
The Role of mTOR and eIF Signaling in Benign Endometrial Diseases



Abstract
:1. Introduction
1.1. Overview of mTOR Signaling and Its Correlated eIFs
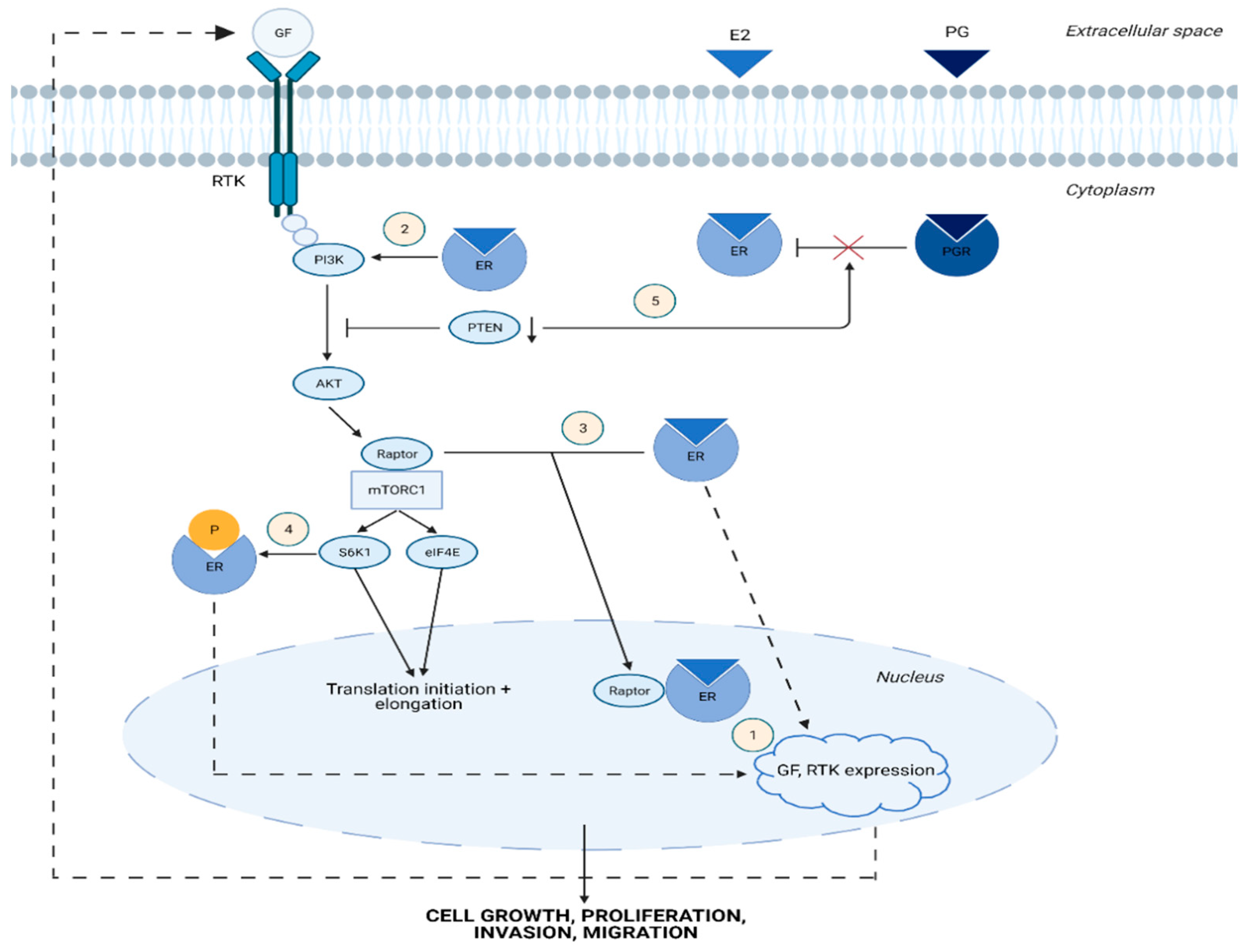
1.2. Cross-Talk between Estrogen and mTOR Signaling Pathway
1.3. Brief Outline of mTOR Pathway Dysregulation in Malignant Endometrial Diseases
2. mTOR and eIF Signaling in Benign Endometrial Diseases
2.1. mTOR Signaling in Adenomyosis
2.2. mTOR-Associated eIFs in Adenomyosis
2.3. mTOR Signaling in Endometriosis
2.4. mTOR-Associated eIFs in Endometriosis
2.5. mTOR Signaling in Endometritis
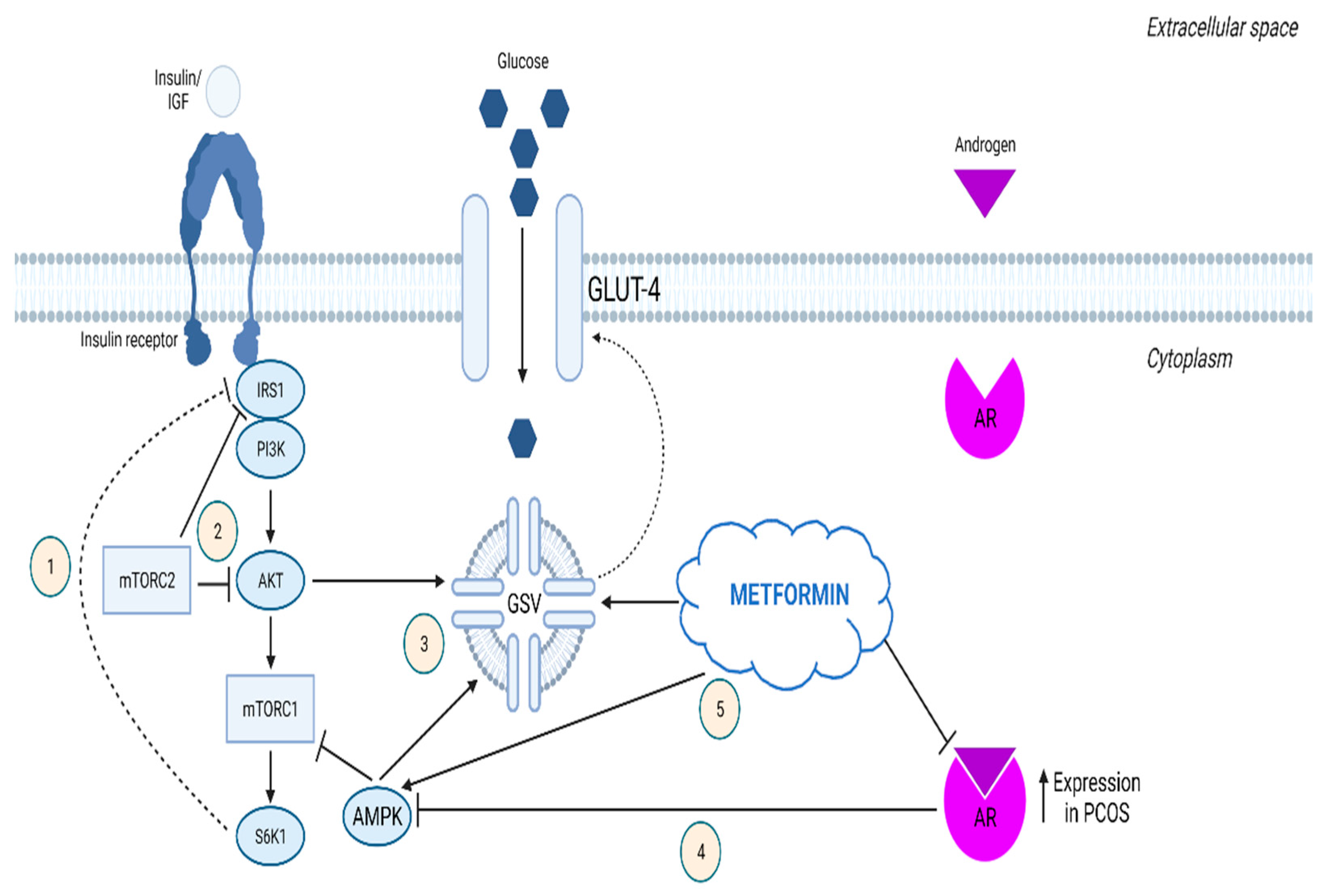
2.6. mTOR Signaling in Typical Endometrial Hyperplasia
2.7. mTOR-Associated eIFs in Typical Endometrial Hyperplasia
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stratopoulou, C.A.; Donnez, J.; Dolmans, M.M. Origin and Pathogenic Mechanisms of Uterine Adenomyosis: What Is Known So Far. Reprod. Sci. 2021, 28, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Kitawaki, J.; Kado, N.; Ishihara, H.; Koshiba, H.; Kitaoka, Y.; Honjo, H. Endometriosis: The Pathophysiology as an Estrogen-Dependent Disease. J. Steroid Biochem. Mol. Biol. 2002, 83, 149–155. [Google Scholar] [CrossRef]
- Chandra, V.; Kim, J.J.; Benbrook, D.M.; Dwivedi, A.; Rai, R. Therapeutic Options for Management of Endometrial Hyperplasia. J. Gynecol. Oncol. 2016, 27, e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habiba, M.; Pluchino, N.; Petignat, P.; Bianchi, P.; Brosens, I.A.; Benagiano, G. Adenomyosis and Endometrial Cancer: Literature Review. Gynecol. Obstet. Investig. 2018, 83, 313–328. [Google Scholar] [CrossRef]
- Melin, A.; Sparén, P.; Persson, I.; Bergqvist, A. Endometriosis and the Risk of Cancer with Special Emphasis on Ovarian Cancer. Hum. Reprod. 2006, 21, 1237–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutter, G.L.; Zaino, R.J.; Baak, J.P.A.; Bentley, R.C.; Robboy, S.J. Benign Endometrial Hyperplasia Sequence and Endometrial Intraepithelial Neoplasia. Int. J. Gynecol. Pathol. 2007, 26, 103–114. [Google Scholar] [CrossRef]
- Fourquet, J.; Báez, L.; Figueroa, M.; Iriarte, R.I.; Flores, I. Quantification of the Impact of Endometriosis Symptoms on Health-Related Quality of Life and Work Productivity. Fertil. Steril. 2011, 96, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Hagerty, K.A.; Skipper, B.; Bocklage, T. Chronic Endometritis: A Combined Histopathologic and Clinical Review of Cases from 2002 to 2007. Int. J. Gynecol. Pathol. 2010, 29, 44–50. [Google Scholar] [CrossRef]
- Montgomery, B.E.; Daum, G.S.; Dunton, C.J. Endometrial Hyperplasia: A Review. Obstet. Gynecol. Surv. 2004, 59, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Levgur, M.; Abadi, M.A.; Tucker, A. Adenomyosis: Symptoms, Histology, and Pregnancy Terminations. Obstet. Gynecol. 2000, 95, 688–691. [Google Scholar] [CrossRef]
- Sobočan, M.; Bračič, S.; Knez, J.; Takač, I.; Haybaeck, J. The Communication between the PI3K/AKT/MTOR Pathway and Y-Box Binding Protein-1 in Gynecological Cancer. Cancers 2020, 12, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobočan, M.; Smolle, M.A.; Schatz, C.; Haybaeck, J. The Interplay of Tumor Stroma and Translational Factors in Endometrial Cancer. Cancers 2020, 12, 2074. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Czapiewski, P.; Lapińska-Szumczyk, S.; Majewska, H.; Supernat, A.; Zaczek, A.; Biernat, W.; Golob-Schwarzl, N.; Haybaeck, J. The Prognostic Significance of Eukaryotic Translation Initiation Factors (EIFs) in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; Luna, J.I.; Marusina, A.I.; Merleev, A.; Kundu-Raychaudhuri, S.; Fiorentino, D.; Raychaudhuri, S.P.; Maverakis, E. Dual MTOR Inhibition Is Required to Prevent TGF-β-Mediated Fibrosis: Implications for Scleroderma. J. Investig. Dermatol. 2015, 135, 2873–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, G.G.; Abraham, R.T. Targeting the MTOR Signaling Network in Cancer. Trends Mol. Med. 2007, 13, 433–442. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR Complexes, Only One of Which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Caron, E.; Ghosh, S.; Matsuoka, Y.; Ashton-Beaucage, D.; Therrien, M.; Lemieux, S.; Perreault, C.; Roux, P.P.; Kitano, H. A Comprehensive Map of the MTOR Signaling Network. Mol. Syst. Biol. 2010, 6, 453. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 Complex: A Molecular Switchboard Controlling Cell Growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.I.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The Mammalian Target of Rapamycin (MTOR) Partner, Raptor, Binds the MTOR Substrates P70 S6 Kinase and 4E-BP1 through Their TOR Signaling (TOS) Motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular Mechanisms of MTOR-Mediated Translational Control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate MTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengrod, J.C.; Gardner, L.B. Cellular Adaptation to Nutrient Deprivation: Crosstalk between the MTORC1 and EIF2α Signaling Pathways and Implications for Autophagy. Cell Cycle 2015, 14, 2571–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilka, R.; Ernst, C.; Mehta, A.K.; Haybaeck, J. Eukaryotic Translation Initiation Factors in Cancer Development and Progression. Cancer Lett. 2013, 340, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. MTOR and S6K1 Mediate Assembly of the Translation Preinitiation Complex through Dynamic Protein Interchange and Ordered Phosphorylation Events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Morris, C.; Durand, S.; Jalinot, P. Decreased Expression of the Translation Factor EIF3e Induces Senescence in Breast Cancer Cells via Suppression of PARP1 and Activation of MTORC1. Oncotarget 2021, 12, 649–664. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR Complex 2 Controls the Actin Cytoskeleton and Is Rapamycin Insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Wang, C.; Sommer, E.; Kozasa, T.; Srinivasula, S.; Alessi, D.; Offermanns, S.; Simon, M.I.; Wu, D. PRR5L Degradation Promotes MTORC2-Mediated PKC-δ Phosphorylation and Cell Migration Downstream of Gα 12. Nat. Cell Biol. 2012, 14, 686–696. [Google Scholar] [CrossRef] [Green Version]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-Induced Insulin Resistance Is Mediated by MTORC2 Loss and Uncoupled from Longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Zhang, H.; Liu, W.; Liu, M.; Shi, M.; Wen, Z.; Li, C. Metformin Inhibits Growth of Eutopic Stromal Cells from Adenomyotic Endometrium via AMPK Activation and Subsequent Inhibition of AKT Phosphorylation: A Possible Role in the Treatment of Adenomyosis. Reproduction 2013, 146, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, K.E.; Rosendahl, A.H.; Olsson, H.; Malmström, P.; Hartman, L.; Fernö, M. Association between Insulin-like Growth Factor-1 Receptor (IGF1R) Negativity and Poor Prognosis in a Cohort of Women with Primary Breast Cancer. BMC Cancer 2014, 14, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciruelos Gil, E.M. Targeting the PI3K/AKT/MTOR Pathway in Estrogen Receptor-Positive Breast Cancer. Cancer Treat. Rev. 2014, 40, 862–871. [Google Scholar] [CrossRef]
- Hou, X.; Zhao, M.; Wang, T.; Zhang, G. Upregulation of Estrogen Receptor Mediates Migration, Invasionand Proliferation of Endometrial Carcinoma Cells by Regulating the PI3K/AKT/MTOR Pathway. Oncol. Rep. 2014, 31, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Alayev, A.; Salamon, R.S.; Berger, S.M.; Schwartz, N.S.; Cuesta, R.; Snyder, R.B.; Holz, M.K. MTORC1 Directly Phosphorylates and Activates ERα upon Estrogen Stimulation. Oncogene 2016, 35, 3535–3543. [Google Scholar] [CrossRef]
- Miller, T.W.; Pérez-Torres, M.; Narasanna, A.; Guix, M.; Stål, O.; Pérez-Tenorio, G.; Gonzalez-Angulo, A.M.; Hennessy, B.T.; Mills, G.B.; Kennedy, J.P.; et al. Loss of Phosphatase and Tensin Homologue Deleted on Chromosome 10 Engages ErbB3 and Insulin-like Growth Factor-I Receptor Signaling to Promote Antiestrogen Resistance in Breast Cancer. Cancer Res. 2009, 69, 4192–4201. [Google Scholar] [CrossRef] [Green Version]
- Yamnik, R.L.; Holz, M.K. MTOR/S6K1 and MAPK/RSK Signaling Pathways Coordinately Regulate Estrogen Receptor α Serine 167 Phosphorylation. FEBS Lett. 2010, 584, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yu, Q. Role of MTOR Signaling in Female Reproduction. Front. Endocrinol. 2019, 10, 692. [Google Scholar] [CrossRef]
- Bell, D.W.; Ellenson, L.H. Molecular Genetics of Endometrial Carcinoma. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 339–367. [Google Scholar] [CrossRef]
- Pavlidou, A.; Vlahos, N.F. Molecular Alterations of PI3K/Akt/MTOR Pathway: A Therapeutic Target in Endometrial Cancer. Sci. World J. 2014, 2014, 709736. [Google Scholar] [CrossRef] [Green Version]
- Eritja, N.; Yeramian, A.; Chen, B.J.; Llobet-Navas, D.; Ortega, E.; Colas, E.; Abal, M.; Dolcet, X.; Reventos, J.; Matias-Guiu, X. Endometrial Carcinoma: Specific Targeted Pathways. Adv. Exp. Med. Biol. 2017, 943, 149–207. [Google Scholar]
- Slomovitz, B.M.; Coleman, R.L. The PI3K/AKT/MTOR Pathway as a Therapeutic Target in Endometrial Cancer. Clin. Cancer Res. 2012, 18, 5856–5864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Stanton, M.L.; Feng, W.; Rodriguez, M.E.; Ramondetta, L.; Chen, L.; Brown, R.E.; Duan, X. Morphoproteomic Analysis Reveals an Overexpressed and Constitutively Activated Phospholipase D1-MTORC2 Pathway in Endometrial Carcinoma. Int. J. Clin. Exp. Pathol. 2010, 4, 13–21. [Google Scholar] [PubMed]
- Lu, K.H.; Wu, W.; Dave, B.; Slomovitz, B.M.; Burke, T.W.; Munsell, M.F.; Broaddus, R.R.; Walker, C.L. Loss of Tuberous Sclerosis Complex-2 Function and Activation of Mammalian Target of Rapamycin Signaling in Endometrial Carcinoma. Clin. Cancer Res. 2008, 14, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.I.; Huang, R.S.P.; Mata, D.A.; Decker, B.; Danziger, N.; Lechpammer, M.; Hiemenz, M.; Ramkissoon, S.H.; Ross, J.S.; Elvin, J.A. Clinicopathological and Genomic Characterization of BCORL1-Driven High-Grade Endometrial Stromal Sarcomas. Mod. Pathol. 2021, 34, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Vasudevaraja, V.; Serrano, J.; Stewart, C.J.R.; Oliva, E.; Momeni-Boroujeni, A.; Jungbluth, A.A.; da Cruz Paula, A.; da Silva, E.M.; Weigelt, B.; et al. TSC2-Mutant Uterine Sarcomas with JAZF1-SUZ12 Fusions Demonstrate Hybrid Features of Endometrial Stromal Sarcoma and PEComa and Are Responsive to MTOR Inhibition. Mod. Pathol. 2022, 35, 117–127. [Google Scholar] [CrossRef]
- Quan, P.; Moinfar, F.; Kufferath, I.; Absenger, M.; Kueznik, T.; Denk, H.; Zatloukal, K.; Haybaeck, J. Effects of Targeting Endometrial Stromal Sarcoma Cells via Histone Deacetylase and PI3K/AKT/MTOR Signaling. Anticancer. Res. 2014, 34, 2883–2897. [Google Scholar]
- García-Solares, J.; Donnez, J.; Donnez, O.; Dolmans, M.M. Pathogenesis of Uterine Adenomyosis: Invagination or Metaplasia? Fertil. Steril. 2018, 109, 371–379. [Google Scholar] [CrossRef]
- Cuesta, R.; Gritsenko, M.A.; Petyuk, V.A.; Shukla, A.K.; Tsai, C.F.; Liu, T.; McDermott, J.E.; Holz, M.K. Phosphoproteome Analysis Reveals Estrogen-ER Pathway as a Modulator of MTOR Activity via Deptor*. Mol. Cell. Proteom. 2019, 18, 1607–1618. [Google Scholar] [CrossRef]
- Guo, Y.; Lang, X.; Lu, Z.; Wang, J.; Li, T.; Liao, Y.; Jia, C.; Zhao, W.; Fang, H. MiR-10b Directly Targets ZEB1 and PIK3CA to Curb Adenomyotic Epithelial Cell Invasiveness via Upregulation of E-Cadherin and Inhibition of Akt Phosphorylation. Cell. Physiol. Biochem. 2015, 35, 2169–2180. [Google Scholar] [CrossRef]
- Guo, J.; Gao, J.; Yu, X.; Luo, H.; Xiong, X.; Huang, O. Expression of DJ-1 and MTOR in Eutopic and Ectopic Endometria of Patients with Endometriosis and Adenomyosis. Gynecol. Obstet. Investig. 2015, 79, 195–200. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Zhang, J.; Qi, Y.-H.; Kong, M.; Liu, S.-A.; Hu, J.-J. Linc-ROR Promotes Endometrial Cell Proliferation by Activating the PI3K-Akt Pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2218–2225. [Google Scholar] [PubMed]
- Hu, H.; Li, H.; He, Y. MicroRNA-17 Downregulates Expression of the PTEN Gene to Promote the Occurrence and Development of Adenomyosis. Exp. Ther. Med. 2017, 14, 3805–3811. [Google Scholar] [CrossRef] [Green Version]
- Goteri, G.; Lucarini, G.; Montik, N.; Zizzi, A.; Stramazzotti, D.; Fabris, G.; Tranquilli, A.L.; Ciavattini, A. Expression of Vascular Endothelial Growth Factor (VEGF), Hypoxia Inducible Factor-1α (HIF-1α), and Microvessel Density in Endometrial Tissue in Women with Adenomyosis. Int. J. Gynecol. Pathol. 2009, 28, 157–163. [Google Scholar] [CrossRef]
- Wang, W.; Taylor, R.N.; Bagchi, I.C.; Bagchi, M.K. Regulation of Human Endometrial Stromal Proliferation and Differentiation by C/EBPβ Involves Cyclin E-Cdk2 and STAT3. Mol. Endocrinol. 2012, 26, 2016–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Sun, Y.; Yang, B.; Yang, Y.; Zhang, Y.; Yu, T.; Huang, H.; Zhang, J.; Xu, H. Transcriptome Sequencing of Adenomyosis Eutopic Endometrium: A New Insight into Its Pathophysiology. J. Cell. Mol. Med. 2019, 23, 8381–8391. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-Coding RNAs in Human Disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, B.; Wang, H.Y.; Chang, A.; Zheng, X.F.S. Emerging Role of MicroRNAs in MTOR Signaling. Cell. Mol. Life Sci. 2017, 74, 2613–2625. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Li, M.; Peng, J.; Shi, Y.; Sun, P. MiR-92a Promotes Progesterone Resistance in Endometriosis through PTEN/AKT Pathway. Life Sci. 2020, 242, 117190. [Google Scholar] [CrossRef]
- Zhao, Q.; Han, B.; Zhang, Y.; Su, K.; Wang, C.; Hai, P.; Bian, A.; Guo, R. Effect of MiR-194-5p Regulating STAT1/MTOR Signaling Pathway on the Biological Characteristics of Ectopic Endometrial Cells from Mice. Am. J. Transl. Res. 2020, 12, 6136–6148. [Google Scholar] [PubMed]
- Zhou, X.; Chen, Z.; Pei, L.; Sun, J. MicroRNA MiR-106a-5p Targets Forkhead Box Transcription Factor FOXC1 to Suppress the Cell Proliferation, Migration, and Invasion of Ectopic Endometrial Stromal Cells via the PI3K/Akt/MTOR Signaling Pathway. Bioengineered 2021, 12, 2203–2213. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. MTORC1 Drives HIF-1α and VEGF-A Signalling via Multiple Mechanisms Involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Pal, R.; Monaghan, S.A.; Schafer, P.; Ouyang, H.; Mapara, M.; Galson, D.L.; Lentzsch, S. IMiD Immunomodulatory Compounds Block C/EBPβ Translation through EIF4E down-Regulation Resulting in Inhibition of MM. Blood 2011, 117, 5157–5165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, V.; Hall, M.N. Reduced C/EBP Β-LIP Translation Improves Metabolic Health. EMBO Rep. 2015, 16, 881–882. [Google Scholar] [CrossRef] [Green Version]
- Calkhoven, C.F.; Müller, C.; Leutz, A. Translational Control of C/EBPalpha and C/EBPbeta Isoform Expression. Genes Dev. 2000, 14, 1920–1932. [Google Scholar] [CrossRef]
- Shen, M.; Liu, X.; Zhang, H.; Guo, S.W. Transforming Growth Factor Β1 Signaling Coincides with Epithelial-Mesenchymal Transition and Fibroblast-to-Myofibroblast Transdifferentiation in the Development of Adenomyosis in Mice. Hum. Reprod. 2016, 31, 355–369. [Google Scholar] [CrossRef]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and MTOR Signaling Pathways in the Induction of Epithelial-Mesenchymal Transition (EMT) Process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Zheng, D.; Duan, H.; Wang, S.; Xu, Q.; Gan, L.; Li, J.; Dong, Q. FAK Regulates Epithelial-Mesenchymal Transition in Adenomyosis. Mol. Med. Rep. 2018, 18, 5461–5472. [Google Scholar] [CrossRef] [Green Version]
- Herndon, C.N.; Aghajanova, L.; Balayan, S.; Erikson, D.; Barragan, F.; Goldfien, G.; Vo, K.C.; Hawkins, S.; Giudice, L.C. Global Transcriptome Abnormalities of the Eutopic Endometrium from Women with Adenomyosis. Reprod. Sci. 2016, 23, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Shen, M.; Liu, X.; Nie, J. The Possible Role of Eukaryotic Translation Initiation Factor 3 Subunit e (EIF3e) in the Epithelial–Mesenchymal Transition in Adenomyosis. Reprod. Sci. 2019, 26, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Pavone, M.E.; Lu, Z.; Wei, J.; Kim, J.J. Increased Activation of the PI3K/AKT Pathway Compromises Decidualization of Stromal Cells from Endometriosis. J. Clin. Endocrinol. Metab. 2012, 97, E35–E43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makker, A.; Goel, M.M.; Das, V.; Agarwal, A. PI3K-Akt-MTOR and MAPK Signaling Pathways in Polycystic Ovarian Syndrome, Uterine Leiomyomas and Endometriosis: An Update. Gynecol. Endocrinol. 2012, 28, 175–181. [Google Scholar] [CrossRef]
- Cinar, O.; Seval, Y.; Uz, Y.H.; Cakmak, H.; Ulukus, M.; Kayisli, U.A.; Arici, A. Differential Regulation of Akt Phosphorylation in Endometriosis. Reprod. BioMed. Online 2009, 19, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Laudanski, P.; Szamatowicz, J.; Kowalczuk, O.; Kuźmicki, M.; Grabowicz, M.; Chyczewski, L. Expression of Selected Tumor Suppressor and Oncogenes in Endometrium of Women with Endometriosis. Hum. Reprod. 2009, 24, 1880–1890. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, M.; Zheng, X.; Sun, Y.; Wen, Z.; Zhao, X. Endometriotic Stromal Cells Lose the Ability to Regulate Cell-Survival Signaling in Endometrial Epithelial Cells in Vitro. Mol. Hum. Reprod. 2009, 15, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Honda, H.; Barrueto, F.F.; Gogusev, J.; Im, D.D.; Morin, P.J. Serial Analysis of Gene Expression Reveals Differential Expression between Endometriosis and Normal Endometrium. Possible Roles for AXL and SHC1 in the Pathogenesis of Endometriosis. Reprod. Biol. Endocrinol. 2008, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Madanes, D.; Bilotas, M.A.; Bastón, J.I.; Singla, J.J.; Meresman, G.F.; Barañao, R.I.; Ricci, A.G. PI3K/AKT Pathway Is Altered in the Endometriosis Patient’s Endometrium and Presents Differences According to Severity Stage. Gynecol. Endocrinol. 2020, 36, 436–440. [Google Scholar] [CrossRef]
- Kim, T.H.; Yu, Y.; Luo, L.; Lydon, J.P.; Jeong, J.W.; Kim, J.J. Activated AKT Pathway Promotes Establishment of Endometriosis. Endocrinology 2014, 155, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Jo, M.W.; Lee, E.Y.; Kim, H.J.; Choi, D.S. Differential Induction of Autophagy by Mtor Is Associated with Abnormal Apoptosis in Ovarianendometriotic Cysts. Mol. Hum. Reprod. 2014, 20, 309–317. [Google Scholar] [CrossRef]
- McKinnon, B.; Mueller, M.; Montgomery, G. Progesterone Resistance in Endometriosis: An Acquired Property? Trends Endocrinol. Metab. 2018, 29, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, R.M.; Kim, T.H.; Shin, J.-H.; Jeong, J.-W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leconte, M.; Nicco, C.; Ng, C.; Chéreau, C.; Chouzenoux, S.; Marut, W.; Guibourdenche, J.; Arkwright, S.; Weill, B.; Chapron, C.; et al. The MTOR/AKT Inhibitor Temsirolimus Prevents Deep Infiltrating Endometriosis in Mice. Am. J. Pathol. 2011, 179, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jo, M.; Lee, E.; Lee, D.Y.; Choi, D. Dienogest Enhances Autophagy Induction in Endometriotic Cells by Impairing Activation of AKT, ERK1/2, and MTOR. Fertil. Steril. 2015, 104, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Wang, Y.; Xu, G.; Dai, L. Effect of Rapamycin on Endometriosis in Mice. Exp. Ther. Med. 2016, 12, 101–106. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Ye, Q.; Zhuang, M.; Xie, S.; Zhong, R.; Cui, J.; Zhou, J.; Zhu, Y.; Zhang, T.; Cao, L. Ginsenoside Rg3 Inhibits Angiogenesis in a Rat Model of Endometriosis through the VEGFR-2-Mediated PI3K/Akt/MTOR Signaling Pathway. PLoS ONE 2017, 12, e0186520. [Google Scholar] [CrossRef] [Green Version]
- Gadducci, A.; Zannoni, G.F. Endometriosis-Associated Extraovarian Malignancies: A Challenging Question for the Clinician and the Pathologist. Anticancer Res. 2020, 40, 2429–2438. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Labidi-Galy, S.I.; Moschetta, M.; Uccello, M.; Kalaitzopoulos, D.R.; Perez-Fidalgo, J.A.; Boussios, S. Endometriosis-Associated Ovarian Carcinomas: Insights into Pathogenesis, Diagnostics, and Therapeutic Targets—A Narrative Review. Ann. Transl. Med. 2020, 8, 1712. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Dedes, K.J.; Fink, D.; Imesch, P. ARID1A Mutations and PI3K/AKT Pathway Alterations in Endometriosis and Endometriosis-Associated Ovarian Carcinomas. Int. J. Mol. Sci. 2013, 14, 18824–18849. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Banno, K.; Okawa, R.; Yanokura, M.; Iijima, M.; Iriekunitomi, H.; Nakamura, K.; Iida, M.; Adachi, M.; Umene, K.; et al. ARID1A Gene Mutation in Ovarian and Endometrial Cancers (Review). Oncol. Rep. 2016, 35, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Rogers-Broadway, K.R.; Kumar, J.; Sisu, C.; Wander, G.; Mazey, E.; Jeyaneethi, J.; Pados, G.; Tsolakidis, D.; Klonos, E.; Grunt, T.; et al. Differential Expression of MTOR Components in Endometriosis and Ovarian Cancer: Effects of Rapalogues and Dual Kinase Inhibitors on MTORC1 and MTORC2 Stoichiometry. Int. J. Mol. Med. 2019, 43, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Shen, M.; Liu, X.; Guo, S.W. Reduced Expression of Eukaryotic Translation Initiation Factor 3 Subunit e and Its Possible Involvement in the Epithelial–Mesenchymal Transition in Endometriosis. Reprod. Sci. 2018, 25, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Jo, M.W.; Lee, E.Y.; Lee, D.Y.; Choi, D.S. Dienogest Regulates Apoptosis, Proliferation, and Invasiveness of Endometriotic Cyst Stromal Cells via Endoplasmic Reticulum Stress Induction. Mol. Hum. Reprod. 2020, 26, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, L.; Shi, H.; Du, W.; Qi, Y.; Qiu, C.; Liang, X.; Shi, W.; Liu, J. Endoplasmic Reticulum-Targeting Photosensitizer Hypericin Confers Chemo-Sensitization towards Oxaliplatin through Inducing pro-Death Autophagy. Int. J. Biochem. Cell Biol. 2017, 87, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, W.; Bazer, F.W.; Song, G. Naringenin Induces Mitochondriamediated Apoptosis and Endoplasmic Reticulum Stress by Regulating MAPK and AKT Signal Transduction Pathways in Endometriosis Cells. Mol. Hum. Reprod. 2017, 23, 842–854. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.; Bazer, F.W.; Lim, W.; Song, G. Chrysin Leads to Cell Death in Endometriosis by Regulation of Endoplasmic Reticulum Stress and Cytosolic Calcium Level. J. Cell. Physiol. 2019, 234, 2480–2490. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, H.; Chen, Z.; Zhang, W.; Liu, X.; Fang, J.; Liu, F.; Kwak-Kim, J. Endometrial TGF-β, IL-10, IL-17 and Autophagy Are Dysregulated in Women with Recurrent Implantation Failure with Chronic Endometritis. Reprod. Biol. Endocrinol. 2019, 17, 2. [Google Scholar] [CrossRef] [Green Version]
- Buzzaccarini, G.; Vitagliano, A.; Andrisani, A.; Santarsiero, C.M.; Cicinelli, R.; Nardelli, C.; Ambrosini, G.; Cicinelli, E. Chronic Endometritis and Altered Embryo Implantation: A Unified Pathophysiological Theory from a Literature Systematic Review. J. Assist. Reprod. Genet. 2020, 37, 2897–2911. [Google Scholar] [CrossRef]
- Ahmadi, M.; Abdolmohamadi-vahid, S.; Ghaebi, M.; Dolati, S.; Abbaspour-Aghdam, S.; Danaii, S.; Berjis, K.; Madadi-Javid, R.; Nouri, Z.; Siahmansouri, H.; et al. Sirolimus as a New Drug to Treat RIF Patients with Elevated Th17/Treg Ratio: A Double-Blind, Phase II Randomized Clinical Trial. Int. Immunopharmacol. 2019, 74, 105730. [Google Scholar] [CrossRef]
- Emons, G.; Beckmann, M.W.; Schmidt, D.; Mallmann, P. New WHO Classification of Endometrial Hyperplasias. Geburtshilfe Frauenheilkd. 2015, 75, 135–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhu, L.; Kuokkanen, S.; Pollard, J.W. Activation of Protein Synthesis in Mouse Uterine Epithelial Cells by Estradiol-17β Is Mediated by a PKC-ERK 1/2-MTOR Signaling Pathway. Proc. Natl. Acad. Sci. USA 2015, 112, E1382–E1391. [Google Scholar]
- Bajwa, P.; Nielsen, S.; Lombard, J.M.; Rassam, L.; Nahar, P.; Rueda, B.R.; Wilkinson, J.E.; Miller, R.A.; Tanwar, P.S. Overactive MTOR Signaling Leads to Endometrial Hyperplasia in Aged Women and Mice. Oncotarget 2017, 8, 7265–7275. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.S.; Lombard, J.M.; Ius, Y.; O’Sullivan, R.; Wood, L.G.; Nahar, P.; Jaaback, K.; Tanwar, P.S. Adipose-Derived VEGF–MTOR Signaling Promotes Endometrial Hyperplasia and Cancer: Implications for Obese Women. Mol. Cancer Res. 2018, 16, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wu, D.C.; Qu, L.H.; Liao, H.Q.; Li, M.X. The Role of MTOR in Ovarian Neoplasms, Polycystic Ovary Syndrome, and Ovarian Aging. Clin. Anat. 2018, 31, 891–898. [Google Scholar] [CrossRef]
- Li, X.; Cui, P.; Jiang, H.-Y.; Guo, Y.-R.; Pishdari, B.; Hu, M.; Feng, Y.; Billig, H.; Shao, R. Reversing the Reduced Level of Endometrial GLUT4 Expression in Polycystic Ovary Syndrome: A Mechanistic Study of Metformin Action. Am. J. Transl. Res. 2015, 7, 574–586. [Google Scholar] [PubMed]
- Sumarac-Dumanovic, M.; Apostolovic, M.; Janjetovic, K.; Jeremic, D.; Popadic, D.; Ljubic, A.; Micic, J.; Dukanac-Stamenkovic, J.; Tubic, A.; Stevanovic, D.; et al. Downregulation of Autophagy Gene Expression in Endometria from Women with Polycystic Ovary Syndrome. Mol. Cell. Endocrinol. 2017, 440, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gressel, G.M.; Parkash, V.; Pal, L. Management Options and Fertility-Preserving Therapy for Premenopausal Endometrial Hyperplasia and Early-Stage Endometrial Cancer. Int. J. Gynecol. Obstet. 2015, 131, 234–239. [Google Scholar] [CrossRef]
- Ong, P.S.; Wang, L.Z.; Dai, X.; Tseng, S.H.; Loo, S.J.; Sethi, G. Judicious Toggling of MTOR Activity to Combat Insulin Resistance and Cancer: Current Evidence and Perspectives. Front. Pharmacol. 2016, 7, 395. [Google Scholar] [CrossRef]
- Nwanodi, O. Progestin Intrauterine Devices and Metformin: Endometrial Hyperplasia and Early Stage Endometrial Cancer Medical Management. In Healthcare; Multidisciplinary Digital Publishing Institute: Basel, Switzerland, 2017; Volume 5, p. 30. [Google Scholar]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The Therapeutic Potential of Metformin in Neurodegenerative Diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef]
- Ohara, M.; Yoshida-Komiya, H.; Ono-Okutsu, M.; Yamaguchi-Ito, A.; Takahashi, T.; Fujimori, K. Metformin Reduces Androgen Receptor and Upregulates Homeobox A10 Expression in Uterine Endometrium in Women with Polycystic Ovary Syndrome. Reprod. Biol. Endocrinol. 2021, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-Q.; Guo, X.-C.; Li, L.; Gao, Z.-H.; Ji, M. Treatment with Metformin and Sorafenib Alleviates Endometrial Hyperplasia in Polycystic Ovary Syndrome by Promoting Apoptosis via Synergically Regulating Autophagy. J. Cell. Physiol. 2020, 235, 1339–1348. [Google Scholar] [CrossRef]
- Erdemoglu, E.; Güney, M.; Giray, S.G.; Take, G.; Mungan, T. Effects of Metformin on Mammalian Target of Rapamycin in a Mouse Model of Endometrial Hyperplasia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 145, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Tas, M.; Kutuk, M.S.; Serin, I.S.; Ozgun, M.T.; Oner, G.; Ozturk, F. Comparison of Antiproliferative Effects of Metformine and Progesterone on Estrogen-Induced Endometrial Hyperplasia in Rats. Gynecol. Endocrinol. 2013, 29, 311–314. [Google Scholar] [CrossRef]
- Sharifzadeh, F.; Aminimoghaddam, S.; Kashanian, M.; Fazaeli, M.; Sheikhansari, N. A Comparison between the Effects of Metformin and Megestrol on Simple Endometrial Hyperplasia. Gynecol. Endocrinol. 2017, 33, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Cruz, H.; Oróstica, L.; Plaza-Parrochia, F.; Torres-Pinto, I.; Romero, C.; Vega, M. The Insulin-Sensitizing Mechanism of Myo-Inositol Is Associated with AMPK Activation and GLUT-4 Expression in Human Endometrial Cells Exposed to a PCOS Environment. Am. J. Physiol. Endocrinol. Metab. 2020, 318, 237–248. [Google Scholar] [CrossRef]
- Ferreira, G.D.; Germeyer, A.; de Barros Machado, A.; do Nascimento, T.L.; Strowitzki, T.; Brum, I.S.; von Eye Corleta, H.; Capp, E. Metformin Modulates PI3K and GLUT4 Expression and Akt/PKB Phosphorylation in Human Endometrial Stromal Cells after Stimulation with Androgen and Insulin. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 157–162. [Google Scholar] [CrossRef]
- Lam, H.C.; Baglini, C.v.; Lope, A.L.; Parkhitko, A.A.; Liu, H.J.; Alesi, N.; Malinowska, I.A.; Ebrahimi-Fakhari, D.; Saffari, A.; Yu, J.J.; et al. P62/SQSTM1 Cooperates with Hyperactive MTORC1 to Regulate Glutathione Production, Maintain Mitochondrial Integrity, and Promote Tumorigenesis. Cancer Res. 2017, 77, 3255–3267. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Li, J.; Yang, L.; Zhu, L.; Huang, X.; Zhang, S.; Luo, L.; Jiang, Z.; Jiang, T.; Xu, W.; et al. Tamoxifen Activates Nrf2-Dependent SQSTM1 Transcription to Promote Endometrial Hyperplasia. Theranostics 2017, 7, 1890–1900. [Google Scholar] [CrossRef]
- Li, Y.; Chen, C.; Ma, Y.; Xiao, J.; Luo, G.; Li, Y.; Wu, D. Multi-System Reproductive Metabolic Disorder: Significance for the Pathogenesis and Therapy of Polycystic Ovary Syndrome (PCOS). Life Sci. 2019, 228, 167–175. [Google Scholar] [CrossRef]
- Harada, M.; Takahashi, N.; Azhary, J.M.; Kunitomi, C.; Fujii, T.; Osuga, Y. Endoplasmic Reticulum Stress: A Key Regulator of the Follicular Microenvironment in the Ovary. Mol. Hum. Reprod. 2021, 27, gaaa088. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ma, Z.; Zhao, H.; Chen, X.; Zhang, Y.; Guo, H.; Zhao, Y.; Chen, Z.J. Activation of EIF2α Signaling Cascade Is Associated with Testosterone-Induced Cell Apoptosis in INS-1 Cells. Horm. Metab. Res. 2014, 46, 574–580. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Endometrial Disease | Non-Coding RNA | Component | Impact on Signaling | Impact on Disease Progression |
|---|---|---|---|---|
| Adenomyosis | Linc-ROR [53] | PTEN downregulation | PI3K/AKT pathway upregulation | promotion of endometrial cell proliferation |
| miR-17 [54] | 3′UTR of PTEN | possible PI3K/AKT pathwayupregulation | promotion of endometrial cell apoptosis | |
| miR-10b [51] | 3′UTR of PIK3CA | PI3K/AKT pathway downregulation | inhibition of endometrial cell invasion and migration | |
| Endometriosis | miR-92a [61] | 3′UTR of PTEN | possible PI3K/AKT pathway upregulation | promotion of progesterone resistance in endometriosis |
| miR-194-5p [62] | 3′UTR of STAT1 | STAT1/mTOR pathway downregulation | inhibition of endometrial cell proliferation and invasion | |
| miR-106a-5p [63] | 3′UTR of FOXC1 | PI3K/AKT/mTOR pathway downregulation | inhibition of ectopic endometrial stromal cell proliferation, migration, and invasion |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Driva, T.S.; Schatz, C.; Sobočan, M.; Haybaeck, J. The Role of mTOR and eIF Signaling in Benign Endometrial Diseases. Int. J. Mol. Sci. 2022, 23, 3416. https://doi.org/10.3390/ijms23073416
Driva TS, Schatz C, Sobočan M, Haybaeck J. The Role of mTOR and eIF Signaling in Benign Endometrial Diseases. International Journal of Molecular Sciences. 2022; 23(7):3416. https://doi.org/10.3390/ijms23073416
Chicago/Turabian StyleDriva, Tatiana S., Christoph Schatz, Monika Sobočan, and Johannes Haybaeck. 2022. "The Role of mTOR and eIF Signaling in Benign Endometrial Diseases" International Journal of Molecular Sciences 23, no. 7: 3416. https://doi.org/10.3390/ijms23073416
APA StyleDriva, T. S., Schatz, C., Sobočan, M., & Haybaeck, J. (2022). The Role of mTOR and eIF Signaling in Benign Endometrial Diseases. International Journal of Molecular Sciences, 23(7), 3416. https://doi.org/10.3390/ijms23073416