
Recycling and Reshaping—E3 Ligases and DUBs in the Initiation of T Cell Receptor-Mediated Signaling and Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Ubiquitination and Deubiquitination—A Basic Introduction
2. Beyond Ubiquitination—Ubiquitin-like Proteins
3. Ubiquitin and Ubiquitin-like Proteins in T-Lymphocytes
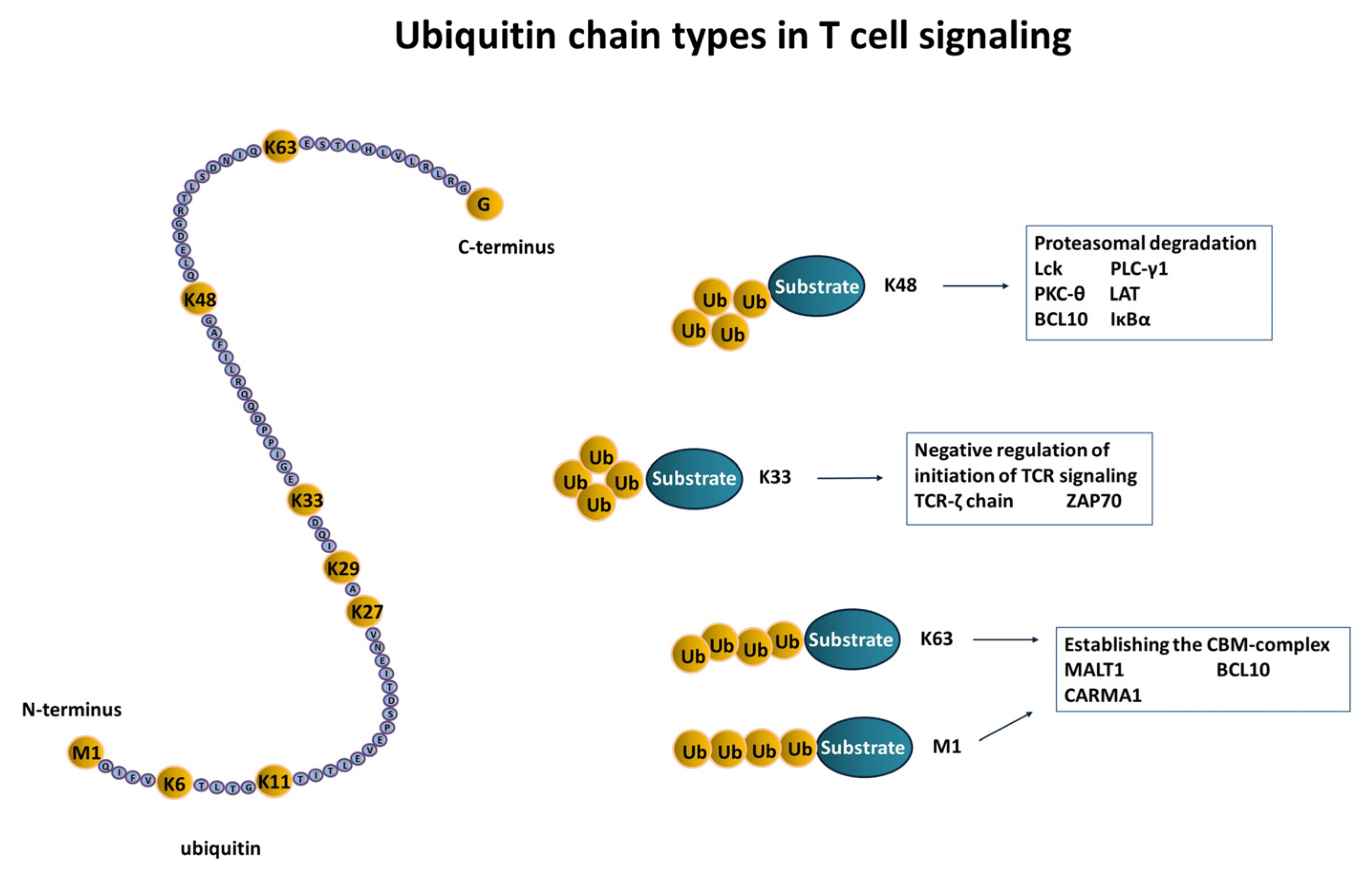
3.1. Initiation of T Cell Signaling
3.2. Transcription Factor Activation in T Cells
3.3. T Cell Development
4. Role of Ubiquitination in Anergy and Autoimmune Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Seifert, U.; Bialy, L.P.; Ebstein, F.; Bech-Otschir, D.; Voigt, A.; Schröter, F.; Prozorovski, T.; Lange, N.; Steffen, J.; Rieger, M.; et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell 2010, 142, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Colberg, L.; Cammann, C.; Greinacher, A.; Seifert, U. Structure and function of the ubiquitin-proteasome system in platelets. J. Thromb. Haemost. 2020, 18, 771–780. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [Green Version]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [Green Version]
- Ishikura, S.; Weissman, A.M.; Bonifacino, J.S. Serine Residues in the Cytosolic Tail of the T-cell Antigen Receptor α-Chain Mediate Ubiquitination and Endoplasmic Reticulum-associated Degradation of the Unassembled Protein. J. Biol. Chem. 2010, 285, 23916–23924. [Google Scholar] [CrossRef] [Green Version]
- Anania, V.G.; Bustos, D.J.; Lill, J.R.; Kirkpatrick, D.; Coscoy, L. A Novel Peptide-Based SILAC Method to Identify the Posttranslational Modifications Provides Evidence for Unconventional Ubiquitination in the ER-Associated Degradation Pathway. Int. J. Proteom. 2013, 2013, 1–12. [Google Scholar] [CrossRef]
- Tait, S.W.; de Vries, E.; Maas, C.; Keller, A.M.; D’Santos, C.S.; Borst, J. Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J. Cell Biol. 2007, 179, 1453–1466. [Google Scholar] [CrossRef] [Green Version]
- Metzger, M.B.; Pruneda, J.N.; Klevit, R.E.; Weissman, A.M. RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim. Biophys. Acta 2014, 1843, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and Quantitative Assessment of the Ubiquitin-Modified Proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplantine, E.; Fontan, E.; Chiaravalli, J.; Lopez, T.; Lakisic, G.; Véron, M.; Agou, F.; Israël, A. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J. 2009, 28, 2885–2895. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Smith, H.; Windheim, M.; Peggie, M.; Carrick, E.; Morrice, N.; Cohen, P. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem. J. 2008, 409, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, M.E.; Koehler, C.F.; Hunter, T. Emerging functions of branched ubiquitin chains. Cell Discov. 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and Cellular Roles of Ubiquitin-Specific Deubiquitinating Enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef] [Green Version]
- Komander, D. Mechanism, Specificity and Structure of the Deubiquitinases. Subcell. Biochem. 2010, 54, 69–87. [Google Scholar] [CrossRef]
- Mevissen, T.E.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [Green Version]
- Eifler, K.; Cuijpers, S.A.G.; Willemstein, E.; Raaijmakers, J.; El Atmioui, D.; Ovaa, H.; Medema, R.; Vertegaal, A.C.O. SUMO targets the APC/C to regulate transition from metaphase to anaphase. Nat. Commun. 2018, 9, 1119. [Google Scholar] [CrossRef]
- Enserink, J.M. Sumo and the cellular stress response. Cell Div. 2015, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zou, T.; Zhang, J. Diverse and pivotal roles of neddylation in metabolism and immunity. FEBS J. 2021, 288, 3884–3912. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation—A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.O.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Pan, Z.-Q.; Kentsis, A.; Dias, D.C.; Yamoah, K.; Wu, K. Nedd8 on cullin: Building an expressway to protein destruction. Oncogene 2004, 23, 1985–1997. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.T.; Ayrault, O.; Hunt, H.W.; Taherbhoy, A.M.; Duda, D.M.; Scott, D.C.; Borg, L.A.; Neale, G.; Murray, P.J.; Roussel, M.F.; et al. E2-RING Expansion of the NEDD8 Cascade Confers Specificity to Cullin Modification. Mol. Cell 2009, 33, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zou, J.; Littlejohn, R.; Liu, J.; Su, H. Neddylation, an Emerging Mechanism Regulating Cardiac Development and Function. Front. Physiol. 2020, 11, 1624. [Google Scholar] [CrossRef]
- Duda, D.M.; Borg, L.A.; Scott, D.C.; Hunt, H.W.; Hammel, M.; Schulman, B.A. Structural Insights into NEDD8 Activation of Cullin-RING Ligases: Conformational Control of Conjugation. Cell 2008, 134, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Baek, K.; Scott, D.C.; Schulman, B.A. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr. Opin. Struct. Biol. 2021, 67, 101–109. [Google Scholar] [CrossRef]
- Santonico, E. New Insights into the Mechanisms Underlying NEDD8 Structural and Functional Specificities. In Ubiquitin Proteasome System—Current Insights into Mechanism Cellular Regulation and Disease; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Von Andrian, U.H.; Mackay, C.R. T-Cell Function and Migration—Two Sides of the Same Coin. N. Engl. J. Med. 2000, 343, 1020–1034. [Google Scholar] [CrossRef]
- Mescher, M.F.; Curtsinger, J.M.; Agarwal, P.; Casey, K.A.; Gerner, M.; Hammerbeck, C.D.; Popescu, F.; Xiao, Z. Signals required for programming effector and memory development by CD8 + T cells. Immunol. Rev. 2006, 211, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, J.; Yan, H.; Huang, J.; Wang, F.; Liu, T.; Zeng, L.; Zhou, F. ISGylation in Innate Antiviral Immunity and Pathogen Defense Responses: A Review. Front. Cell Dev. Biol. 2021, 9, 3196. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, K.; Moura, R.F.; Granvik, M.; Igoillo-Esteve, M.; Hohmeier, H.E.; Hendrickx, N.; Newgard, C.B.; Waelkens, E.; Cnop, M.; Schuit, F. Ubiquitin Fold Modifier 1 (UFM1) and Its Target UFBP1 Protect Pancreatic Beta Cells from ER Stress-Induced Apoptosis. PLoS ONE 2011, 6, e18517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.-R.; Byeon, Y.; Kim, D.; Park, S.-G. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 2020, 52, 750–761. [Google Scholar] [CrossRef]
- Cammann, C.; Schraven, B.; Lindquist, J.A. T cell Metabolism—Regulating Energy. J. Clin. Cell. Immunol. 2013, 01, S12. [Google Scholar] [CrossRef] [Green Version]
- Cammann, C.; Rath, A.; Reichl, U.; Lingel, H.; Brunner-Weinzierl, M.; Simeoni, L.; Schraven, B.; Lindquist, J.A. Early changes in the metabolic profile of activated CD8(+) T cells. BMC Cell Biol. 2016, 17, 28. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.C.; Iwashima, M.; Turck, C.W.; Weiss, A. ZAP-70: A 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell 1992, 71, 649–662. [Google Scholar] [CrossRef]
- Bachmaier, K.; Krawczyk, C.M.; Kozieradzki, I.; Kong, Y.-Y.; Sasaki, T.; Oliveira-Dos-Santos, A.; Mariathasan, S.; Bouchard, D.; Wakeham, A.; Itie, A.; et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature 2000, 403, 211–216. [Google Scholar] [CrossRef]
- Chiang, Y.J.; Kole, H.K.; Brown, K.; Naramura, M.; Fukuhara, S.; Hu, R.-J.; Jang, I.K.; Gutkind, J.S.; Shevach, E.M.; Gu, H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 2000, 403, 216–220. [Google Scholar] [CrossRef]
- Venuprasad, K. Cbl-b and Itch: Key Regulators of Peripheral T-cell Tolerance. Cancer Res. 2010, 70, 3009–3012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, N.; Miyake, S.; Reddi, A.L.; Douillard, P.; Ghosh, A.K.; Dodge, I.L.; Zhou, P.; Fernandes, N.D.; Band, H. Negative regulation of Lck by Cbl ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2002, 99, 3794–3799. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Elly, C.; Qiu, L.; Altman, A.; Liu, Y.-C. A direct interaction between the adaptor protein Cbl-b and the kinase Zap-70 induces a positive signal in T cells. Curr. Biol. 1999, 9, 203–210. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Jeon, M.-S.; Liao, L.; Yang, C.; Elly, C.; Yates, J.R., III; Liu, Y.-C. K33-Linked Polyubiquitination of T Cell Receptor-Zeta Regulates Proteolysis-Independent T Cell Signaling. Immunity 2010, 33, 60–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Chen, T.; Li, X.; Yu, Z.; Tang, S.; Wang, C.; Gu, Y.; Liu, Y.; Xu, S.; Li, W.; et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8+ T cell activation. Nat. Immunol. 2015, 16, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bárdos, T.; Li, D.; Gál, I.; Vermes, C.; Xu, J.; Mikecz, K.; Finnegan, A.; Lipkowitz, S.; Glant, T.T. Cutting edge: Regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J. Immunol. 2002, 169, 2236–2240. [Google Scholar] [CrossRef] [Green Version]
- Balagopalan, L.; Barr, V.A.; Sommers, C.L.; Barda-Saad, M.; Goyal, A.; Isakowitz, M.S.; Samelson, L.E. c-Cbl-Mediated Regulation of LAT-Nucleated Signaling Complexes. Mol. Cell. Biol. 2007, 27, 8622–8636. [Google Scholar] [CrossRef] [Green Version]
- Reiley, W.W.; Zhang, M.; Jin, W.; Losiewicz, M.; Donohue, K.B.; Norbury, C.C.; Sun, S.-C. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat. Immunol. 2006, 7, 411–417. [Google Scholar] [CrossRef]
- Naik, E.; Webster, J.D.; Devoss, J.; Liu, J.; Suriben, R.; Dixit, V.M. Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J. Exp. Med. 2014, 211, 1947–1955. [Google Scholar] [CrossRef]
- Jahan, A.S.; Lestra, M.; Swee, L.K.; Fan, Y.; Lamers, M.M.; Tafesse, F.G.; Theile, C.S.; Spooner, E.; Bruzzone, R.; Ploegh, H.L.; et al. Usp12 stabilizes the T-cell receptor complex at the cell surface during signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E705–E714. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Wang, H.; Xiao, Y.; Jin, J.; Chang, J.-H.; Zou, Q.; Xie, X.; Cheng, X.; Sun, S.-C. Otud7b facilitates T cell activation and inflammatory responses by regulating Zap70 ubiquitination. J. Exp. Med. 2016, 213, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.-L.; Liang, J.-Q.; Gong, B.-N.; Xie, J.-J.; Yi, Y.-T.; Lan, X.; Li, Y. T Cell Receptor (TCR)-Induced PLC-γ1 Sumoylation via PIASxβ and PIAS3 SUMO E3 Ligases Regulates the Microcluster Assembly and Physiological Function of PLC-γ1. Front. Immunol. 2019, 10, 314. [Google Scholar] [CrossRef]
- Xiong, Y.; Yi, Y.; Wang, Y.; Yang, N.; Rudd, C.E.; Liu, H. Ubc9 Interacts with and SUMOylates the TCR Adaptor SLP-76 for NFAT Transcription in T Cells. J. Immunol. 2019, 203, 3023–3036. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The Immunological Synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-D.; Gong, Y.; Chen, Z.-L.; Gong, B.-N.; Xie, J.-J.; Zhong, C.-Q.; Wang, Q.-L.; Diao, L.-H.; Xu, A.; Han, J.; et al. TCR-induced sumoylation of the kinase PKC-θ controls T cell synapse organization and T cell activation. Nat. Immunol. 2015, 16, 1195–1203. [Google Scholar] [CrossRef]
- Best, S.; Lam, V.; Liu, T.; Bruss, N.; Kittai, A.; Danilova, O.V.; Murray, S.; Berger, A.; Pennock, N.D.; Lind, E.F.; et al. Immunomodulatory effects of pevonedistat, a NEDD8-activating enzyme inhibitor, in chronic lymphocytic leukemia-derived T cells. Leukemia 2021, 35, 156–168. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Gerondakis, S.; Siebenlist, U. Roles of the NF-kappaB Pathway in Lymphocyte Development and Function. Cold Spring Harb. Perspect. Biol. 2010, 2, a000182. [Google Scholar] [CrossRef] [Green Version]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 2012, 246, 77–94. [Google Scholar] [CrossRef]
- Yaron, A.; Hatzubai, A.; Davis, M.; Lavon, I.; Amit, S.; Manning, A.M.; Andersen, J.S.; Mann, M.; Mercurio, F.; Ben-Neriah, Y. Identification of the receptor component of the IκBα-ubiquitin ligase. Nature 1998, 396, 590–594. [Google Scholar] [CrossRef]
- Ruefli-Brasse, A.A.; French, D.M.; Dixit, V.M. Regulation of NF-κB-Dependent Lymphocyte Activation and Development by Paracaspase. Science 2003, 302, 1581–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egawa, T.; Albrecht, B.; Favier, B.; Sunshine, M.J.; Mirchandani, K.; O’Brien, W.; Thome, M.; Littman, D.R. Requirement for CARMA1 in Antigen Receptor-Induced NF-κB Activation and Lymphocyte Proliferation. Curr. Biol. 2003, 13, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Scharschmidt, E.; Wegener, E.; Heissmeyer, V.; Rao, A.; Krappmann, D. Degradation of Bcl10 Induced by T-Cell Activation Negatively Regulates NF-κB Signaling. Mol. Cell. Biol. 2004, 24, 3860–3873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Jin, H.-S.; Liu, Y.-C. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9433–9438. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wang, P.; Zhao, J.; Tan, Y.; Sheng, J.; He, S.; Du, X.; Huang, Y.; Yang, Y.; Li, J.; et al. USP12 promotes CD4(+) T cell responses through deubiquitinating and stabilizing BCL10. Cell Death Differ. 2021, 28, 2857–2870. [Google Scholar] [CrossRef]
- Sun, L.; Deng, L.; Ea, C.-K.; Xia, Z.-P.; Chen, Z.J. The TRAF6 Ubiquitin Ligase and TAK1 Kinase Mediate IKK Activation by BCL10 and MALT1 in T Lymphocytes. Mol. Cell 2004, 14, 289–301. [Google Scholar] [CrossRef]
- Zhou, H.; Wertz, I.; O’Rourke, K.; Ultsch, M.; Seshagiri, S.; Eby, M.; Xiao, W.; Dixit, V.M. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 2003, 427, 167–171. [Google Scholar] [CrossRef]
- King, C.G.; Kobayashi, T.; Cejas, P.J.; Kim, T.; Yoon, K.; Kim, G.K.; Chiffoleau, E.; Hickman, S.P.; Walsh, P.T.; Turka, L.A.; et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat. Med. 2006, 12, 1088–1092. [Google Scholar] [CrossRef]
- Düwel, M.; Welteke, V.; Oeckinghaus, A.; Baens, M.; Kloo, B.; Ferch, U.; Darnay, B.G.; Ruland, J.; Marynen, P.; Krappmann, D. A20 Negatively Regulates T Cell Receptor Signaling to NF-κB by Cleaving Malt1 Ubiquitin Chains. J. Immunol. 2009, 182, 7718–7728. [Google Scholar] [CrossRef]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces MALT1 paracaspase–mediated cleavage of the NF-κB inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef]
- Hu, J.; Yi, S.; Wang, C.; Zhang, Y.; Tang, J.; Huang, X.; Yang, L.; Yang, J.; Li, H. A20 Inhibits Intraocular Inflammation in Mice by Regulating the Function of CD4+T Cells and RPE Cells. Front. Immunol. 2021, 11, 603939. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Chang, J.-H.; Xiao, Y.; Brittain, G.C.; Yu, J.; Zhou, X.; Wang, Y.-H.; Cheng, X.; Li, P.; et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat. Immunol. 2011, 12, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Tournier, C.; Davis, R.J.; Flavell, R.A. Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J. 1999, 18, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Q.; Jin, J.; Hu, H.; Li, H.S.; Romano, S.; Xiao, Y.; Nakaya, M.; Zhou, X.; Cheng, X.; Yang, P.; et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat. Immunol. 2014, 15, 562–570. [Google Scholar] [CrossRef] [Green Version]
- Hetfeld, B.K.; Helfrich, A.; Kapelari, B.; Scheel, H.; Hofmann, K.; Guterman, A.; Glickman, M.; Schade, R.; Kloetzel, P.-M.; Dubiel, W. The Zinc Finger of the CSN-Associated Deubiquitinating Enzyme USP15 Is Essential to Rescue the E3 Ligase Rbx1. Curr. Biol. 2005, 15, 1217–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, A.; Glöckner-Pagel, J.; Vaeth, M.; Schumann, J.E.; Buttmann, M.; Bopp, T.; Schmitt, E.; Serfling, E.; Berberich-Siebelt, F. Sumoylation of the Transcription Factor NFATc1 Leads to Its Subnuclear Relocalization and Interleukin-2 Repression by Histone Deacetylase. J. Biol. Chem. 2009, 284, 10935–10946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Qureischi, M.; Dietz, L.; Vaeth, M.; Vallabhapurapu, S.D.; Klein-Hessling, S.; Klein, M.; Liang, C.; König, A.; Serfling, E.; et al. Lack of NFATc1 SUMOylation prevents autoimmunity and alloreactivity. J. Exp. Med. 2021, 218, e20181853. [Google Scholar] [CrossRef]
- Fang, D.; Elly, C.; Gao, B.; Fang, N.; Altman, Y.; Joazeiro, C.; Hunter, T.; Copeland, N.; Jenkins, N.; Liu, Y.-C. Dysregulation of T lymphocyte function in itchy mice: A role for Itch in TH2 differentiation. Nat. Immunol. 2002, 3, 281–287. [Google Scholar] [CrossRef]
- Jin, H.-S.; Liao, L.; Park, Y.; Liu, Y.-C. Neddylation pathway regulates T-cell function by targeting an adaptor protein Shc and a protein kinase Erk signaling. Proc. Natl. Acad. Sci. USA 2012, 110, 624–629. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Liu, J.; Pei, Y.; Zhang, Y.; Zhou, D.; Pan, W.; Zhang, J. Neddylation contributes to CD4+ T cell-mediated protective immunity against blood-stage Plasmodium infection. PLoS Pathog. 2018, 14, e1007440. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.X.; Pan, W.; Zheng, L.; Zhong, X.-P.; Tan, L.; Liang, Z.; He, J.; Feng, P.; Zhao, Y.; Qiu, Y.-R. Thymic Epithelial Cells Contribute to Thymopoiesis and T Cell Development. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Weih, F.; Carrasco, D.; Durham, S.K.; Barton, D.S.; Rizzo, C.A.; Ryseck, R.-P.; Lira, S.A.; Bravo, R. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-κB/Rel family. Cell 1995, 80, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Shimo, Y.; Yanai, H.; Qin, J.; Ohshima, D.; Maruyama, Y.; Asaumi, Y.; Kitazawa, J.; Takayanagi, H.; Penninger, J.; et al. The Tumor Necrosis Factor Family Receptors RANK and CD40 Cooperatively Establish the Thymic Medullary Microenvironment and Self-Tolerance. Immunity 2008, 29, 423–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkly, L.; Hession, C.; Ogata, L.; Reilly, C.; Marconl, L.A.; Olson, D.; Tizard, R.; Gate, R.; Lo, D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature 1995, 373, 531–536. [Google Scholar] [CrossRef]
- Naito, A.; Azuma, S.; Tanaka, S.; Miyazaki, T.; Takaki, S.; Takatsu, K.; Nakao, K.; Nakamura, K.; Katsuki, M.; Yamamoto, T.; et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis inTRAF6-deficient mice. Genes Cells 1999, 4, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Uchida, D.; Hatakeyama, S.; Matsushima, A.; Han, H.; Ishido, S.; Hotta, H.; Kudoh, J.; Shimizu, N.; Doucas, V.; Nakayama, K.I.; et al. AIRE Functions as an E3 Ubiquitin Ligase. J. Exp. Med. 2004, 199, 167–172. [Google Scholar] [CrossRef]
- Perniola, R. Twenty Years of AIRE. Front. Immunol. 2018, 9, 98. [Google Scholar] [CrossRef]
- Lukasiak, S.; Schiller, C.; Oehlschlaeger, P.; Schmidtke, G.; Krause, P.; Legler, D.F.; Autschbach, F.; Schirmacher, P.; Breuhahn, K.; Groettrup, M. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene 2008, 27, 6068–6074. [Google Scholar] [CrossRef] [Green Version]
- Buerger, S.; Herrmann, V.L.; Mundt, S.; Trautwein, N.; Groettrup, M.; Basler, M. The Ubiquitin-like Modifier FAT10 Is Selectively Expressed in Medullary Thymic Epithelial Cells and Modifies T Cell Selection. J. Immunol. 2015, 195, 4106–4116. [Google Scholar] [CrossRef] [Green Version]
- Sanna, S.; Pitzalis, M.; Zoledziewska, M.; Zara, I.; Sidore, C.; Murru, R.; Whalen, M.B.; Busonero, F.; Maschio, A.; Costa, G.; et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat. Genet. 2010, 42, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Fujiwara, Y.; Wang, H.-Y.; Kitao, M.; Hayashi, C.; Someya, T.; Kanamori, M.; Oiso, Y.; Tajima, N.; Yamada, Y.; et al. Identification and functional analysis of CBLB mutations in type 1 diabetes. Biochem. Biophys. Res. Commun. 2008, 368, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Eggebrecht, N.; Staufner, C.; Kathemann, S.; Elgizouli, M.; Kopajtich, R.; Prokisch, H.; Lainka, E. Mutation in ITCH Gene Can Cause Syndromic Multisystem Autoimmune Disease with Acute Liver Failure. Pediatrics 2019, 143, e20181554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.T.; Wang, Z.-E.; Shen, L.; Schroeder, A.; Eckalbar, W.; Weiss, A. Cbl-b deficiency prevents functional but not phenotypic T cell anergy. J. Exp. Med. 2021, 218, e20202477. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Anstadt, E.J.; Khanna, K.M.; Clark, R.B. Cbl-b-deficient mice express alterations in trafficking-related molecules but retain sensitivity to the multiple sclerosis therapeutic agent, FTY720. Clin. Immunol. 2015, 158, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Martín, D.; Ibarra-Sánchez, M.; Romo-Tena, J.; Cruz-Ruíz, J.; Esparza-López, J.; Galindo-Campos, M.; Díaz-Zamudio, M.; Alcocer-Varela, J. Casitas B Lineage Lymphoma b Is a Key Regulator of Peripheral Tolerance in Systemic Lupus Erythematosus. Arthritis Care Res. 2012, 65, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Kobashigawa, Y.; Tomitaka, A.; Kumeta, H.; Noda, N.N.; Yamaguchi, M.; Inagaki, F. Autoinhibition and phosphorylation-induced activation mechanisms of human cancer and autoimmune disease-related E3 protein Cbl-b. Proc. Natl. Acad. Sci. USA 2011, 108, 20579–20584. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Qiao, G.; Tang, J.; Tang, R.; Guo, H.; Warwar, S.; Langdon, W.Y.; Tao, L.; Zhang, J. Protein Tyrosine Phosphatase SHP-1 Modulates T Cell Responses by Controlling Cbl-b Degradation. J. Immunol. 2015, 195, 4218–4227. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.P.; Vieyra-Garcia, P.A.; Wagner, K.; Penninger, J.; Wolf, P. Cbl-b deficiency provides protection against UVB-induced skin damage by modulating inflammatory gene signature. Cell Death Dis. 2018, 9, 835. [Google Scholar] [CrossRef] [Green Version]
- Loeser, S.; Loser, K.; Bijker, M.S.; Rangachari, M.; Van Der Burg, S.H.; Wada, T.; Beissert, S.; Melief, C.J.; Penninger, J.M. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J. Exp. Med. 2007, 204, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, P.; Han, L.; Guo, Z.; Wang, X.; Chen, Z.; Nie, J.; Yin, S.; Piccioni, M.; Tsun, A.; et al. Cutting Edge: Ubiquitin-Specific Protease 4 Promotes Th17 Cell Function under Inflammation by Deubiquitinating and Stabilizing RORγt. J. Immunol. 2015, 194, 4094–4097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, J.; Schlüter, D.; Wang, X. Deubiquitinating enzymes (DUBs): DoUBle-edged swords in CNS autoimmunity. J. Neuroinflamm. 2020, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Brackett, C.M.; Blagg, B.S.J. Current Status of SUMOylation Inhibitors. Curr. Med. Chem. 2020, 27, 3892–3912. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Langston, S.P.; Grossman, S.; England, D.; Afroze, R.; Bence, N.; Bowman, D.; Bump, N.; Chau, R.; Chuang, B.-C.; Claiborne, C.; et al. Discovery of TAK-981, a First-in-Class Inhibitor of SUMO-Activating Enzyme for the Treatment of Cancer. J. Med. Chem. 2021, 64, 2501–2520. [Google Scholar] [CrossRef]
- Yoshimura, C.; Muraoka, H.; Ochiiwa, H.; Tsuji, S.; Hashimoto, A.; Kazuno, H.; Nakagawa, F.; Komiya, Y.; Suzuki, S.; Takenaka, T.; et al. TAS4464, A Highly Potent and Selective Inhibitor of NEDD8-Activating Enzyme, Suppresses Neddylation and Shows Antitumor Activity in Diverse Cancer Models. Mol. Cancer Ther. 2019, 18, 1205–1216. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cammann, C.; Israel, N.; Slevogt, H.; Seifert, U. Recycling and Reshaping—E3 Ligases and DUBs in the Initiation of T Cell Receptor-Mediated Signaling and Response. Int. J. Mol. Sci. 2022, 23, 3424. https://doi.org/10.3390/ijms23073424
Cammann C, Israel N, Slevogt H, Seifert U. Recycling and Reshaping—E3 Ligases and DUBs in the Initiation of T Cell Receptor-Mediated Signaling and Response. International Journal of Molecular Sciences. 2022; 23(7):3424. https://doi.org/10.3390/ijms23073424
Chicago/Turabian StyleCammann, Clemens, Nicole Israel, Hortense Slevogt, and Ulrike Seifert. 2022. "Recycling and Reshaping—E3 Ligases and DUBs in the Initiation of T Cell Receptor-Mediated Signaling and Response" International Journal of Molecular Sciences 23, no. 7: 3424. https://doi.org/10.3390/ijms23073424