diabetic Nephropathy in Fat-Fed ApoE/Mice. PLoS ONE 2014, 9, e92952. [Google Scholar] [CrossRef] [Green Version]
- Threatt, J.; Williamson, J.F.; Huynh, K.; Davis, R.M. Ocular Disease, Knowledge and Technology Applications in Patients with Diabetes. Am. J. Med. Sci. 2013, 345, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Hubert, K.E.; Davies, M.H.; Stempel, A.J.; Griffith, T.S.; Powers, M.R. TRAIL-Deficient Mice Exhibit Delayed Regression of Retinal Neovascularization. Am. J. Pathol. 2009, 175, 2697–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secchiero, P.; Perri, P.; Melloni, E.; Martini, A.; Lamberti, G.; Sebastiani, A.; Zauli, G. Decreased Levels of Soluble TNF-Related Apoptosis-Inducing Ligand (TRAIL) in the Conjunctival Sac Fluid of Patients with Diabetes Affected by Proliferative Retinopathy. Diabet. Med. J. Br. Diabet. Assoc. 2011, 28, 1277–1278. [Google Scholar] [CrossRef]
- Anand, A.; Sharma, N.K.; Singh, R.; Gupta, A.; Prabhakar, S.; Jindal, N.; Bhatt, A.K.; Sharma, S.K.; Gupta, P.K. Does DcR1 (TNF-Related Apoptosis-Inducing-Ligand Receptor 3) Have Any Role in Human AMD Pathogenesis? Sci. Rep. 2014, 4, 4114. [Google Scholar] [CrossRef] [PubMed]
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The Role of Apoptosis in Age-Related Macular Degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| First Author (Year of Publication) | Experimental Model or Study Population | TRAIL-Related Intervention (If Applicable) | Study Methods | Key Findings |
|---|---|---|---|---|
| Animal data | ||||
| Lamhamedi-Cherradi (2003) | NOD mice challenged with cyclophosphamide Normal and TRAIL-deficient C57BL/6 mice treated with multiple low doses of streptozotocin | Soluble TRAIL receptor (sDR5) to block TRAIL function TRAIL gene knockout | Induction of diabetes, production of recombinant human sDR5, ELISA, histochemistry, quantification of islet inflammatory lesions, cell cultures, analyses of cell viability and apoptosis | Accelerated diabetes onset ↑ severity of autoimmune insulitis in pancreatic islets ↑ GAD65-specific immune responses ↑ incidence and extent of islet inflammation in TRAIL-deficient mice |
| Mi (2003) | NOD mice challenged with cyclophosphamide NOD mice receiving diabetogenic spleen T-cells from newly-diagnosed diabetic NOD mice | Soluble TRAIL receptor (sDR5) to block TRAIL function | Induction of diabetes, production of recombinant human sDR5, splenic T-cell isolation and proliferation assays, T-cell adoptive transfer, cell cultures, gene expression profiling of pancreatic islets, analyses of cell viability and apoptosis, ELISA, immunoblotting | ↑ incidence of cyclophosphamide-induced T1DM ↑ incidence and earlier onset of T1DM post-transfer of diabetogenic T-cells |
| Dirice (2009) | Rats treated with multiple low doses of streptozotocin | Adenovirus-mediated TRAIL gene delivery into pancreatic islets (Ad5hTRAIL) | Ex vivo genetic engineering of pancreatic β-cells, transplantation of genetically modified pancreatic islets in streptozotocin-induced diabetic rats, metabolic assays, ELISA, pancreas histology | Prolonged normoglycemia ↓ severity of insulitis Extended islet graft survival and function |
| Zauli (2010) | C57BL/6 mice treated with multiple low doses of streptozotocin | Recombinant TRAIL treatment (intraperitoneal injections) for 5 days In vitro exposure of human/mouse PBMCs and isolated human islets to recombinant TRAIL | Islet isolation, cell cultures, RNA and protein analyses, metabolic assays, ELISA, pancreas histology | ↓ hyperglycemia ↑ body weight ↑ insulin secretion Partially preserved islet morphology and function ↓ TNF-α, ↓ OPG, ↓ VCAM-1 expression in TRAIL-treated mice ↑ SOCS1 expression in PBMCs and human islets exposed in vitro to TRAIL |
| Kang (2010) | NOD mice | Adenovirus-mediated systemic human TRAIL gene delivery (iv injection) | Metabolic assays, cell cultures, RNA extraction and RT-PCR in pancreas and liver, pancreatic islet isolation and histopathological analysis, cell viability and flow cytometry apoptosis assays, Western blot analysis, ELISA for plasma cytokine and TIMP-1 measurements, gelatin zymography for the inhibition of MMPs | ↓ hyperglycemia ↑ TIMP-1 expression ↓ pancreatic MMP activity ↓ cytokine-induced insulitis and apoptosis Prevention of T1DM development |
| Clinical data | ||||
| Tornese (2014) | 507 pediatric subjects n = 387 patients with T1DM n = 98 healthy controls n = 22 healthy AA-positive subjects | NA | Retrospective study ELISA for serum soluble TRAIL measurements | ↓ serum soluble TRAIL levels in T1DM vs. other groups ↓ serum soluble TRAIL levels in T1DM patients presenting with DKA at onset (vs. those without DKA) Inverse correlation between serum TRAIL levels at diagnosis and insulin requirements up to 2 years of follow-up |
| Tornese (2015) | n = 11 pediatric patients with newly diagnosed T1DM complicated by DKA and secondary DKA | NA | Pilot study ELISA for serum soluble TRAIL measurements at sequential time points after admission, blood gas analysis for metabolic status assessment | ↑ serum soluble TRAIL levels shortly after insulin administration and metabolic stabilization Inverse correlation between serum TRAIL levels and the degree of metabolic decompensation |
| First Author (Year of Publication) | Experimental Model or Study Population | TRAIL-Related Intervention (If Applicable) | Study Methods | Key Findings |
|---|---|---|---|---|
| Animal data | ||||
| Di Bartolo (2011) | ApoE (−/−) HFD-fed mice | TRAIL gene knockout | Metabolic assays, RNA extraction and gene expression analysis, pancreatic islet histology, immunohistochemistry, morphometric analysis of atherosclerotic plaques | ↑ body weight ↑ glycemia ↓ insulinemia ↓ islet insulin ↑ serum lipids ↑ pancreatic islet inflammation/apoptosis IGT Β-cell dysfunction Exacerbated atherosclerosis and plaque instability |
| Bernardi (2012) | HFD-fed C57BL/6 mice | Weekly intraperitoneal injections of recombinant human TRAIL for 12 weeks | Metabolic assays, gene expression analysis in adipose tissue, ELISA for cytokine measurements | ↓ weight gain ↓ hyperglycemia ↓ hyperinsulinemia ↑ peripheral insulin sensitivity ↑ SM FFA oxidation ↓ proinflammatory cytokines ↓ adipogenic gene expression |
| Cartland (2017) | HFD-fed mice n = 9 healthy humans n = 10 obese patients n = 10 patients with hepatic steatosis n = 10 patients with NASH | TRAIL gene knockout | Plasma biochemistry, glucose and insulin tolerance tests, ex vivo glucose uptake studies, liver histology, tissue cultures, RNA extraction and RT-PCR for gene expression analysis, protein extraction and Western blotting, ELISA for serum soluble TRAIL measurements | In TRAIL-deficient mice: ↑ plasma lipids ↑ plasma glucose and insulin levels ↑ systemic insulin resistance ↓ Akt phosphorylation, GLUT4 expression and glucose uptake in SM ↑ hepatic steatosis, inflammation and fibrosis ↑ hepatic gene expression related to lipogenesis and gluconeogenesis ↑ expression of proinflammatory cytokines In patients with NASH: ↓ serum soluble TRAIL levels (vs. steatosis and obese) |
| Bernardi (2018) | HFD-fed C57BL/6 mice | Weekly injections of recombinant human TRAIL for 8 weeks | Metabolic assays, tissue collection and histology, in vitro studies on HepG2 cells and mouse primary hepatocytes | ↓ body weight ↓ adipocyte hypertrophy ↓ FFAs ↓ inflammatory markers ↓ liver fat content ↑ hepatic PGC-1α expression Improved IGT Improved NAFLD |
| Toffoli (2021) | HFD-fed C57BL/6 and db/db mice | Intraperitoneal injections of recombinant human TRAIL for 8–12 weeks | Production of recombinant human TRAIL, SM extraction (quadriceps), glucose uptake studies, FFA oxidation experiments, gene expression quantification by RT-PCR, DR5 silencing, immunofluorescence, Western blot analysis, histology + in vitro studies on mouse C2C12 myoblasts | Effects on SM: ↑ Akt phosphorylation ↑ insulin-stimulated glucose uptake ↑ myofiber size ↑ myogenin and PGC-1α expression ↑ myogenesis (muscle differentiation) No effect on lipid accumulation in skeletal myotubes |
| Clinical data | ||||
| Bisgin (2012) | n = 22 newly diagnosed drug-naïve patients with T2DM | NA | ELISA for serum soluble TRAIL measurements | ↓ serum soluble TRAIL levels in T2DM patients (vs. controls) |
| Arik (2013) | n = 22 insulin-treated patients with T2DM, DN (macroalbuminuria) and foot ulcers | NA | ELISA for serum soluble TRAIL measurements | ↓ serum soluble TRAIL levels in patients with DN and foot ulcers (vs. non-diabetic controls) No correlation between serum TRAIL levels and HbA1c or fasting glucose levels |
| Xiang (2014) | n = 55 newly diagnosed patients with T2DM | NA | ELISA for serum soluble TRAIL measurements | ↓ serum soluble TRAIL levels in T2DM patients (vs. non-diabetic controls) ↑ serum soluble TRAIL levels after 6 months of antidiabetic treatment Absolute change in serum TRAIL levels ~ absolute change in HbA1c, fasting and postprandial glycemia before and after treatment |
| Chang (2018) | n = 42 patients with T2DM n = 42 patients with DN n = 42 healthy controls | NA | Real-time RT-PCR for TRAIL mRNA levels in PBMCs ELISA for serum cytokine and TRAIL measurements | ↓ TRAIL mRNA in PBMCs and ↓ serum soluble TRAIL levels in patients with T2DM and DN (vs. controls) ↑ proinflammatory cytokines in patients with DN (vs. controls) |
| Choi (2018) | n = 112 patients with T2DM | NA | ELISA for serum soluble TRAIL measurements | ↓ serum soluble TRAIL levels in T2DM patients with microalbuminuria (vs. controls) Inverse correlation between serum TRAIL levels and HbA1c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koliaki, C.; Katsilambros, N. Repositioning the Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) on the TRAIL to the Development of Diabetes Mellitus: An Update of Experimental and Clinical Evidence. Int. J. Mol. Sci. 2022, 23, 3225. https://doi.org/10.3390/ijms23063225
Koliaki C, Katsilambros N. Repositioning the Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) on the TRAIL to the Development of Diabetes Mellitus: An Update of Experimental and Clinical Evidence. International Journal of Molecular Sciences. 2022; 23(6):3225. https://doi.org/10.3390/ijms23063225
Chicago/Turabian StyleKoliaki, Chrysi, and Nicholas Katsilambros. 2022. "Repositioning the Role of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) on the TRAIL to the Development of Diabetes Mellitus: An Update of Experimental and Clinical Evidence" International Journal of Molecular Sciences 23, no. 6: 3225. https://doi.org/10.3390/ijms23063225