
The Role of MicroRNAs in Proteostasis Decline and Protein Aggregation during Brain and Skeletal Muscle Aging

 , , and
, , and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Biogenesis and Target Binding of miRNAs
3. Target-Directed miRNA Degradation and Its Role in Fundamental Cellular Processes
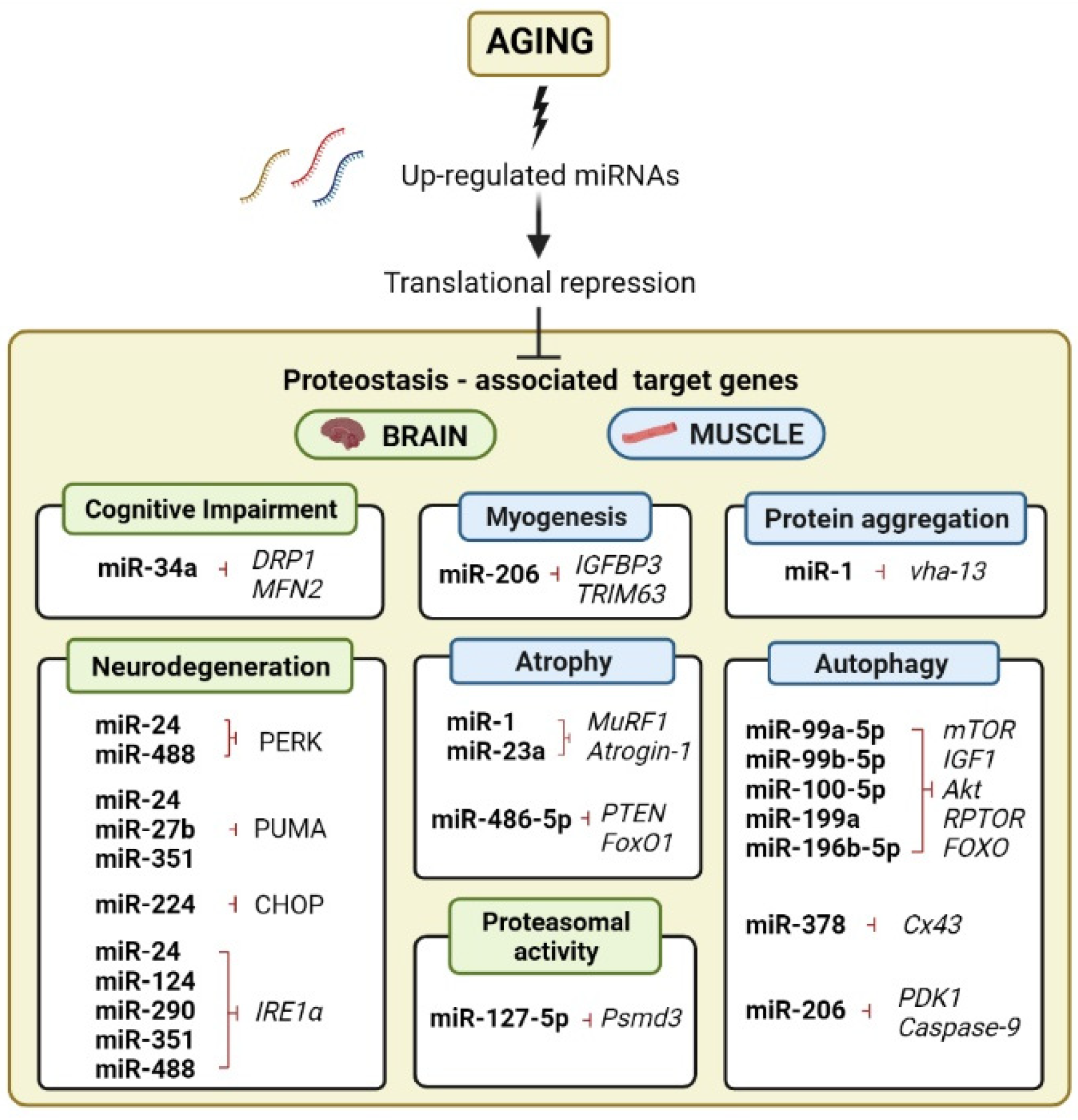
4. Expression Profiles of miRNA Are Dynamic and Are Tissue-Specific throughout Aging
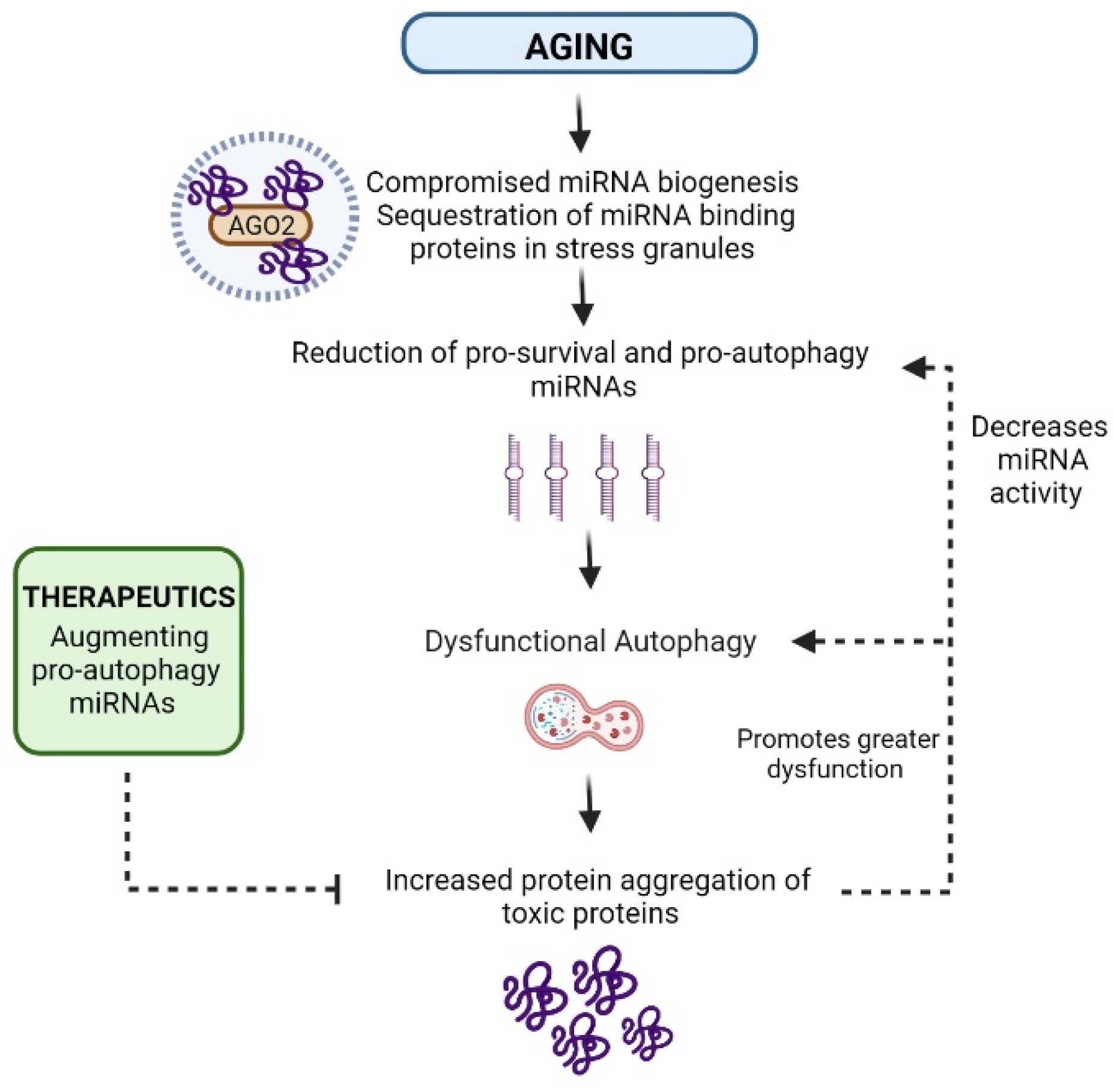
5. The Role of miRNA Regulation in Age-Related Autophagic Decline in Brain and Skeletal Muscle
6. miRNAs Mediate the Ubiquitin-Proteosome System in Brain Aging and Muscle Atrophy
7. miRNAs as UPR and Protein-Aggregation Regulators
8. miRNA-Binding Proteins also Regulate Lifespan through Proteostasis Network Components
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| AGE-BSA | Advanced glycation end products of bovine serum albumin |
| AGEs | Advanced glycation of end products |
| AGO-2 | Argonaute-2 |
| Akt | Protein kinase b |
| alg1 | Argonaute-1 |
| Alg2 | Argonaute-2 |
| ALP | Autophagy lysosomal pathway |
| ApoE | Apolipoprotein E |
| ATF6 | Activating transcription factor 6 |
| AWC | Amphid wing “C” |
| Aβ | Amyloid-β |
| BCl-2 | B-cell lymphoma 2 |
| c-miRNA | Circulating miRNA |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum-associated degradation |
| FOXO | Forkhead box class O |
| HSR | Heat shock response |
| IGF1 | Insulin-like growth factor 1 |
| Ire1α | Inositol-requiring enzyme-1 alpha |
| Keap1 | Kelch-like ECH-associated protein 1 |
| MAFbx | Muscle atrophy F-box |
| mHTT | Mutant huntingtin |
| miRISC | miRNA-induced silencing complex |
| miRNA | microRNA |
| mRNA | Messenger RNA |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MuRF1 | Muscle ring finger 1 |
| nt | nucleotide |
| PDK1 | Phosphoinositide-dependent protein kinase 1 |
| PERK | Protein kinase RNA-like ER kinase |
| PN | Proteostasis network |
| PolyQ35 | Polyglutamine 35 |
| Pre-miRNA | Precursor microRNAs |
| Pri-miRNA | Primary microRNAs |
| Psmd3 | 26S proteasome non-ATPase regulatory subunit 3 |
| PTEN | phosphatase and tensin homolog |
| RAGE | Receptor of AGEs |
| RISC | RNA-induced silencing complex |
| RPTOR | Regulatory-associated protein of MTOR complex 1 |
| SAMP8 | Senescence-accelerated mice prone 8 |
| SAMR1 | Senescence-accelerated mouse resistant |
| TBC1D15 | Tre-2/Bub2/CDC16(TBC) Rab GTPase-activating protein |
| TIR-1 | Toll-like receptor 1 |
| ULK-1 | unc-51-like autophagy activating kinase 1 |
| UPRER | Endoplasmic reticulum unfolded protein response |
| UPRmt | Mitochondrial unfolded protein response |
| UPS | Ubiquitin–proteasome system |
| UTR | Untranslated region |
| XBP1 | X-box binding protein 1 |
References
- Beard, J.R.; Officer, A.; De Carvalho, I.A.; Sadana, R.; Pot, A.M.; Michel, J.P.; Lloyd-Sherlock, P.; Epping-Jordan, J.E.; Peeters, G.M.E.E.; Mahanani, W.R.; et al. The World Report on Ageing and Health: A Policy Framework for Healthy Ageing. Lancet 2016, 387, 2145–2154. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Luna, E.; Decker, S.C.; Riddle, D.M.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.X.; Lee, V.M.Y.; Luk, K.C. Differential α-Synuclein Expression Contributes to Selective Vulnerability of Hippocampal Neuron Subpopulations to Fibril-Induced Toxicity. Acta Neuropathol. 2018, 135, 855–875. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A Common Mechanism of Proteasome Impairment by Neurodegenerative Disease-Associated Oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.G.; Penney, J.; Tsai, L.-H. The Road to Restoring Neural Circuits for the Treatment of Alzheimer’s Disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Mealer, R.G.; Murray, A.J.; Shahani, N.; Subramaniam, S.; Snyder, S.H. Rhes, a Striatal-Selective Protein Implicated in Huntington Disease, Binds Beclin-1 and Activates Autophagy. J. Biol. Chem. 2014, 289, 3547–3554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease and Related Neurodegenerative Disorders. Mol. Neurobiol. 2012, 47, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Dillin, A. Aging as an Event of Proteostasis Collapse. Cold Spring Harb. Perspect. Biol. 2011, 3, a004440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbadia, J.; Brielmann, R.M.; Neto, M.F.; Lin, Y.-F.; Haynes, C.M.; Morimoto, R.I. Mitochondrial Stress Restores the Heat Shock Response and Prevents Proteostasis Collapse during Aging. Cell Rep. 2017, 21, 1481–1494. [Google Scholar] [CrossRef] [Green Version]
- Labbadia, J.; Morimoto, R.I. Repression of the Heat Shock Response Is a Programmed Event at the Onset of Reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Cui, J.; Bai, X.Y.; Shi, S.; Cui, S.; Hong, Q.; Cai, G.; Chen, X. Age-Related Changes in the Function of Autophagy in Rat Kidneys. Age 2012, 34, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Lu, H.; Li, D.; Xiong, X.; Gao, L.; Wu, Z.; Lu, Y. Aberrant Expression of FBXO2 Disrupts Glucose Homeostasis Through Ubiquitin-Mediated Degradation of Insulin Receptor in Obese Mice. Diabetes 2017, 66, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.S.; Romeo, F.; Reis, R.J.S.; Mehta, J.L. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanase, M.; Urbanska, A.M.; Zolla, V.; Clement, C.C.; Huang, L.; Morozova, K.; Follo, C.; Goldberg, M.; Roda, B.; Reschiglian, P.; et al. Role of Carbonyl Modifications on Aging-Associated Protein Aggregation. Sci. Rep. 2016, 6, 19311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Lee, H.; Dominguez, D.; Heiman, M.; Burge, C.B. Widespread Accumulation of Ribosome-Associated Isolated 3′ UTRs in Neuronal Cell Populations of the Aging Brain. Cell Rep. 2018, 25, 2447–2456.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered Proteins in the Aging Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, S.; Pincus, Z.; De Lencastre, A.; Slack, F.J. A MicroRNA Feedback Loop Regulates Global MicroRNA Abundance during Aging. RNA 2018, 24, 159–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.H.; Chiou, G.Y.; Chen, Y.W.; Li, H.Y.; Chiou, S.H. MicroRNA and Aging: A Novel Modulator in Regulating the Aging Network. Ageing Res. Rev. 2010, 9, S59–S66. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs Both Promote and Antagonize Longevity in C. elegans. Curr. Biol. 2010, 20, 2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, S.; de Lencastre, A.; Turner, M.; Slack, F. Novel MicroRNAs Differentially Expressed during Aging in the Mouse Brain. PLoS ONE 2012, 7, e40028. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Slack, F.J. Ageing and the Small, Non-Coding RNA World. Ageing Res. Rev. 2013, 12, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Kinser, H.E.; Pincus, Z. MicroRNAs as Modulators of Longevity and the Aging Process. Hum. Genet. 2020, 139, 291–308. [Google Scholar] [CrossRef]
- Olivieri, F.; Capri, M.; Bonafè, M.; Morsiani, C.; Jung, H.J.; Spazzafumo, L.; Viña, J.; Suh, Y. Circulating MiRNAs and MiRNA Shuttles as Biomarkers: Perspective Trajectories of Healthy and Unhealthy Aging. Mech. Ageing Dev. 2017, 165, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Morsiani, C.; Terlecki-Zaniewicz, L.; Skalicky, S.; Giulia Bacalini, M.; Collura, S.; Conte, M.; Sevini, F.; Garagnani, P.; Salvioli, S.; Hackl, M.; et al. Circulating MiR-19a-3p and MiR-19b-3p Characterize the Human Aging Process and Their IsomiRs Associate with Healthy Status at Extreme Ages. Aging Cell 2021, 20, e13409. [Google Scholar] [CrossRef] [PubMed]
- Borrás, C.; Serna, E.; Gambini, J.; Inglés, M.; Vina, J. Centenarians Maintain MiRNA Biogenesis Pathway While It Is Impaired in Octogenarians. Mech. Ageing Dev. 2017, 168, 54–57. [Google Scholar] [CrossRef]
- Schiffer, I.; Gerisch, B.; Kawamura, K.; Laboy, R.; Hewitt, J.; Denzel, M.S.; Mori, M.A.; Vanapalli, S.; Shen, Y.; Symmons, O.; et al. Mir-1 Coordinately Regulates Lysosomal v-Atpase and Biogenesis to Impact Proteotoxicity and Muscle Function during Aging. Elife 2021, 10, e66768. [Google Scholar] [CrossRef]
- Pircs, K.; Petri, R.; Madsen, S.; Brattås, P.L.; Vuono, R.; Ottosson, D.R.; St-Amour, I.; Hersbach, B.A.; Matusiak-Brückner, M.; Lundh, S.H.; et al. Huntingtin Aggregation Impairs Autophagy, Leading to Argonaute-2 Accumulation and Global MicroRNA Dysregulation. Cell Rep. 2018, 24, 1397–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntzinger, E.; Izaurralde, E. Gene Silencing by MicroRNAs: Contributions of Translational Repression and MRNA Decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Victoria, B.; Nunez Lopez, Y.O.; Masternak, M.M. MicroRNAs and the Metabolic Hallmarks of Aging. Mol. Cell. Endocrinol. 2017, 455, 131. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNA Target Recognition and Regulatory Functions. Cell 2009, 136, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, R.; Doehle, B.P.; Qin, Y.I.; Macara, I.G.; Cullen, B.R. Overexpression of Exportin 5 Enhances RNA Interference Mediated by Short Hairpin RNAs and MicroRNAs. RNA 2005, 11, 220–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medley, J.C.; Panzade, G.; Zinovyeva, A.Y. MicroRNA Strand Selection: Unwinding the Rules. Wiley Interdiscip. Rev. RNA 2021, 12, e1627. [Google Scholar] [CrossRef] [PubMed]
- Córdova-Rivas, S.; Fraire-Soto, I.; Torres, A.M.C.; Servín-González, L.S.; Granados-López, A.J.; López-Hernández, Y.; Reyes-Estrada, C.A.; Gutiérrez-Hernández, R.; Castañeda-Delgado, J.E.; Ramírez-Hernández, L.; et al. 5p and 3p Strands of MiR-34 Family Members Have Differential Effects in Cell Proliferation, Migration, and Invasion in Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 545. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Ge, F.; Du, L.; Zhang, Z.; Liu, D. MiR-34b-3p Represses Cell Proliferation, Cell Cycle Progression and Cell Apoptosis in Non-small-cell Lung Cancer (NSCLC) by Targeting CDK4. J. Cell. Mol. Med. 2019, 23, 5282. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Ajay, S.S.; Yook, J.I.; Kim, H.S.; Hong, S.H.; Kim, N.H.; Dhanasekaran, S.M.; Chinnaiyan, A.M.; Athey, B.D. New Class of MicroRNA Targets Containing Simultaneous 5′-UTR and 3′-UTR Interaction Sites. Genome Res. 2009, 19, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, G.A.; Fabbri, M.; Eiring, A.M. A Non-Canonical Landscape of the MicroRNA System. Front. Genet. 2014, 5, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehlmann, T.; Lehallier, B.; Schaum, N.; Hahn, O.; Kahraman, M.; Li, Y.; Grammes, N.; Geffers, L.; Backes, C.; Balling, R.; et al. Common Diseases Alter the Physiological Age-Related Blood MicroRNA Profile. Nat. Commun. 2020, 11, 5958. [Google Scholar] [CrossRef] [PubMed]
- Bofill-De Ros, X.; Yang, A.; Gu, S. IsomiRs: Expanding the MiRNA Repression Toolbox Beyond the Seed. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194373. [Google Scholar] [CrossRef]
- Dhanoa, J.K.; Verma, R.; Sethi, R.S.; Arora, S.; Mukhopadhyay, C.S. Biogenesis and Biological Implications of IsomiRs in Mammals—A Review. ExRNA 2019, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Neilsen, C.T.; Goodall, G.J.; Bracken, C.P. IsomiRs—the Overlooked Repertoire in the Dynamic MicroRNAome. Trends Genet. 2012, 28, 544–549. [Google Scholar] [CrossRef]
- Wu, K.J. The Role of MiRNA Biogenesis and DDX17 in Tumorigenesis and Cancer Stemness. Biomed. J. 2020, 43, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 668648. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, Y.; Li, M.; Tu, J.; Lu, Z. Dysregulation of MiRNA Isoform Level at 5′ End in Alzheimer’s Disease. Gene 2016, 584, 167–172. [Google Scholar] [CrossRef]
- Telonis, A.G.; Magee, R.; Loher, P.; Chervoneva, I.; Londin, E.; Rigoutsos, I. Knowledge about the Presence or Absence of MiRNA Isoforms (IsomiRs) Can Successfully Discriminate amongst 32 TCGA Cancer Types. Nucleic Acids Res. 2017, 45, 2973–2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliani, A.; Londin, E.; Ferracin, M.; Menss, E.; Prattichizzo, F.; Ramini, D.; Marcheselli, F.; Recchioni, R.; Rippo, M.R.; Bonafè, M.; et al. Long-Term Exposure of Human Endothelial Cells to Metformin Modulates MiRNAs and IsomiRs. RNA 2020, 10, 21782. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of MiRNA Expression across Human Tissues. Nucleic Acids Res. 2016, 44, 3865. [Google Scholar] [CrossRef] [PubMed]
- Alberti, C.; Manzenreither, R.A.; Sowemimo, I.; Burkard, T.R.; Wang, J.; Mahofsky, K.; Ameres, S.L.; Cochella, L. Cell-Type Specific Sequencing of MicroRNAs from Complex Animal Tissues. Nat. Methods 2018, 15, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.K.; Pak, T.R. MiRNA Degradation in the Mammalian Brain. Am. J. Physiol. Cell Physiol. 2020, 319, C624–C629. [Google Scholar] [CrossRef] [PubMed]
- Pawlica, P.; Sheu-Gruttadauria, J.; Macrae, I.J.; Steitz, J.A. How Complementary Targets Expose the MicroRNA 3 0 End for Tailing and Trimming during Target-Directed MicroRNA Degradation. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Sheu-Gruttadauria, J.; Pawlica, P.; Klum, S.M.; Wang, S.; Yario, T.A.; Schirle Oakdale, N.T.; Steitz, J.A.; MacRae, I.J. Structural Basis for Target-Directed MicroRNA Degradation. Mol. Cell 2019, 75, 1243–1255.e7. [Google Scholar] [CrossRef] [PubMed]
- Ameres, S.L.; Horwich, M.D.; Hung, J.-H.; Xu, J.; Ghildiyal, M.; Weng, Z.; Zamore, P.D. Target RNA-Directed Trimming and Tailing of Small Silencing RNAs. Science 2010, 328, 1534–1539. [Google Scholar] [CrossRef] [Green Version]
- Marzi, M.J.; Ghini, F.; Cerruti, B.; De Pretis, S.; Bonetti, P.; Giacomelli, C.; Gorski, M.M.; Kress, T.; Pelizzola, M.; Muller, H.; et al. Degradation Dynamics of MicroRNAs Revealed by a Novel Pulse-Chase Approach. Genome Res. 2016, 26, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghini, F.; Rubolino, C.; Climent, M.; Simeone, I.; Marzi, M.J.; Nicassio, F. Endogenous Transcripts Control MiRNA Levels and Activity in Mammalian Cells by Target-Directed MiRNA Degradation. Nat. Commun. 2018, 9, 3119. [Google Scholar] [CrossRef] [PubMed]
- Bitetti, A.; Mallory, A.C.; Golini, E.; Carrieri, C.; Carreño Gutiérrez, H.; Perlas, E.; Pérez-Rico, Y.A.; Tocchini-Valentini, G.P.; Enright, A.J.; Norton, W.H.J.; et al. MicroRNA Degradation by a Conserved Target RNA Regulates Animal Behavior. Nat. Struct. Mol. Biol. 2018, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.A.; Raghavan, P.; Thomou, T.; Boucher, J.; Robida-Stubbs, S.; MacOtela, Y.; Russell, S.J.; Kirkland, J.L.; Blackwell, T.K.; Kahn, C.R. Role of MicroRNA Processing in Adipose Tissue in Stress Defense and Longevity. Cell Metab. 2012, 16, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Finger, F.; Ottens, F.; Springhorn, A.; Drexel, T.; Proksch, L.; Metz, S.; Cochella, L.; Hoppe, T. Olfaction Regulates Organismal Proteostasis and Longevity via MicroRNA-Dependent Signalling. Nat. Metab. 2019, 1, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy Impairment in Muscle Induces Neuromuscular Junction Degeneration and Precocious Aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, D.; He, Y.; Meléndez, A.; Feng, Z.; Hong, Q.; Bai, X.; Li, Q.; Cai, G.; Wang, J.; et al. MiR-34 Modulates Caenorhabditis Elegans Lifespan via Repressing the Autophagy Gene Atg9. Age 2013, 35, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Landreh, M.; Cao, K.; Abe, M.; Hendriks, G.-J.; Kennerdell, J.R.; Zhu, Y.; Wang, L.-S.; Bonini, N.M. The MicroRNA MiR-34 Modulates Ageing and Neurodegeneration in Drosophila. Nature 2012, 482, 519–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. MicroRNA-34c Is a Novel Target to Treat Dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef] [PubMed]
- Fenn, A.M.; Smith, K.M.; Lovett-Racke, A.E.; Guerau-de-Arellano, M.; Whitacre, C.C.; Godbout, J.P. Increased Micro-RNA 29b in the Aged Brain Correlates with the Reduction of Insulin-like Growth Factor-1 and Fractalkine Ligand. Neurobiol. Aging 2013, 34, 2748–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano-Tárraga, C.; Jiménez-Conde, J.; Roquer, J. Epigenetics and Aging. Handb. Nutr. Diet Epigenet. 2019, 2, 1413–1433. [Google Scholar] [CrossRef]
- Kirby, T.J.; Chaillou, T.; Mccarthy, J.J. The Role of MicroRNAs in Skeletal Muscle Health and Disease. Front. Biosci. 2015, 20, 37–77. [Google Scholar]
- Li, Y.; Jiang, J.; Liu, W.; Wang, H.; Zhao, L.; Liu, S.; Li, P.; Zhang, S.; Sun, C.; Wu, Y.; et al. MicroRNA-378 Promotes Autophagy and Inhibits Apoptosis in Skeletal Muscle. Proc. Natl. Acad. Sci. USA 2018, 115, E10849–E10858. [Google Scholar] [CrossRef] [Green Version]
- Rabajdova, M.; Spakova, I.; Zelko, A.; Rosenberger, J.; Kolarcik, P.; Sobolova, V.; Pella, D.; Marekova, M.; Madarasova Geckova, A. The Role of Physical Activity and MiRNAs in the Vascular Aging and Cardiac Health of Dialysis Patients. Physiol. Rep. 2021, 9, e14879. [Google Scholar] [CrossRef] [PubMed]
- Zacharewicz, E.; Della Gatta, P.; Reynolds, J.; Garnham, A.; Crowley, T.; Russell, A.P.; Lamon, S. Identification of MicroRNAs Linked to Regulators of Muscle Protein Synthesis and Regeneration in Young and Old Skeletal Muscle. PLoS ONE 2014, 9, e114009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Ma, R.; Fu, W.; Zhang, C.; Du, X. LncRNA UCA1 Sponges MiR-206 to Exacerbate Oxidative Stress and Apoptosis Induced by Ox-LDL in Human Macrophages. J. Cell. Physiol. 2019, 234, 14154–14160. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xiang, Y.; Fan, L.-J.; Zhang, X.-Y.; Li, J.-P.; Yu, C.-X.; Bao, L.-Y.; Cao, D.-S.; Xing, W.-B.; Liao, X.-H.; et al. Myocardin Inhibited the Gap Protein Connexin 43 via Promoted MiR-206 to Regulate Vascular Smooth Muscle Cell Phenotypic Switch. Gene 2017, 616, 22–30. [Google Scholar] [CrossRef]
- Jin, P.; Gu, W.; Lai, Y.; Zheng, W.; Zhou, Q.; Wu, X. The Circulating MicroRNA-206 Level Predicts the Severity of Pulmonary Hypertension in Patients with Left Heart Diseases. Cell. Physiol. Biochem. 2017, 41, 2150–2160. [Google Scholar] [CrossRef]
- Li, P.; Ma, Y.; Yu, C.; Wu, S.; Wang, K.; Yi, H.; Liang, W. Autophagy and Aging: Roles in Skeletal Muscle, Eye, Brain and Hepatic Tissue. Front. Cell Dev. Biol. 2021, 9, 2925. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Tokarz, P.; Koskela, A.; Paterno, J.; Blasiak, J. Autophagy Regulates Death of Retinal Pigment Epithelium Cells in Age-Related Macular Degeneration. Cell Biol. Toxicol. 2017, 33, 113–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kou, X.; Li, J.; Liu, X.; Chang, J.; Zhao, Q.; Jia, S.; Fan, J.; Ning Chen, X. Swimming Attenuates D-Galactose-Induced Brain Aging via Suppressing MiR-34a-Mediated Autophagy Impairment and Abnormal Mitochondrial Dynamics Swimming Attenuates D-Galactose-Induced Brain Aging via Suppressing MiR-34a-Mediated Autophagy Impairment and Abnormal Mitochon-Drial Dynamics. J. Appl. Physiol. 2017, 122, 1462–1469. [Google Scholar] [CrossRef]
- Nehammer, C.; Ejlerskov, P.; Gopal, S.; Handley, A.; Ng, L.; Moreira, P.; Lee, H.; Issazadeh-Navikas, S.; Rubinsztein, D.C.; Pocock, R. Interferon-b-Induced MiR-1 Alleviates Toxic Protein Accumulation by Controlling Autophagy. elife 2019, 8, e49930. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The Proteasome: Overview of Structure and Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saez, I.; Vilchez, D. The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases. Curr. Genom. 2014, 15, 38–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccarducci, R.; Daniele, S.; Polini, B.; Carpi, S.; Chico, L.; Fusi, J.; Baldacci, F.; Siciliano, G.; Bonuccelli, U.; Nieri, P.; et al. Apolipoprotein e Polymorphism and Oxidative Stress in Human Peripheral Blood Cells: Can Physical Activity Reactivate the Proteasome System through Epigenetic Mechanisms? Oxid. Med. Cell. Longev. 2021, 2021, 8869849. [Google Scholar] [CrossRef] [PubMed]
- Loppi, S.; Korhonen, P.; Bouvy-Liivrand, M.; Caligola, S.; Turunen, T.A.; Turunen, M.P.; de Sande, A.H.; Kołosowska, N.; Scoyni, F.; Rosell, A.; et al. Peripheral Inflammation Preceeding Ischemia Impairs Neuronal Survival through Mechanisms Involving MiR-127 in Aged Animals. Aging Cell 2021, 20, e13287. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal Muscle Atrophy and the E3 Ubiquitin Ligases MuRF1 and MAFbx/Atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469. [Google Scholar] [CrossRef] [Green Version]
- Clavel, S.; Coldefy, A.S.; Kurkdjian, E.; Salles, J.; Margaritis, I.; Derijard, B. Atrophy-Related Ubiquitin Ligases, Atrogin-1 and MuRF1 Are up-Regulated in Aged Rat Tibialis Anterior Muscle. Mech. Ageing Dev. 2006, 127, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Eddins, M.J.; Marblestone, J.G.; Suresh Kumar, K.G.; Leach, C.A.; Sterner, D.E.; Mattern, M.R.; Nicholson, B. Targeting the Ubiquitin E3 Ligase MuRF1 to Inhibit Muscle Atrophy. Cell Biochem. Biophys. 2011, 60, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Wada, S.; Kato, Y.; Okutsu, M.; Miyaki, S.; Suzuki, K.; Yan, Z.; Schiaffino, S.; Asahara, H.; Ushida, T.; Akimoto, T. Translational Suppression of Atrophic Regulators by MicroRNA-23a Integrates Resistance to Skeletal Muscle Atrophy. J. Biol. Chem. 2011, 286, 38456–38465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukreti, H.; Amuthavalli, K.; Harikumar, A.; Sathiyamoorthy, S.; Feng, P.Z.; Anantharaj, R.; Tan, S.L.K.; Lokireddy, S.; Bonala, S.; Sriram, S.; et al. Muscle-Specific MicroRNA1 (MiR1) Targets Heat Shock Protein 70 (HSP70) during Dexamethasone-Mediated Atrophy. J. Biol. Chem. 2013, 288, 6663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Liu, H.W.; Chan, Y.C.; Hu, S.H.; Liu, M.Y.; Chang, S.J. The Green Tea Polyphenol Epigallocatechin-3-Gallate Attenuates Age-Associated Muscle Loss via Regulation of MiR-486-5p and Myostatin. Arch. Biochem. Biophys. 2020, 692, 108511. [Google Scholar] [CrossRef]
- Martínez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER Proteostasis Control of Neuronal Physiology and Synaptic Function. Trends Neurosci. 2018, 41, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Dillin, A. XBP-1 Is a Cell-Nonautonomous Regulator of Stress Resistance and Longevity. Cell 2013, 153, 1435–1447. [Google Scholar] [CrossRef] [Green Version]
- Logue, S.E.; Cleary, P.; Saveljeva, S.; Samali, A. New Directions in ER Stress-Induced Cell Death. Apoptosis 2013, 18, 537–546. [Google Scholar] [CrossRef]
- Byrd, A.E.; Aragon, I.V.; Brewer, J.W. MicroRNA-30c-2* Limits Expression of Proadaptive Factor XBP1 in the Unfolded Protein Response. J. Cell Biol. 2012, 196, 689–698. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Read, D.E.; Deepti, A.; Cawley, K.; Gupta, A.; Oommen, D.; Verfaillie, T.; Matus, S.; Smith, M.A.; Mott, J.L.; et al. Perk-Dependent Repression of MiR-106b-25 Cluster Is Required for ER Stress-Induced Apoptosis. Cell Death Dis. 2012, 3, e333. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, P.; Koshy, L.; Sudhakaran, P.R.; Nair, G.M.; Gangaprasad, A.; Nair, A.J. Dysregulation of MiRNA and Its Reversal with L-Ascorbic Acid during AGE-BSA Induced ER Stress in Mus Musculus Neuronal Cells. Gene Rep. 2020, 21, 100841. [Google Scholar] [CrossRef]
- Ruberti, F.; Pezzola, S.; Barbato, C. Advances in MicroRNAs and Alzheimer’s Disease Research. In Alzheimer’s Disease Pathogenesis-Core Concepts, Shifting Paradigms and Therapeutic Targets; InTech: Houston, TX, USA, 2011; ISBN 978-953-307-690-4. [Google Scholar] [CrossRef] [Green Version]
- Aalto, A.P.; Nicastro, I.A.; Broughton, J.P.; Chipman, L.B.; Schreiner, W.P.; Chen, J.S.; Pasquinelli, A.E. Opposing Roles of MicroRNA Argonautes during Caenorhabditis Elegans Aging. PLoS Genet. 2018, 14, e1007379. [Google Scholar] [CrossRef] [PubMed]
- Reis, F.C.G.; Branquinho, J.L.O.; Brandão, B.B.; Guerra, B.A.; Silva, I.D.; Frontini, A.; Thomou, T.; Sartini, L.; Cinti, S.; Ronald Kahn, C.; et al. Fat-Specific Dicer Deficiency Accelerates Aging and Mitigates Several Effects of Dietary Restriction in Mice. Aging 2016, 8, 1201–1222. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P.; Konovalova, J.; Najam, S.S.; Alter, H.; Piepponen, T.P.; Erfle, H.; Sonntag, K.C.; Schütz, G.; Vinnikov, I.A.; Domanskyi, A. Dicer and MicroRNAs Protect Adult Dopamine Neurons. Cell Death Dis. 2017, 8, e2813. [Google Scholar] [CrossRef] [Green Version]
- Rybak-Wolf, A.; Jens, M.; Murakawa, Y.; Herzog, M.; Landthaler, M.; Rajewsky, N. A Variety of Dicer Substrates in Human and C. Elegans. Cell 2014, 159, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Knuckles, P.; Vogt, M.A.; Lugert, S.; Milo, M.; Chong, M.M.W.; Hautbergue, G.M.; Wilson, S.A.; Littman, D.R.; Taylor, V. Drosha Regulates Neurogenesis by Controlling Neurogenin 2 Expression Independent of MicroRNAs. Nat. Neurosci. 2012, 15, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, J.S.; Schmid, S.; Aguado, L.C.; Sabin, L.R.; Yasunaga, A.; Shim, J.V.; Sachs, D.; Cherry, S.; Tenoever, B.R. Drosha as an Interferon-Independent Antiviral Factor. Proc. Natl. Acad. Sci. USA 2014, 111, 7108–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, J.S.; Langlois, R.A.; Pham, A.M.; Tenoever, B.R. Evidence for a Cytoplasmic Microprocessor of Pri-MiRNAs. RNA 2012, 18, 1338–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Li, W.; She, H.; Dou, J.; Duong, D.M.; Du, Y.; Yang, S.H.; Seyfried, N.T.; Fu, H.; Gao, G.; et al. Stress Induces P38 MAPK-Mediated Phosphorylation and Inhibition of Drosha-Dependent Cell Survival. Mol. Cell 2015, 57, 721–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Jeong, K.; Kim, V.N. Genome-Wide Mapping of DROSHA Cleavage Sites on Primary MicroRNAs and Noncanonical Substrates. Mol. Cell 2017, 66, 258–269.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francisco, S.; Martinho, V.; Ferreira, M.; Reis, A.; Moura, G.; Soares, A.R.; Santos, M.A.S. The Role of MicroRNAs in Proteostasis Decline and Protein Aggregation during Brain and Skeletal Muscle Aging. Int. J. Mol. Sci. 2022, 23, 3232. https://doi.org/10.3390/ijms23063232
Francisco S, Martinho V, Ferreira M, Reis A, Moura G, Soares AR, Santos MAS. The Role of MicroRNAs in Proteostasis Decline and Protein Aggregation during Brain and Skeletal Muscle Aging. International Journal of Molecular Sciences. 2022; 23(6):3232. https://doi.org/10.3390/ijms23063232
Chicago/Turabian StyleFrancisco, Stephany, Vera Martinho, Margarida Ferreira, Andreia Reis, Gabriela Moura, Ana Raquel Soares, and Manuel A. S. Santos. 2022. "The Role of MicroRNAs in Proteostasis Decline and Protein Aggregation during Brain and Skeletal Muscle Aging" International Journal of Molecular Sciences 23, no. 6: 3232. https://doi.org/10.3390/ijms23063232
APA StyleFrancisco, S., Martinho, V., Ferreira, M., Reis, A., Moura, G., Soares, A. R., & Santos, M. A. S. (2022). The Role of MicroRNAs in Proteostasis Decline and Protein Aggregation during Brain and Skeletal Muscle Aging. International Journal of Molecular Sciences, 23(6), 3232. https://doi.org/10.3390/ijms23063232