1. Introduction
Proteinic L-lysine (Lys) residues undergo multiple post-translational modifications (PTM) including glycation of the ε-amine (
Nε) group of Lys with various species. The advanced glycation end-products (AGEs) of these reactions are
Nε-(1-carboxymethyl)-L-lysine (CML) and
Nε-(1-carboxyethyl)-L-lysine (CEL). CML and CEL belong to the best-investigated AGEs. AGEs are widely used as markers for carbohydrate metabolism [
1]. The concentration of circulating and excretory CML and CEL serves as a measure for the extent of the glycation of Lys residues in generally unknown proteins. In healthy humans, free glyoxal (GO) and methylglyoxal (MGO) occur in blood in the mid-to-upper nM-range [
2,
3,
4] and are excreted in the urine in the very low range of 0.1 to 5.6 pmol/mmol creatinine [
3,
5]. Glucose as an origin of MGO is questionable [
3]. The circulating and excretory concentrations of CML and CEL in healthy humans are of the order of 2 µM, and their creatinine-corrected excretion rate is of the order of 2 µmol/mmol creatinine [
6]. Thus, the concentrations of MGO and GO in human biological fluids are considerably lower than those of CEL and CML. Based on the concentration of CML and CEL rather than of GO and MGO,
Nε-glycation of proteins is considered to play a significant role in various diseases, notably including diabetes and cardiovascular diseases [
7,
8].
Over the past five decades, the formation of CML, CEL and of other Lys, arginine (Arg) and cysteine (Cys) derived AGEs and their precursors in enzymes, peptides and proteins including human and bovine albumin were often investigated using various glycating species such as reducing sugars and their glycolytic products GO and MGO. Free Lys, Arg and Cys are relatively abundant amino acids. Yet, unlike with proteins, the reactions of MGO and GO with soluble Lys and other amino acids are much less often investigated, presumably because of the general thought that only proteins are glycated. Pioneering work found that MGO was polymerized in the presence of Lys [
9,
10]. Moreover, MGO and GO were found to react with many amino acids in aqueous buffers in part with remarkable differences [
11]. Lys was among the amino acids, which react with MGO and GO slowly, with MGO being more reactive than GO [
11]. Yet, in that study, neither CML nor CEL were measured. Being highly reactive, aldehydes, MGO, GO and other carbonylic compounds may react with all of their functionalities of soluble amino acids in aqueous solutions [
11]. Arg and Cys were found to be much more reactive than Lys, suggesting that not only the nucleophilic strength, for instance of the SH group of Cys, but also other structural features, for instance the formation of cyclic compounds with the guanidine group of Arg, may influence their reactions with MGO and GO.
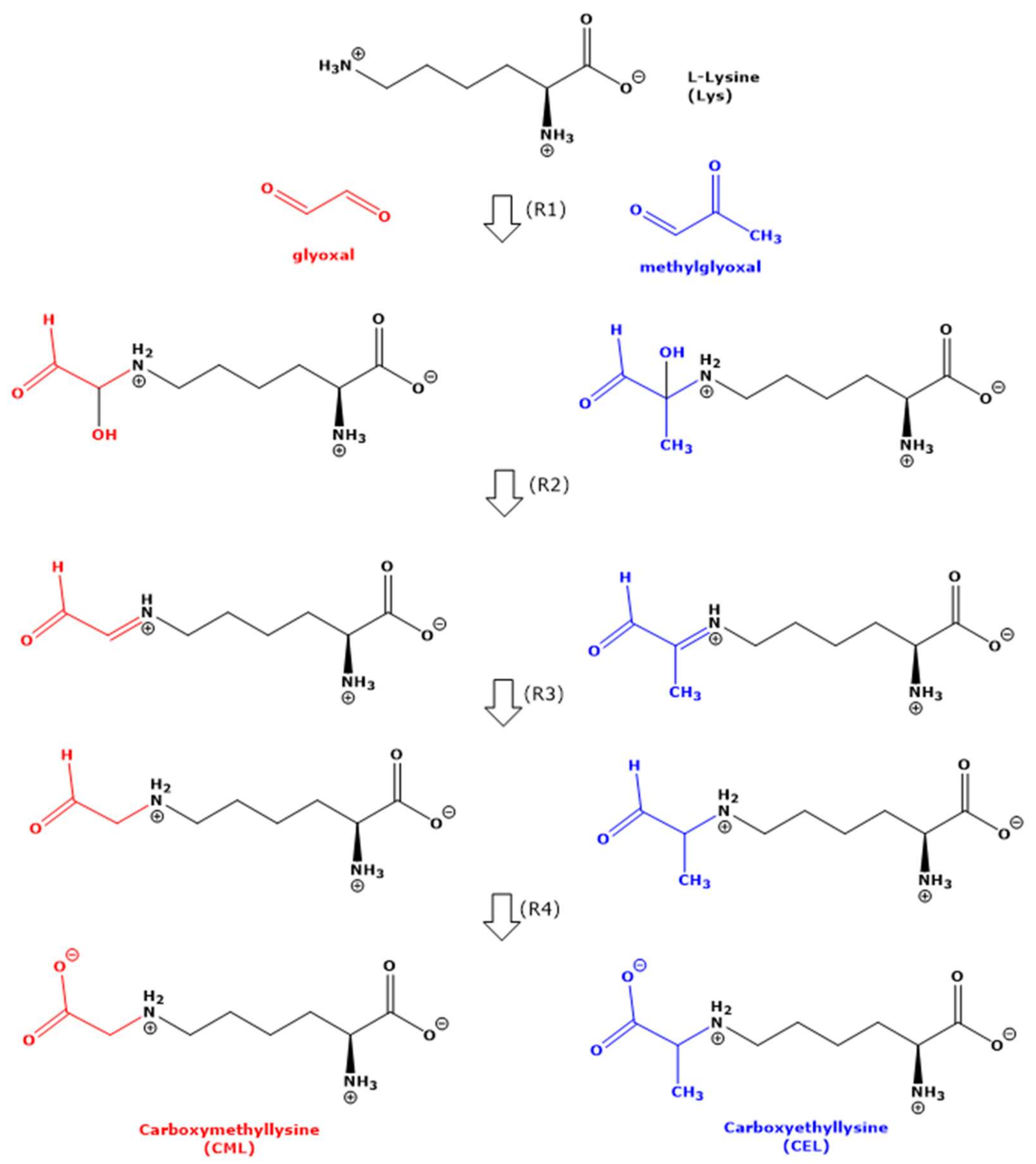
The first reaction product of aldehydes such as MGO and GO with amine groups including those of amino acids is considered to be a hydroxyl group-containing intermediate (i.e., -CH(OH)-NH-), which subsequently loses a molecule of water to form a Schiff base (-CH=N-) [
12] (
Figure 1). Schiff bases are assumed to undergo additional reactions and rearrangements finally to deliver low-molecular-mass reaction products, i.e., CML in the case of Lys with GO, and CEL in the case of Lys with MGO (
Figure 1). CML and CEL are not Schiff bases but contain -CH
2-NH- and -CH(CH
3)-NH- as terminal groups, respectively. In vitro, the imine group of Schiff bases is chemically reduced using reagents such as NaBH
4 [
13]. In vivo, the reduction in the imine group of Schiff bases are likely to be reduced by enzymes that use NAD(P)H as the electron donor. Finally, the terminal aldehyde group needs to be oxidized to form the terminal carboxylic group of the amino acid (
Figure 1). The formation of Schiff bases from the reaction of Lys with MGO is not always formulated and discussed [
4].
In the present study, we investigated the reaction of free, non-proteinic Lys with GO alone, with MGO alone and with mixtures of GO and MGO in phosphate buffer of neutral pH at two different conditions. A temperature of 37 °C was used to simulate in vivo conditions, and the temperature of 80 °C was used to simulate in vitro conditions, which are often in the context of nutrients [
12]. In our study, we used a recently reported gas chromatographic-mass spectrometric (GC-MS) method to study these reactions and to determine the concentration of remaining Lys and the concentrations of CML and CEL. GC-MS was also used to detect potential formation of intermediates including alcohols and Schiff bases of Lys and GO or MGO. Reactions involving GO and MGO were investigated using spectroscopic methods [
14,
15,
16]. In the present study, we also used spectrophotometry in a few kinetic investigations.
3. Discussion
The aim of the present study was to investigate the formation of CML and CEL from free non-proteinic Lys, GO and MGO in an aqueous buffer of pH 7.4 without the use of additional chemical and enzymatic treatments which are commonly used in the research area of AGEs. Thus, in our study, we did not use reducing reagents such as NaBH
4 to reduce intermediate Schiff bases of Lys with GO and MGO. We also did not use any enzymes to oxidize their terminal aldehyde groups to form the dicarboxylic acids CML and CEL. We used a previously reported GC-MS method [
6] for the specific measurement of CML, CEL and Lys in their reaction mixtures.
The absence of the expected aldehydic Schiff bases and the formation of CML and CEL under the experimental conditions of our study suggest that disproportionation and hydride transfer are likely to have occurred in the expected Schiff bases. In the case of Lys (residue R), this could involve a reduction in OCH-CH=N-R (with GO) and in -C(CH
3)=N-R (with MGO) to the OCH-CH
2-NH-R and OCH-C(CH
3)-NH-R groups, respectively, and the oxidation of their terminal OCH-groups to their carboxylic acids HOOC-CH
2-NH-R in the case of CML and HOOC-C(CH
3)-NH-R in the case of CEL. Such a disproportionation was described for free glyoxal to glycolic acid [
14]). Formation of CEL from the reaction of Lys residues of bovine serum albumin (BSA) with MGO was reported in the absence of any reductive and oxidative treatment of reaction products [
17]. To our knowledge, the requirement of reductive and oxidative treatments to convert the aldehydic Schiff bases formed by free Lys and of Lys residues in proteins with GO or GMO is not reported thus far. Yet, our study cannot exclude the formation of the expected aldehydic Schiff bases because the GC-MS method has not been developed for their analyses. The formation of a Schiff base from the reaction of Lys with MGO is rarely addressed, explicitly formulated or discussed in the literature [
4].
Inorganic polyvalent anions including phosphate and carbonate were shown to catalyze the formation of MGO from glyceraldehyde and dihydroxyacetone [
18]. In our study, we used sodium phosphate (100 mM, pH 7.4) to prepare the buffer. It is, therefore, possible that under the experimental conditions of our study, phosphate anions were involved in the formation of CEL and possibly of CML. Yet, we consider that possible effects of phosphate on the reaction of Lys with MGO and GO were constant.
GO and MGO exist in aqueous solutions in their free forms, as monohydrates and dehydrates. In addition, MGO and possibly GO may form polymers in the presence of Lys albeit at much higher concentrations and temperatures than those used in the present study [
9,
10]. In our study, we did not measure MGO and GO, but only qualitatively by spectrophotometry.
Loss of Lys in our study could also be due to the formation of ε-caprolactam from esterified Lys [
19]. Lys could also react with GO and MGO via its α-amine group as well as with its carboxylic group, with the latter presumably yielding lysinate derivatives [
20]. In our study, we did not analyze formation of such Lys derivatives. In some experiments, Lys concentration increased again at higher reaction times. Decomposition of unknown Lys derivatives to Lys may have resulted in such increases. CEL is less stable than CML and decomposes to Lys [
6]. This may have decreased the extent of the actual yield of CEL and increased the concentration of Lys. Lys-containing samples treated with MGO become stronger brownish compared to GO presumably due to the formation of polymeric melanoids [
10]. In our study, we did not further investigate this issue.
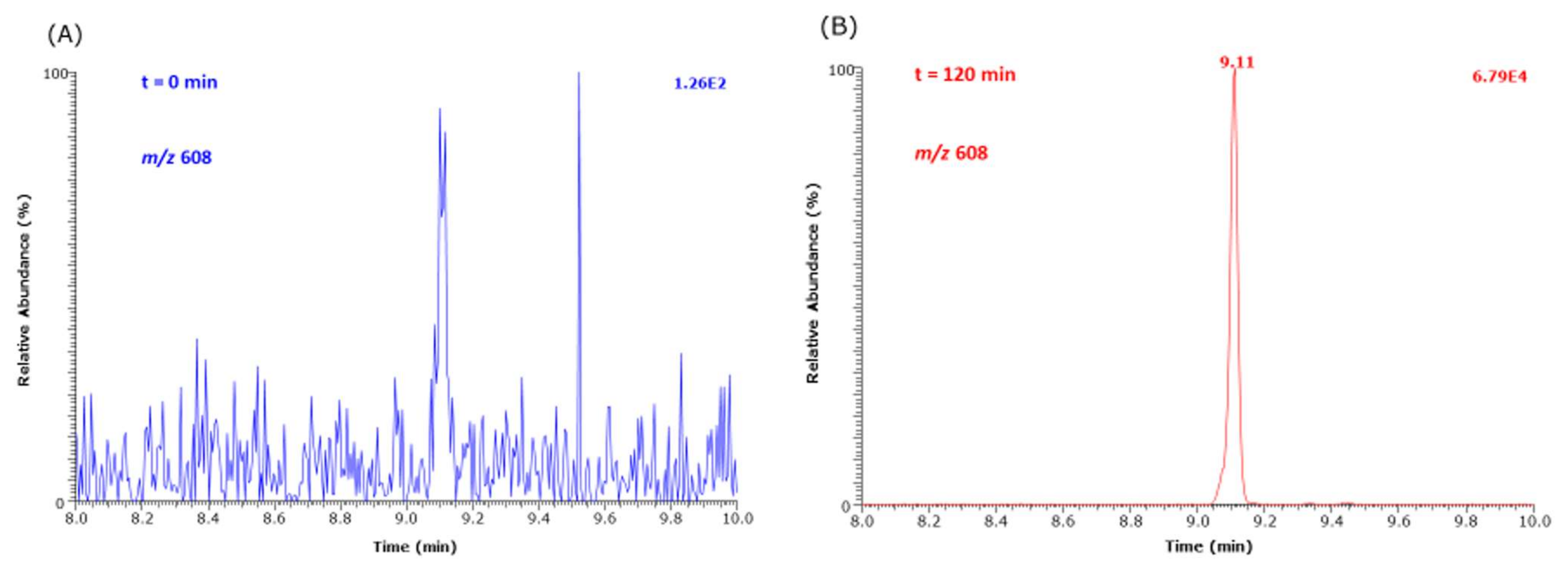
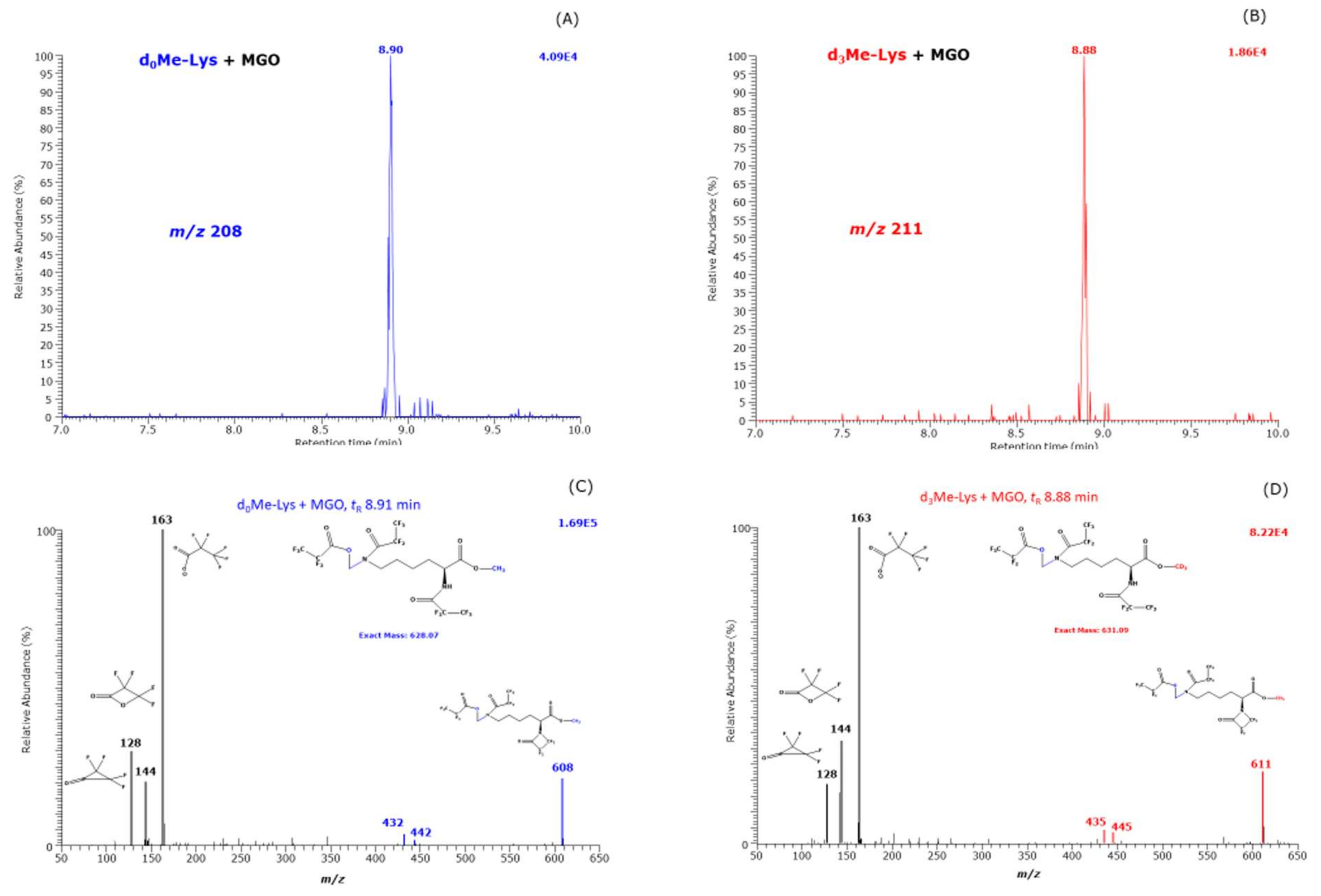
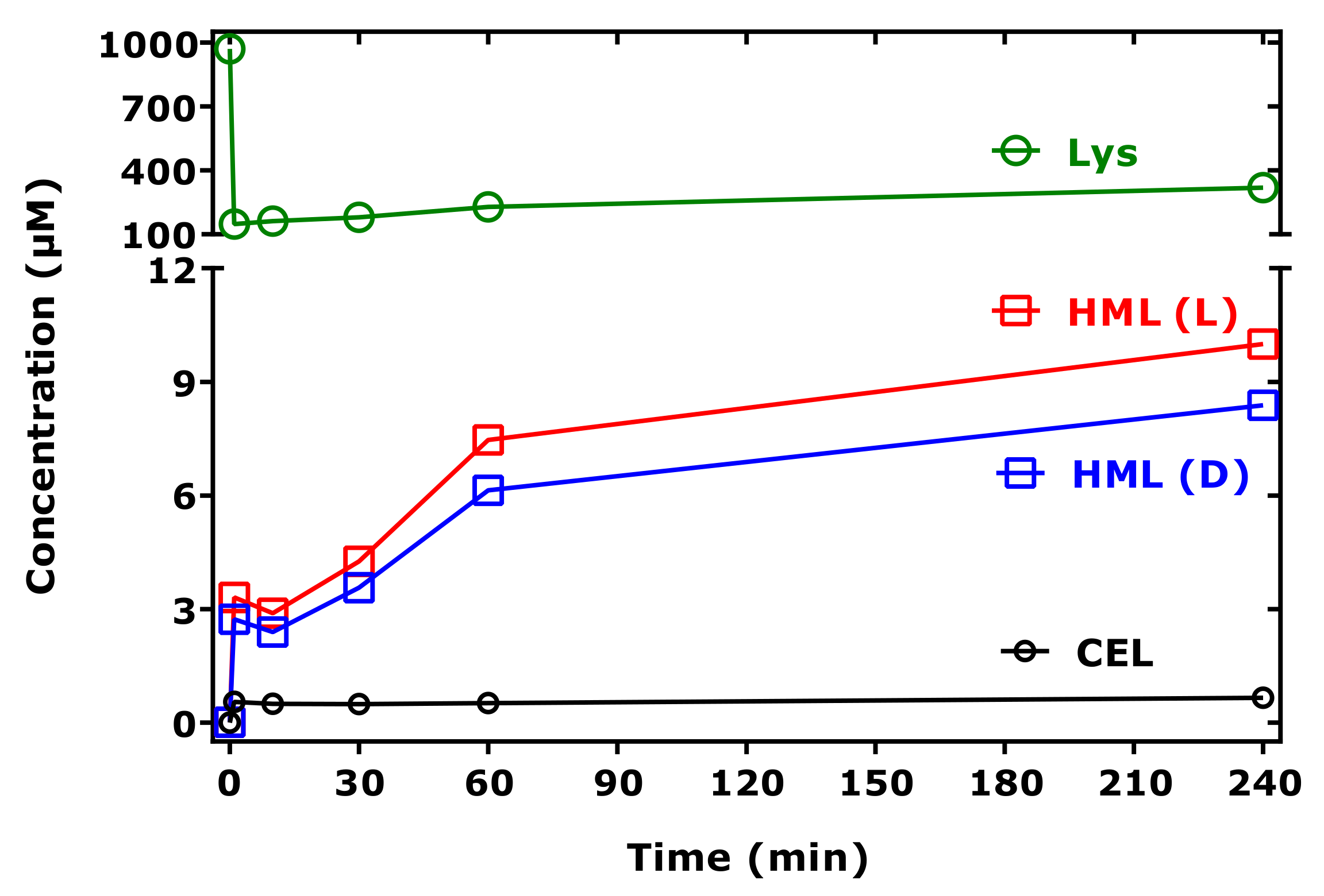
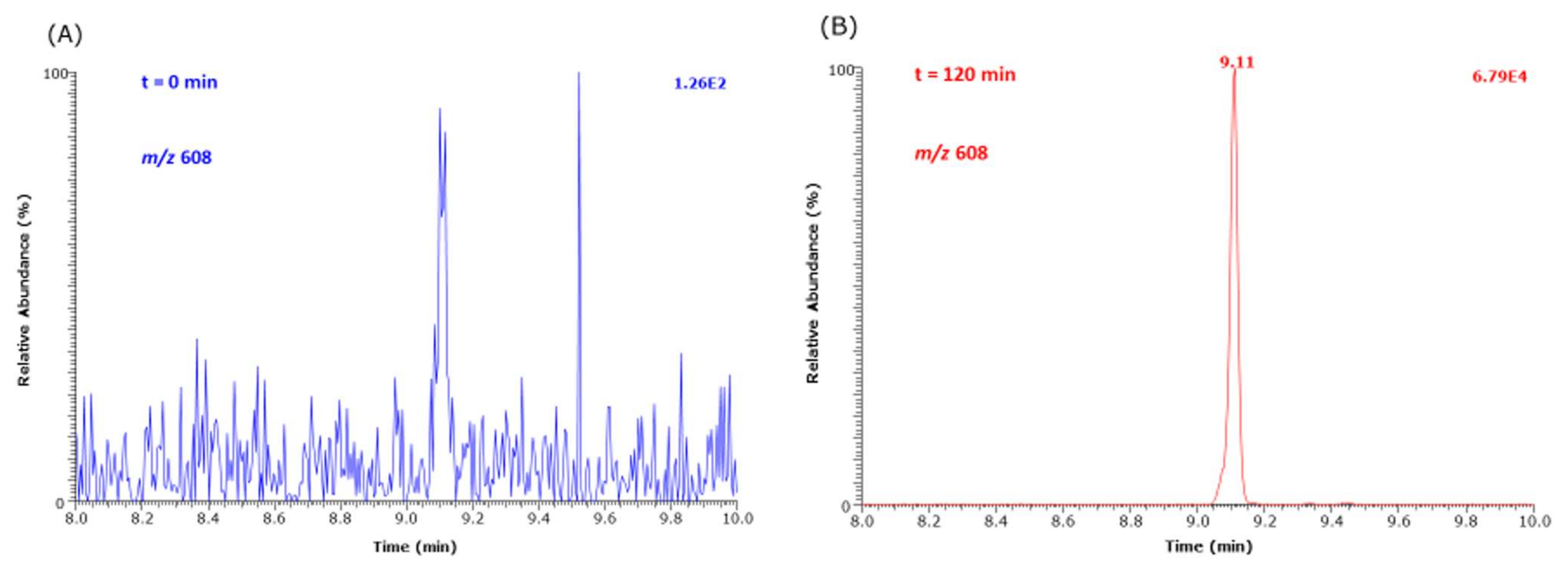
Our study revealed, for the first time, strong evidence of the formation of hydroxymethyl-lysine (HML) in reaction mixtures of Lys and MGO. First, semi-quantitative analyses using
d,
l-5-hydroxy-lysine as a surrogate internal standard suggest that HML is formed at higher yields than CML and CEL. HML is rarely reported in the literature, mostly in the context of formyl-lysine [
21] and hydroxylation of histone-derived monomethyl-lysine [
22]. Synthetic HML was found to be a source of bioavailable Lys in ruminants [
23]. HML can be easily obtained by the reaction of Lys with formaldehyde in Ca(OH)
2-containing aqueous solution [
23]. MGO is considered to form spontaneously formaldehyde and acetaldehyde [
10]. In our study, we did not test this possibility. Due to the presumably large number of chemically reactive species from decomposed, hydrated or polymerized MGO [
24], other reactions in Lys- and MGO-containing aqueous solutions may also occur.
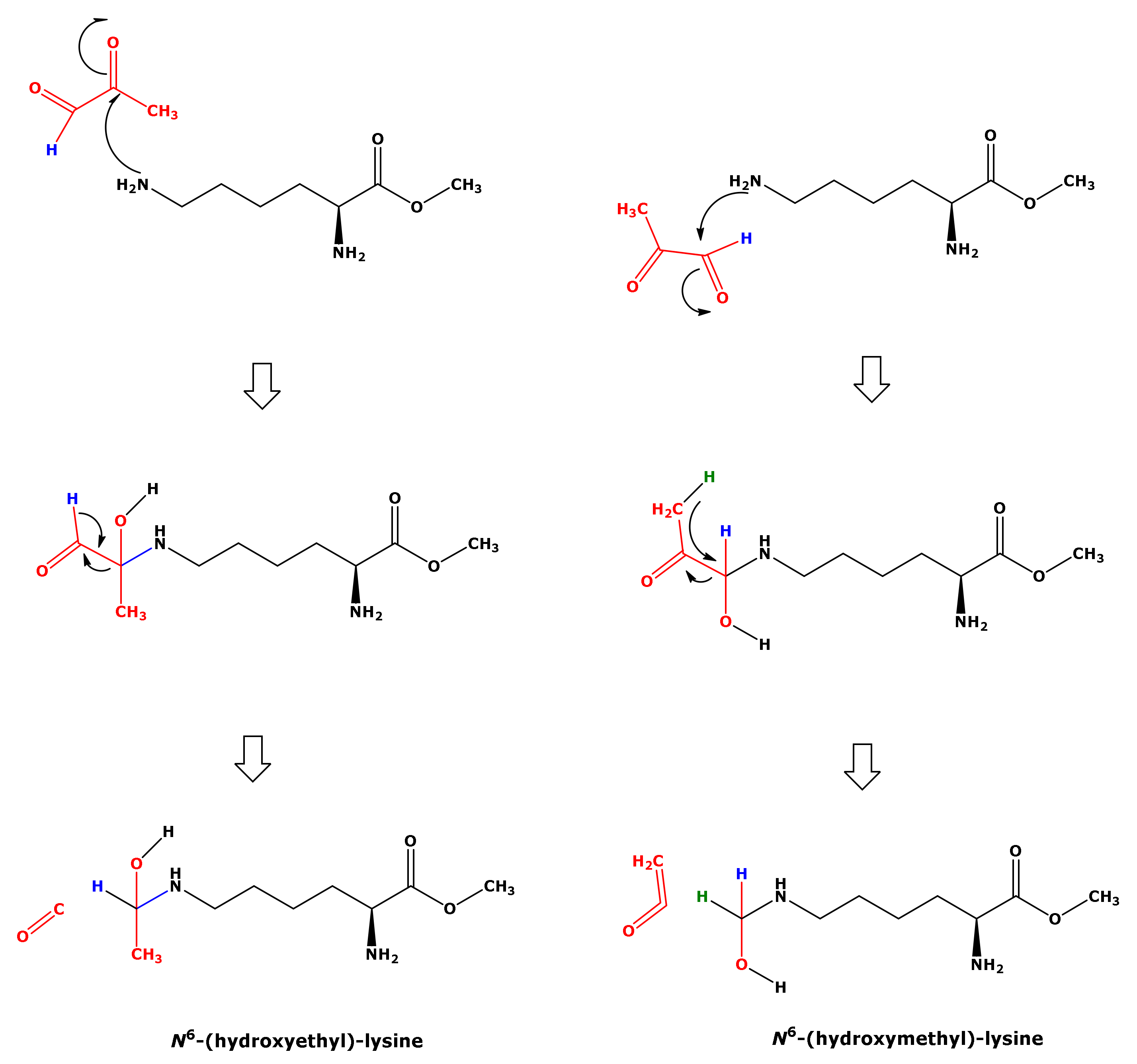
In our study, the reaction of Lys with MGO did not result in the formation of hydroxyethyl-lysine (HEL). We propose a mechanism for the formation of HML from Lys and MGO (
Figure 8). This mechanism includes nucleophilic attack of the terminal amine group of Lys on the aldehyde group of MGO and intramolecular hydride transfer. That Lys and GO did not form HML, analogous to Lys and MGO (
Figure 8), could be due to differences in the leaving groups. The nucleophilic attack of the terminal amine group of Lys on the carbonyl groups of GO and MGO is considered to form hydroxylic compounds as intermediates that subsequently form the corresponding Schiff bases (
Figure 1). In our study, we did not identify any Schiff bases. Yet, this does not exclude their possible formation.
The identification of HML as a reaction product of Lys and MGO may suggest that the first reaction product between Lys and MGO is more stable than that of Lys and GO. This may be an explanation for the formation of CEL with lower yield compared to CML.
GO and MGO react with Lys residues via crosslinking in proteins to form GO-Lys dimer (GOLD) and MGO-Lys dimer (MOLD) [
25]. GOLD and MOLD are permanently positively charged imidazolium compounds and not accessible to GC-MS analysis. We therefore did not analyze such species in the present study.
Our in vitro study may have some limitations. We used very high concentrations of GO and MGO (range, 0.5–10 mM), which are, however, commonly used in similar studies. We performed the experiments at two temperatures to simulate physiological conditions and those often used in the context of nutrients including milk and dairy products. In proteolysates, free Lys is found in mM-concentrations, yet the concentration of GO and MGO are expected to be very low and to lie rather in the lower µM-range. Preliminary experiments using physiologically relevant concentrations of GO and MGO did not allow demonstration of a formation of CML and CEL despite the use of a very sensitive GC-MS method. At high concentrations of Lys, GO and MGO, our study demonstrates the formation of CML, CEL and HML in 100 mM phosphate buffer (pH 7.4) without the need of additional chemicals and enzymes. Yet, the yield of CML, CEL and HML is very low. Whether CML, CEL and HML are formed at higher concentrations from reactions of GO and GMO with Lys in the presence of reductors and oxidants should be a subject of further investigations.
In summary, our study shows that, in absence of any reductors and oxidants, free Lys reacts with GO to form CML, and with MGO to form CML at a very low extent. We demonstrate for the first time that the reaction of Lys methyl ester with MGO, but not the reaction of Lys methyl ester with GO, generates HML. HML seems to be formed at higher concentrations than CML and CEL. We propose mechanisms for the formation of CML, CEL and HML, which include disproportionation and hydride transfer. These mechanisms are supported by the literature but remain to be investigated using appropriate chemicals such as deuterated GO and MGO. Chemical synthesis of HML will facilitate investigations on the physiological occurrence of HML in biological samples and its action on the receptors of AGEs (RAGEs) [
26]. It is worth mentioning that CML and CEL have a very low affinity to RAGEs [
26]. The hydroxy group of HML could increase its affinity to RAGEs. The significance of HML in health and disease, notably in diabetes, remains to be investigated. In case of the occurrence of HML in biological samples at higher concentrations than CML and CEL, HML could turn out to be a better biomarker of glycation.
4. Materials and Methods
4.1. Chemicals, Materials and Reagents
Glyoxal and methylglyoxal were obtained from Sigma-Aldrich (Steinheim, Germany) each as 40 wt.% solution in water. L-Lysine, Nε-(1-carboxymethyl)-L-lysine (chemical purity, 95%) and Nε-(1-carboxyethyl)-L-lysine (chemical purity, 95%) were purchased from Cayman (Ann Arbor, Michigan, USA). Tetradeuterated methanol (CD3OD, 99% at 2H) and pentafluoropropionic anhydride (PFPA) were supplied by Sigma-Aldrich (Steinheim, Germany). Methanol was obtained from Chemsolute (Renningen, Germany). Ethyl acetate (EA) was obtained from Merck (Darmstadt, Germany). Hydrochloric acid (37 wt.%) was purchased from Baker (Deventer, The Netherlands) and was used to prepare the esterification reagent (2 M HCl in CH3OH or 2 M HCl in CD3OD). Glassware for GC-MS (1.5-mL autosampler vials and 0.2-mL microvials) and the fused-silica capillary column Optima 17 (15 m × 0.25 mm I.D., 0.25-µm film thickness) were purchased from Macherey-Nagel (Düren, Germany). Spectrophotometric analyses were performed at room temperature on the spectrophotometer model Specord 50 from Analytik Jena (Jena, Germany) using 1-cm cuvettes (UVetten, Eppendorf, Hamburg, Germany). Scans were performed in the range 190–500 nm (1 s per cycle).
4.2. Procedure for the Reaction of Lys with GO and MGO
The kinetic studies were performed in 100 mM sodium phosphate buffer, pH 7.4, in triplicate on three different days at either 37 °C in a thermostate (model MBT250; Kleinfeld, Gehrden, Germany) or 80 °C in a drying oven (model T6; Heraeus, Hanau, Germany). Lys (100 mM in distilled water), GO and MGO (each 100 mM in phosphate buffer daily prepared) stock solutions were prepared and used to reach desired concentrations in reaction mixtures. The incubation mixtures (500 µL final volume) in Eppendorf tubes contained Lys, GO, MGO or an equimolar mixture of GO and MGO at varying concentrations (0.5, 2.5, 5, 7.5, 10 mM) as specified in the Results section. If not otherwise specified, Lys was first added to the reaction mixtures which were then left to equilibrate at 37 °C of 80 °C for 30 min. Reactions were started with the addition of GO, MGO or GO plus MGO (varying volumes in the range 2.5 to 50-µL aliquots of their 100 mM solutions). The first samples were taken immediately after the Lys addition, then transferred into autosampler glass vials, and the liquid was evaporated to dryness under a stream of nitrogen glass. The incubation time of this sample corresponds to time zero (0 min). Subsequently, each 10-µL sample was taken from the incubation mixtures at 1, 10, 30, 60 and 240 min, transferred to glass vials and evaporated therein to dryness under a stream of nitrogen.
The residues obtained by solvent evaporation were subjected to a two-step derivatization procedure as described elsewhere [
6]. They were reconstructed in 100-µL aliquots of a 2 M HCl/CH
3OH solution; the glass vials were tightly sealed and heated for 60 min at 80 °C to prepare methyl esters of amino acids. After cooling to room temperature, the samples were spiked with 10-µL aliquots of newly synthesized trideuteromethyl esters of Lys, CML and CEL using a 2 M HCl/CD
3OD solution used as the internal standards. In quantitative analyses, the final concentrations of the internal standards were 500 µM for d
3Me-Lys, 10 µM for d
6Me-CEL and 10 µM for d
6Me-CML. After cooling to room temperature and sample evaporation to dryness under a stream of nitrogen, 100-µL aliquots of a freshly prepared PFPA solution in ethyl acetate (PFPA-EA, 1:4, v/v) were added; the glass vials were tightly sealed and heated for 30 min at 65 °C to prepare
N-pentafluoropropionic amides of the methyl esters (Me-PFP) of amino acids. After cooling to room temperature and sample evaporation to dryness under a stream of nitrogen gas, the residues were treated first with 200-µL aliquots of 400 mM borate buffer, pH 8.5, and immediately thereafter with 200-µL aliquots of toluene. The mixtures were immediately vortex-mixed for 60 s and centrifuged (4000×
g, 5 min, 18 °C) to remove acidic components and to extract the Me-PFP derivatives of the reaction products into toluene. Finally, aliquots (150 µL) of the upper organic phase were transferred into 1.5-mL autosampler glass vials equipped with 200-µL microinserts; the samples were sealed and subjected to quantitative GC-MS analysis as described below.
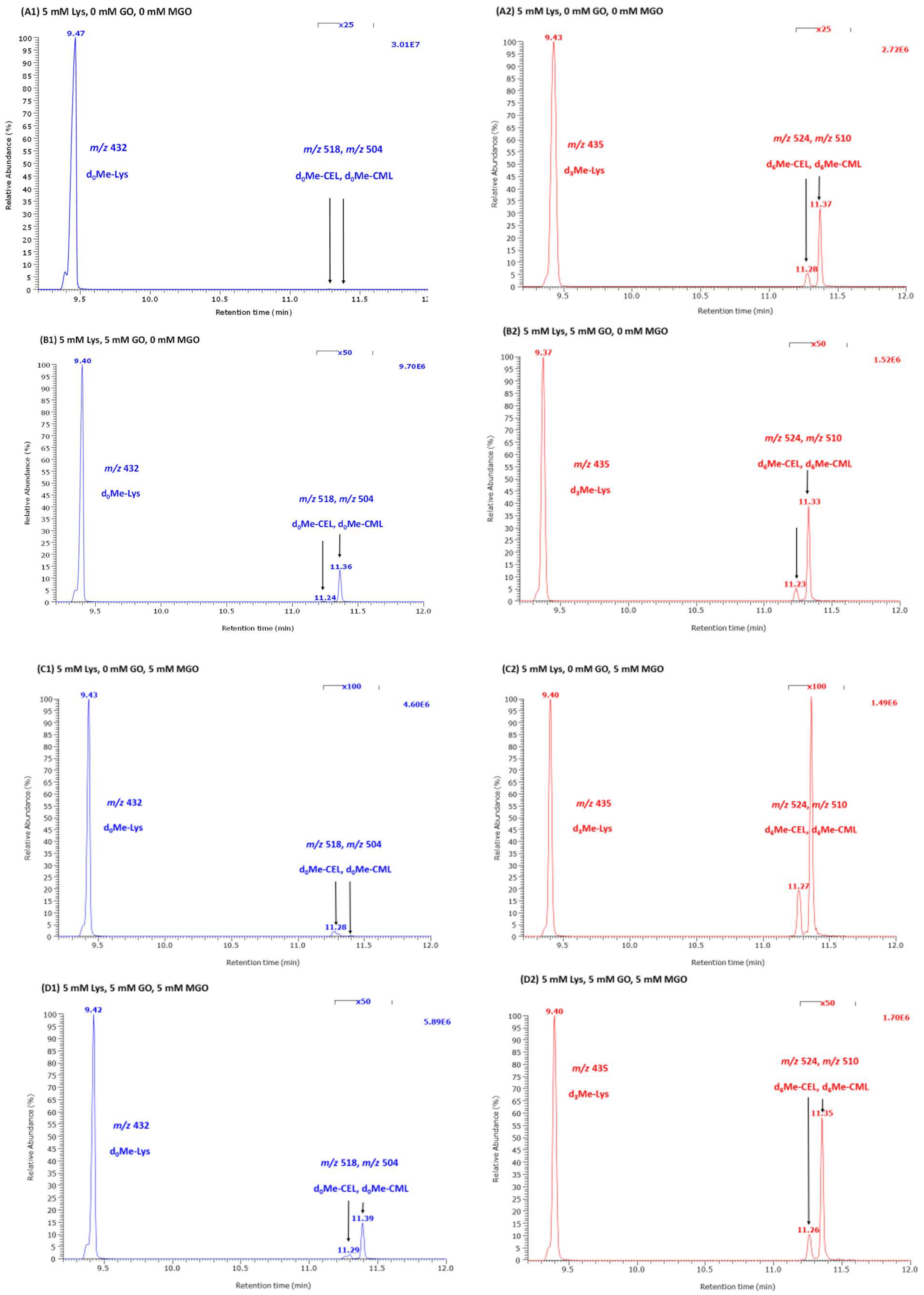
4.3. GC-MS Conditions and Quantitative Analyses of Lys, CML, CEL and HML in Reaction Mixtures of Lys with GO and MGO
The concentration of Lys, CML, CEL was determined simultaneously by GC-MS using their respective internal standards as described elsewhere [
6]. The concentration of HML was determined by GC-MS using
d,
l-5-hydroxy-lysine as the internal standard. Analyses were performed on a GC-MS apparatus model ISQ. Toluene aliquots (1 µL) were injected in the splitless mode. The 10-µL Hamilton needle of the autosampler was cleaned automatically three times with toluene (5 µL) after each injection. Injector temperature was kept at 280 °C. Helium was used as the carrier gas at a constant flow rate of 1.0 mL/min. The oven temperature was held at 40 °C for 0.5 min and ramped to 210 °C at a rate of 15 °C/min and then to 320 °C at a rate of 35 °C/min. Interface and ion-source temperatures were set to 300 °C and 250 °C, respectively. Electron energy was 70 eV and electron current 50 µA. Methane was used as the reagent gas for negative-ion chemical ionization at a constant flow rate of 2.4 mL/min. Quantitative analyses were performed in the selected-ion monitoring (SIM) mode at an electron multiplier voltage of 1400 V. The ions with mass-to-charge (
m/
z) were
m/
z 432 for Lys,
m/
z 504 for CML and
m/
z 518 for CEL, and
m/
z 435,
m/
z 510 and
m/
z 524 for the respective internal standards. The dwell time was 50 ms for each ion. In qualitative analyses, the ions with
m/
z 430,
m/
z 502 and
m/
z 516 were monitored for the putative Schiff bases of Lys, CML and CEL. Mass spectra of reaction products of Lys with GO or MGO were generated after their derivatization by scanning the quadrupole in the
m/
z-range 50 to 1000. The retention times of the internal standards observed in a series of quantitative analyses (
n = 148) were (mean ± standard deviation (SD)) 9.406 ± 0.023 min (relative standard deviation (RSD), 0.25%) for Lys, 11.36 ± 0.022 min (0.19%) for CEL and 11.27 ± 0.022 min (0.20%) for CML. The mean relative retention time was 1.19 for d
3Me-CEL/d
3Me-Lys, 1.199 for d
3Me-CML/d
3Me-Lys and 1.001 for d
3Me-CML/d
3Me-CEL and varied by 8.3% each. The corresponding peak area values (arbitrary units) of the internal standards in these analyses were 4.2 × 10
6 (RSD, 28%), 1.19 × 10
4 (RSD, 51%) and 4.47 × 10
4 (RSD, 46%).
4.4. Statistical Analysis and Data Presentation
Data analyses were performed using GraphPad Prism 7 for Windows (GraphPad Software, San Diego, CA, USA). Data are presented as mean ± SD or mean ± SEM.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}