
Diversity-Oriented Synthesis Catalyzed by Diethylaminosulfur-Trifluoride—Preparation of New Antitumor Ecdysteroid Derivatives

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
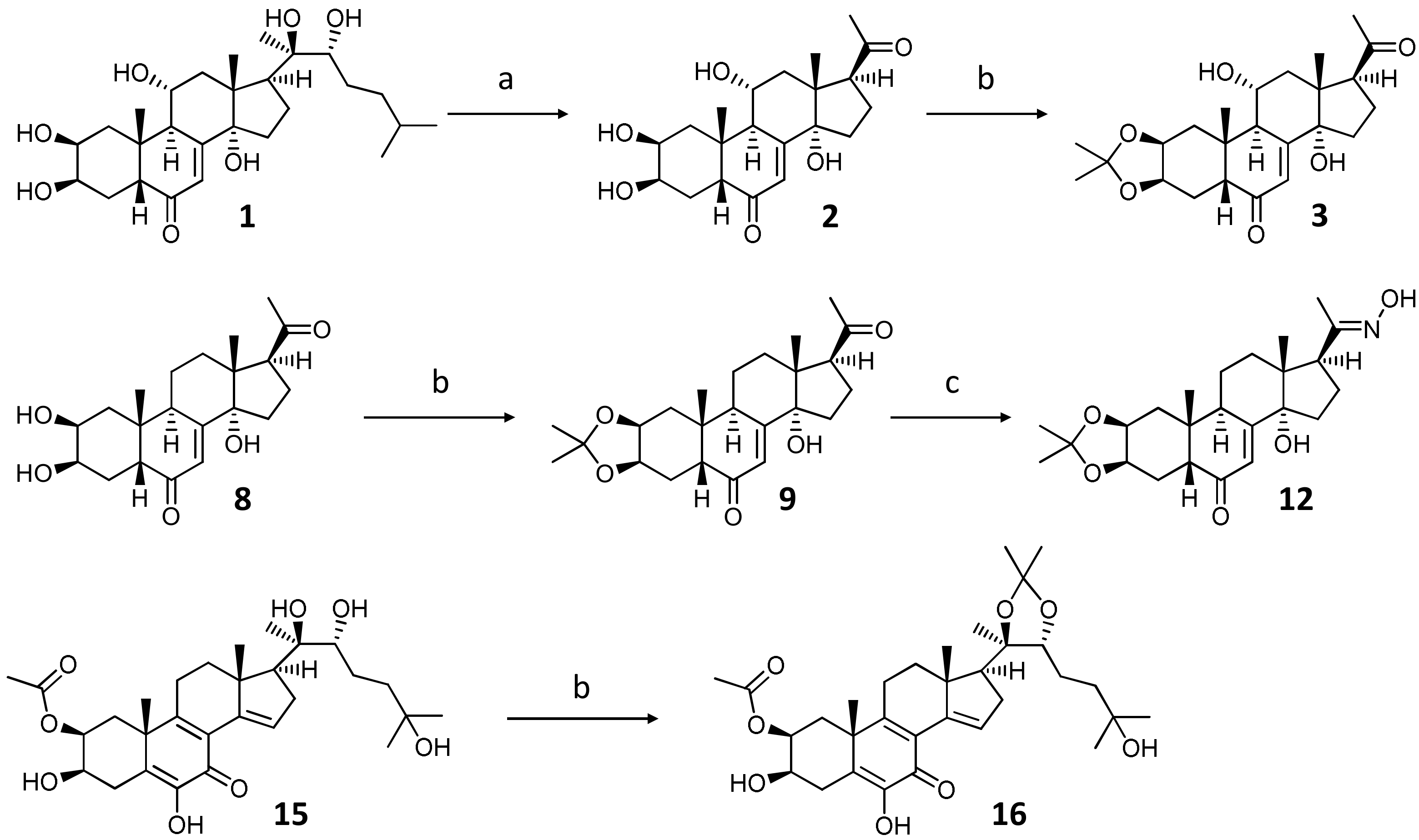
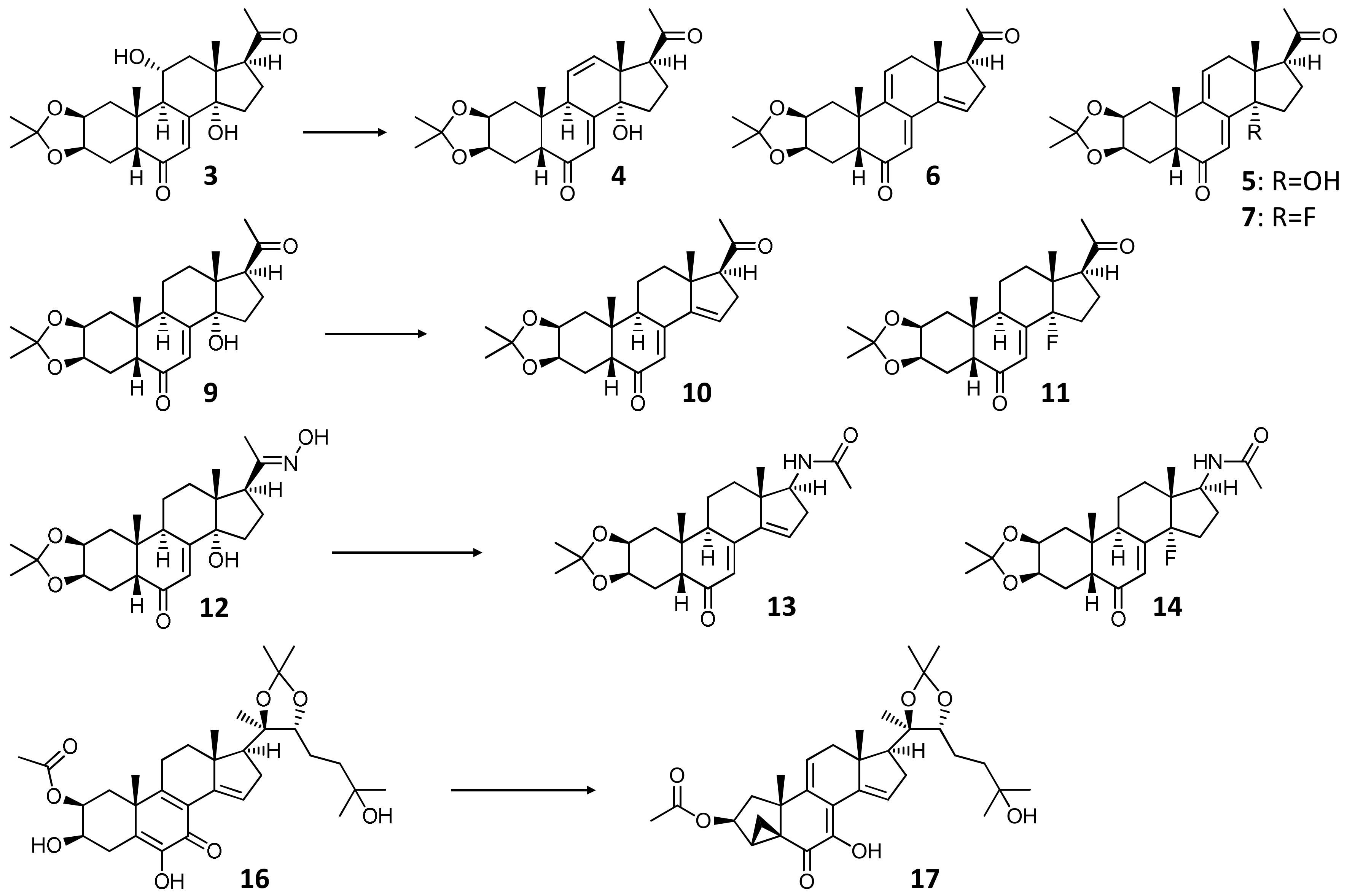
2.1.1. Preparation of Starting Materials for Reactions with DAST
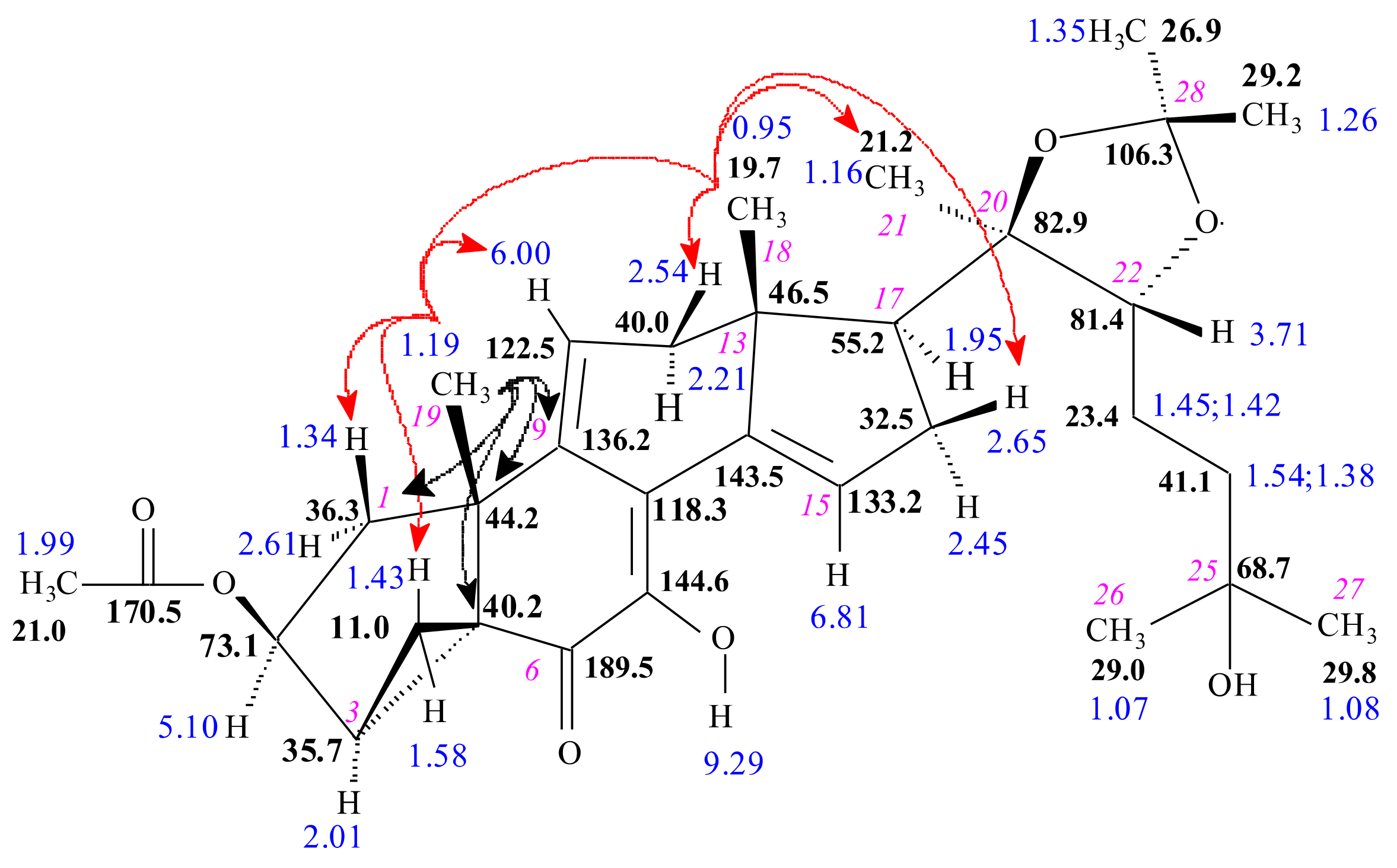
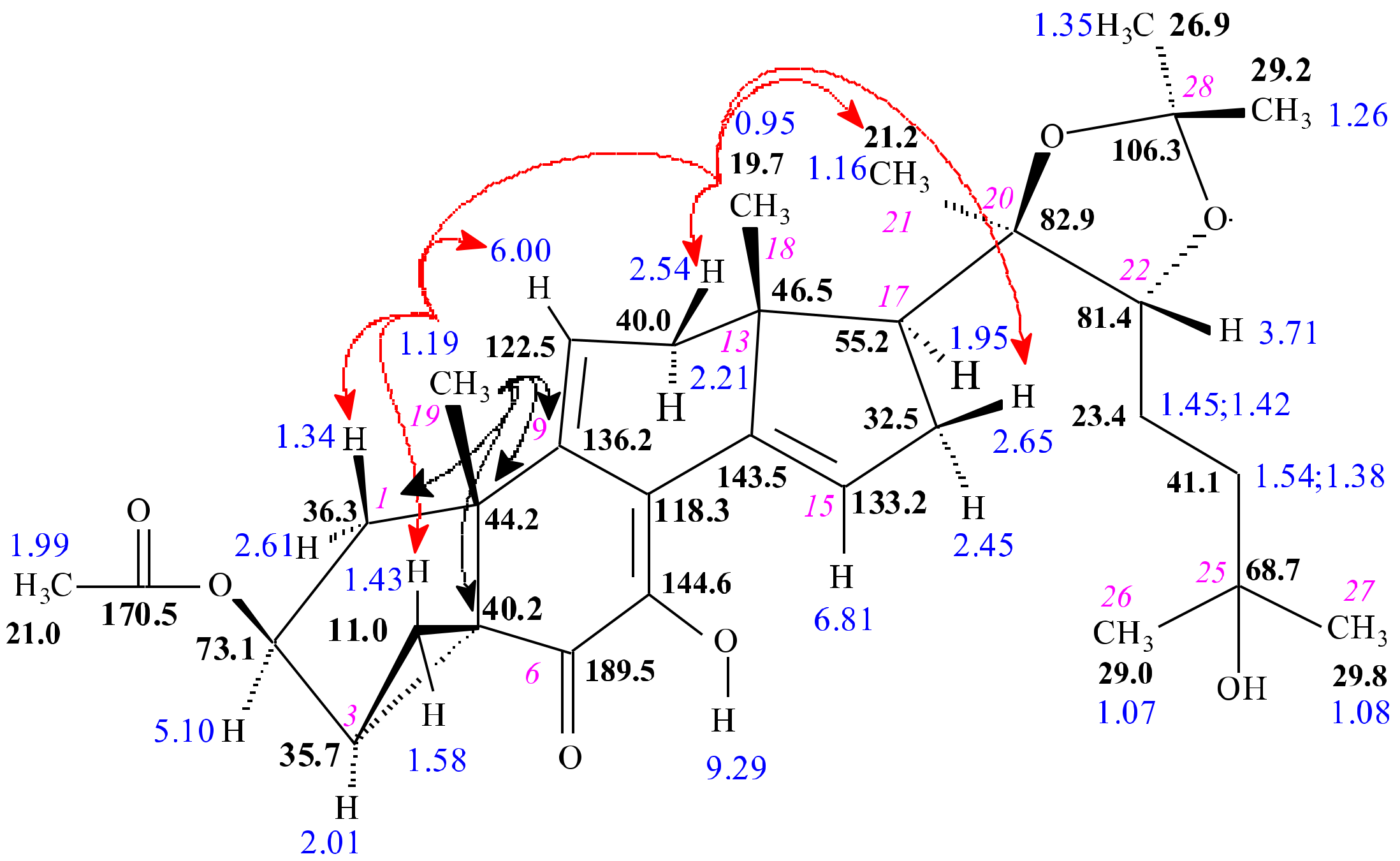
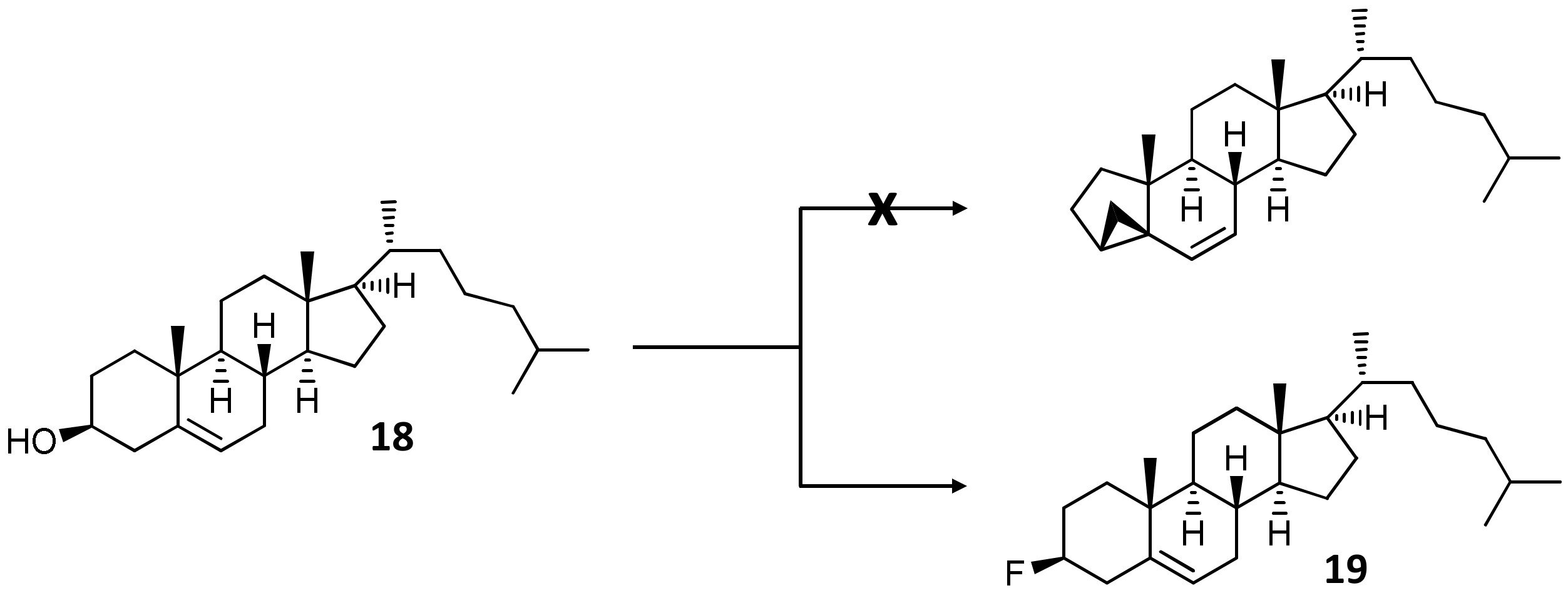
2.1.2. DAST-Mediated Transformation of Compounds 3, 9, 12, 16, 18, and Structure Elucidation of the Products
2.2. Biology
2.2.1. Cytotoxicity, Anti-Proliferative, and ABCB1 Inhibitory Activities
2.2.2. Combination Assays
3. Materials and Methods
3.1. Synthesis and Chromatographic Purification
3.2. Preparation of Ecdysteroid Derivatives
3.2.1. Natural Ecdysteroids Isolated from Plants or Semi-Synthesized before
3.2.2. Preparation of Sidechain Cleaved Ajugasterone C Derivative (2)
3.2.3. General Procedure for the Preparation of Ecdysteroid Acetonides 3, 9, 16
3.2.4. Preparation of Poststerone 2,3-acetonide 20-oxime (12)
3.2.5. General Procedure for the DAST-Catalyzed Transformation of Ecdysteroids
3.3. Structure Elucidation
3.4. Cell Lines
3.5. Cell Viability Assay for Determination of Cytotoxicity and Antiproliferative Activity
3.6. ABCB1 Inhibition Assay
3.7. Combination Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gouverneur, V.; Seppelt, K. Introduction: Fluorine Chemistry. Chem. Rev. 2015, 115, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Baecker, D.; Obermoser, V.; Kirchner, E.A.; Hupfauf, A.; Kircher, B.; Gust, R. Fluorination as tool to improve bioanalytical sensitivity and COX-2-selective antitumor activity of cobalt alkyne complexes. Dalton Trans. 2019, 48, 15856–15868. [Google Scholar] [CrossRef]
- Hong, H.; Zhang, L.; Xie, F.; Zhuang, R.; Jiang, D.; Liu, H.; Li, J.; Yang, H.; Zhang, X.; Nie, L.; et al. Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors. Nat. Commun. 2019, 10, 989. [Google Scholar] [CrossRef] [Green Version]
- Kaźmierczak, M.; Bilska-Markowska, M. Diethylaminosulfur Trifluoride (DAST) Mediated Transformations Leading to Valuable Building Blocks and Bioactive Compounds. Eur. J. Org. Chem. 2021, 2021, 5585–5604. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.Y.; Shi, M. Metal-Free Cross-Coupling of Arylboronic Acids and Derivatives with DAST-Type Reagents for Direct Access to Diverse Aromatic Sulfinamides and Sulfonamides. Angew. Chem. Int. Ed. Engl. 2016, 55, 10811–10815. [Google Scholar] [CrossRef]
- Jiang, H.; Shen, H.; Zhu, S.; Wang, B.; Yang, Y.; Nong, Z.; Yi, M.; Tang, S.; Gui, Q.-W. Diethylaminosulfur Trifluoride: A Novel, Low-Cost, Stable Double Thiolation Reagent for Imidazo[1,2-α]pyridines. ACS Omega 2021, 6, 26273–26281. [Google Scholar] [CrossRef]
- Csábi, J.; Martins, A.; Sinka, I.; Csorba, A.; Molnár, J.; Zupkó, I.; Tóth, G.; Tillekeratne, L.M.V.; Hunyadi, A. Synthesis and in vitro evaluation of the antitumor potential and chemo-sensitizing activity of fluorinated ecdysteroid derivatives. MedChemComm 2016, 7, 2282–2289. [Google Scholar] [CrossRef] [Green Version]
- Balázs, A.; Hunyadi, A.; Csábi, J.; Tillekeratne, L.M.V.; Martins, A.; Tóth, G. New cyclic 2,3-sulfite ester derivatives of poststerone—Discriminating diastereomers and probing spatial proximities by NMR and DFT calculations. Magn. Reson. Chem. 2017, 55, 1102–1107. [Google Scholar] [CrossRef]
- Hunyadi, A.; Csábi, J.; Martins, A.; Molnár, J.; Balázs, A.; Tóth, G. Backstabbing P-gp: Side-Chain Cleaved Ecdysteroid 2,3-Dioxolanes Hyper-Sensitize MDR Cancer Cells to Doxorubicin without Efflux Inhibition. Molecules 2017, 22, 199. [Google Scholar] [CrossRef]
- Martins, A.; Tóth, N.; Ványolós, A.; Béni, Z.; Zupkó, I.; Molnár, J.; Báthori, M.; Hunyadi, A. Significant Activity of Ecdysteroids on the Resistance to Doxorubicin in Mammalian Cancer Cells Expressing the Human ABCB1 Transporter. J. Med. Chem. 2012, 55, 5034–5043. [Google Scholar] [CrossRef] [PubMed]
- Vágvölgyi, M.; Martins, A.; Kulmány, Á.; Zupkó, I.; Gáti, T.; Simon, A.; Tóth, G.; Hunyadi, A. Nitrogen-containing ecdysteroid derivatives vs. multi-drug resistance in cancer: Preparation and antitumor activity of oximes, oxime ethers and a lactam. Eur. J. Med. Chem. 2018, 144, 730–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savchenko, R.G.; Nové, M.; Spengler, G.; Hunyadi, A.; Parfenova, L.V. In vitro adjuvant antitumor activity of various classes of semi-synthetic poststerone derivatives. Bioorg. Chem. 2021, 106, 104485. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-Involving Synthesis and Reactions of Oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef] [PubMed]
- Mirjafary, Z.; Abdoli, M.; Saeidian, H.; Boroon, S.; Kakanejadifard, A. Oxime ethers as versatile precursors in organic synthesis: A review. RSC Adv. 2015, 5, 79361–79384. [Google Scholar] [CrossRef]
- Bogdán, D.; Haessner, R.; Vágvölgyi, M.; Passarella, D.; Hunyadi, A.; Gáti, T.; Tóth, G. Stereochemistry and complete 1H and 13C NMR signal assignment of C-20-oxime derivatives of posterone 2,3-acetonide in solution state. Magn. Reson. Chem. 2018, 56, 859–866. [Google Scholar] [CrossRef]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab. J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef]
- Ley, S.; Baumann, M.; Baxendale, I. The Use of Diethylaminosulfur Trifluoride (DAST) for Fluorination in a Continuous-Flow Microreactor. Synlett 2008, 2008, 2111–2114. [Google Scholar] [CrossRef] [Green Version]
- Duddeck, H.; Dietrich, W.; Tóth, G. Structure Elucidation by Modern NMR; Steinkopff: Darmstadt, Germany, 1998. [Google Scholar] [CrossRef]
- Pretsch, E.; Tóth, G.; Munk, E.M.; Badertscher, M. Computer-Aided Structure Elucidation; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Csábi, J.; Hsieh, T.-J.; Hasanpour, F.; Martins, A.; Kele, Z.; Gáti, T.; Simon, A.; Tóth, G.; Hunyadi, A. Oxidized Metabolites of 20-Hydroxyecdysone and Their Activity on Skeletal Muscle Cells: Preparation of a Pair of Desmotropes with Opposite Bioactivities. J. Nat. Prod. 2015, 78, 2339–2345. [Google Scholar] [CrossRef]
- Li, H.; Qin, J.; Yang, Z.; Guan, X.; Zhang, L.; Liao, P.; Li, X. DAST-promoted Beckmann rearrangement/intramolecular cyclization of acyclic ketoximes: Access to 2-oxazolines, benzimidazoles and benzoxazoles. Chem. Commun. 2015, 51, 8637–8639. [Google Scholar] [CrossRef]
- Kim, D.; Lim, H.N. Synthesis of Acyl Fluorides via DAST-Mediated Fluorinative C-C Bond Cleavage of Activated Ketones. Org. Lett. 2020, 22, 7465–7469. [Google Scholar] [CrossRef] [PubMed]
- Lafont, R.; Serova, M.; Didry-Barca, B.; Raynal, S.; Guibout, L.; Dinan, L.; Veillet, S.; Latil, M.; Dioh, W.; Dilda, P.J. 20-Hydroxyecdysone activates the protective arm of the RAAS via the MAS receptor. J Mol Endocrinol 2021, 68, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Fan, J.; Wu, F.; Huang, Q.; Guo, M.; Lv, Z.; Han, J.; Duan, L.; Hu, G.; Chen, L.; et al. The ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Pleiotropic Roles in Cancer. Front Physiol. 2017, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Simões e Silva, A.C.; Sampaio, W.O. The Role of Angiotensin–(1-7) in Cancer. In Angiotensin-(1-7); Springer: Cham, Switzerland, 2019; pp. 219–229. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chung, F.S.; Santiago, J.S.; Jesus, M.F.M.D.; Trinidad, C.V.; See, M.F.E. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583–1598. [Google Scholar]
- Hunyadi, A.; Herke, I.; Lengyel, K.; Báthori, M.; Kele, Z.; Simon, A.; Tóth, G.; Szendrei, K. Ecdysteroid-containing food supplements from Cyanotis arachnoidea on the European market: Evidence for spinach product counterfeiting. Sci. Rep. 2016, 6, 37322. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4 | 5 | 6 | 7 | 10 | 11 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| no. | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C |
| 1β | 1.37 | 37.8 | 1.56 | 37.0 | 1.48 | 37.8 | 1.55 | 37.1 | 1.28 | 37.5 | 1.30 | 37.3 |
| α | 2.04 | 2.19 | 2.25 | 2.22 | 1.96 | 1.98 | ||||||
| 2 | 4.28 | 71.5 | 4.18 | 71.9 | 4.19 | 71.5 | 4.17 | 71.8 | 4.18 | 71.9 | 4.23 | 72.0 |
| 3 | 4.31 | 71.8 | 4.15 | 71.2 | 4.16 | 71.5 | 4.17 | 71.2 | 4.28 | 71.6 | 4.29 | 71.4 |
| 4β | 2.17 | 26.9 | 2.05 | 28.2 | 2.17 | 29.0 | 2.08 | 28.2 | 2.17 | 26.7 | 2.09 | 28.2 |
| α | 1.86 | 1.86 | 1.58 | 1.86 | 1.77 | 1.91 | ||||||
| 5 | 2.44 | 49.7 | 2.45 | 50.6 | 2.47 | 50.2 | 2.48 | 50.7 | 2.36 | 50.7 | 2.39 | 50.6 |
| 6 | 201.6 | 202.4 | 202.3 | 202.2 | 201.9 | 201.9 | ||||||
| 7 | 5.88 | 121.2 | 5.82 | 119.5 | 6.12 | 117.5 | 5.91 | 121.8 d 7 Hz | 6.10 | 121.5 | 5.91 | 124.1 d 6.7 Hz |
| 8 | 158.1 | 152.2 | 141.8 | 149.5 d 19 Hz | 152.7 | 155.0 d 20 Hz | ||||||
| 9 | 3.39 | 38.5 | 135.7 | 135.9 | 135.4 | 2.41 | 38.6 | 2.69 | 35.7 | |||
| 10 | 38.9 | 39.4 | 40.4 | 39.4 | 38.4 | 37.6 | ||||||
| 11β | 5.78 | 124.5 | 6.17 | 130.8 | 6.17 | 130.1 | 6.18 | 130.6 | 1.72 | 20.5 | 1.63 | 20.6 |
| α | 1.84 | 1.84 | ||||||||||
| 12β | 6.33 | 133.7 | 2.41 | 36.0 | 2.75 | 40.9 | 2.46 | 35.9 d 6 Hz | 2.31 | 38.7 | 1.91 | 30.4 d 4.5 Hz |
| α | 2.87 | 2.57 | 2.81 | 1.74 | 2.13 | |||||||
| 13 | 51.0 | 46.9 | 46.0 | 46.9 d 20 Hz | 47.3 | 48.2 d 19.5 Hz | ||||||
| 14 | 84.6 | 83.7 | 142.9 | 106.0 d 168 Hz | 146.1 | 107.3 d 166 Hz | ||||||
| 15β | 2.08 | 28.6 | 2.05 | 31.1 | 6.28 | 128.4 | 2.04 | 28.5 d 25 Hz | 5.97 | 128.2 | 1.98 | 28.4 d 28 Hz |
| α | 1.75 | 1.86 | 2.14 | |||||||||
| 16β | 2.30 | 21.8 | 2.31 | 21.5 | 2.98 | 32.0 | 2.34 | 21.5 | 2.93 | 31.6 | 2.32 | 21.1 |
| α | 2.09 | 2.01 | 2.48 | 2.03 | 2.36 | 1.95 | ||||||
| 17 | 3.37 | 55.2 | 3.34 | 58.4 | 3.08 | 64.1 | 3.22 | 58.6 | 3.06 | 64.9 | 3.17 | 58.8 |
| 18 | 0.73 | 21.1 | 0.64 | 17.4 | 0.84 | 19.4 | 0.67 | 16.9 d 5 Hz | 0.85 | 18.9 | 0.65 | 16.5 d 4 Hz |
| 19 | 0.89 | 23.9 | 1.23 | 29.6 | 1.14 | 30.1 | 1.20 | 29.7 | 0.98 | 23.2 | 1.00 | 23.7 |
| 20 | 208.9 | 209.2 | 207.6 | 208.9 | 207.8 | 208.3 | ||||||
| 21 | 2.22 | 31.0 | 2.17 | 31.0 | 2.22 | 31.0 | 2.19 | 31.0 | 2.20 | 31.2 | 2.17 | 31.3 |
| 22 | 108.4 | 108.5 | 108.5 | 108.3 | 108.3 | |||||||
| βMe | 1.51 | 1.52 | 28.5 | 1.52 | 28.5 | 1.53 | 28.5 | 1.50 | 28.5 | 1.50 | 28.5 | |
| αMe | 1.35 | 1.32 | 26.3 | 1.32 | 26.4 | 1.33 | 26.4 | 1.34 | 26.3 | 1.34 | 26.4 | |
| HO-14 | 2.37 | 1.92 | 1.92 | |||||||||
| 13 | 14 | 15 | 17 | 19 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| no. | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C | 1H | 13C |
| 1β | 1.25 | 37.5 | 1.28 | 37.3 | 2.47 | 40.5 | 1.48 | 36.3 | 1.89 | 36.4 d 11 Hz |
| α | 1.95 | 1.98 | 1.47 | 2.25 | 1.07 | |||||
| 2β | 71.9 | 72.0 | 73.0 | 73.1 | 1.70 | 28.8 d 18 Hz | ||||
| α | 4.16 | 4.23 | 5.20 | 4.19 | 1.99 | |||||
| 3 | 4.26 | 71.6 | 4.29 | 71.3 | 3.64 | 71.8 | 2.01 | 35.7 | 4.39 | 92.8 d 174 Hz |
| 4β | 2.14 | 26.7 | 2.08 | 26.27 | 2.52 | 28.3 | 1.43 | 11.0 | 2.45 | 39.4 d 19 Hz |
| α | 1.76 | 1.92 | 3.24 | 1.58 | 2.45 | |||||
| 5 | 2.34 | 50.7 | 2.39 | 50.7 | 131.3 | 40.2 | 139.4 d 12 Hz | |||
| 6 | 202.2 | 202.2 | 144.8 | 189.5 | 5.40 | 123.0 | ||||
| 7 | 6.06 | 120.9 | 5.88 | 123.3 7 Hz | 181.4 | 144.6 | 1.99;1.54 | 31.9 | ||
| 8 | 153.3 | 155.4 d 20 Hz | 125.0 | 118.3 | 1.46 | 31.87 | ||||
| 9 | 2.38 | 38.7 | 2.69 | 35.8 | 164.5 | 136.2 | 0.93 | 50.0 | ||
| 10 | 38.5 | 37.7 | 41.8 | 44.2 | 36.5 | |||||
| 11β | 1.60 | 20.3 | 1.55 | 20.2 | 2.60 | 25.4 | 6.00 | 122.5 | 1.50 | 21.1 |
| α | 1.77 | 1.80 | 2.60 | 1.50 | ||||||
| 12β | 2.02 | 37.0 | 1.61 | 28.7 d 5 Hz | 2.28 | 37.8 | 2.54 | 40.0 | 2.03 | 39.7 |
| α | 1.59 | 2.05 | 1.52 | 2.21 | 1.18 | |||||
| 13 | 46.9 | 47.9 d 19 Hz | 47.9 | 46.5 | 42.3 | |||||
| 14 | 147.5 | 105.5 d 166 Hz | 142.4 | 143.5 | 1.00 | 56.7 | ||||
| 15β | 5.93 | 126.5 | 2.09 | 27.7 d 28 Hz | 6.85 | 128.2 | 6.81 | 133.2 | 1.08 | 24.3 |
| α | 1.95 | 1.58 | ||||||||
| 16β | 2.21 | 36.0 | 1.50 | 26.9 | 2.71 | 32.7 | 2.65 | 32.5 | 1.27 | 28.2 |
| α | 2.67 | 2.41 | 2.25 | 2.45 | 1.85 | |||||
| 17 | 4.41 | 60.3 | 4.60 | 55.4 | 1.99 | 56.5 | 1.95 | 55.2 | 1.10 | 56.2 |
| 18 | 0.89 | 17.2 | 0.71 | 15.3 d 4 Hz | 1.07 | 18.3 | 0.95 | 19.7 | 0.69 | 11.9 |
| 19 | 0.96 | 23.2 | 1.00 | 23.4 | 1.44 | 27.3 | 1.19 | 26.4 | 1.04 | 19.3 |
| 20 | 170.2 | 170.1 | 77.3 | 82.9 | 1.38 | 35.8 | ||||
| 21 | 2.02 | 23.4 | 2.02 | 23.6 | 1.26 | 20.5 | 1.16 | 21.2 | 0.93 | 18.7 |
| 22 | 108.3 | 108.3 | 3.38 | 78.7 | 3.71 | 81.4 | 1.35;1.00 | 36.2 | ||
| βMe | 1.49 | 28.5 | 1.50 | 28.6 | 1.52 | 28.5 | 1.52 | |||
| αMe | 1.33 | 26.3 | 1.34 | 26.3 | 1.32 | 26.4 | 1.32 | |||
| 23 | 1.62;1.31 | 27.3 | 1.45;1.42 | 23.4 | 1.34;1.15 | 23.8 | ||||
| 24 | 1.81;1.43 | 42.4 | 1.54;1.38 | 41.4 | 1.14 | 39.5 | ||||
| 25 | 71.4 | 68.7 | 28.0 | |||||||
| 26 | 1.17 | 28.9 | 29.0 | 0.87 | 22.6 | |||||
| 27 | 1.20 | 30.0 | 29.8 | 0.88 | 22.8 | |||||
| 28 | 106.3 | |||||||||
| βMe | 1.26 | 29.2 | ||||||||
| αMe | 1.35 | 26.9 | ||||||||
| NH | 5.80 | 5.57 | ||||||||
| HO-7 | 9.29 | |||||||||
| Cytotoxicity (IC50 ± SD [µM]) | Antiproliferative Effect (IC50 ± SD [µM]) | ABCB1 Inhibition (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| L5178Y | L5178YMDR | MRC-5 | L5178Y | L5178YMDR | MRC-5 | 2 µM | 20 µM | |
| 3 | >100 | >100 | >100 | 75.5 ± 4.1 | >100 | 21.4 ± 0.2 | −0.03 | −0.35 |
| 5 | >100 | >100 | >100 | 56.5 ± 6.0 | 80.2 ± 2.1 | 20.9 ± 3.6 | 0.20 | −0.41 |
| 6 | 91.1 ± 6.8 | >100 | >100 | 31.2 ± 3.9 | 54.1 ± 2.3 | >100 | −0.38 | 0.13 |
| 7 | 38.6 ± 1.6 | 57.5 ± 6.3 | >100 | 15.2 ± 1.9 | 20.0 ± 2.4 | >100 | −0.24 | 0.29 |
| 9 | >100 | >100 | >100 | 49.2 ± 3.4 | 85.2 ± 5.1 | >100 | −0.40 | −0.42 |
| 10 | 72.9 ± 5.6 | 83.0 ± 5 | >100 | 27.7 ± 0.6 | 42.3 ± 1.5 | >100 | −0.38 | −0.16 |
| 11 | 91.9 ± 9.3 | 81.9 ± 2.9 | >100 | 31.8 ± 2.1 | 55.7 ± 5.9 | 10.0 ± 2.1 | −0.57 | −0.51 |
| 13 | >100 | >100 | 35.0 ± 3.2 | 4.6 ± 0.8 | 4.8 ± 0.6 | 12.0 ± 1.9 | 0.09 | –0.01 |
| 14 | >100 | >100 | >100 | 71.6 ± 1.5 | 65.9 ± 2.1 | >100 | 0.20 | −0.12 |
| 16 | 63.0 ± 4.4 | 78.7 ± 6.7 | >100 | 50.3 ± 0.2 | 40.0 ± 3.4 | 67.2 ± 14.6 | 0.71 | 15.7 |
| 17 | 27.2 ± 1 | 35.9 ± 0.6 | 5. 9 ± 1.2 | 20.3 ± 1 | 14.3 ± 0.5 | 5.9 ± 1.2 | 1.08 | 45.5 |
| Doxorubicin | 0.30 ± 0.10 | 8.1 ± 2.8 | 1.7 ± 0.5 | 0.014 ± 0.002 | 0.71 ± 0.2 | 0.45 ± 0. 13 | - | - |
| Drug Ratio | CI at | Dm | m | r | CIavg | |||
|---|---|---|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | ||||||
| 7 | 11.6:1 | 0.98 | 0.76 | 0.59 | 4.05 | 2.18 | 0.940 | 0.71 |
| 23.1:1 | 1.16 | 1.26 | 1.37 | 7.83 | 1.32 | 0.971 | 1.30 | |
| 46.4:1 | 1.17 | 0.84 | 0.60 | 11.56 | 2.64 | 0.974 | 0.78 | |
| 10 | 23.2:1 | 0.75 | 0.50 | 0.33 | 6.50 | 2.20 | 0.988 | 0.46 |
| 46.4:1 | 0.87 | 0.51 | 0.30 | 10.99 | 2.76 | 0.983 | 0.46 | |
| 92.8:1 | 0.86 | 0.55 | 0.35 | 14.02 | 2.14 | 0.999 | 0.50 | |
| 185.6:1 | 1.02 | 0.61 | 0.36 | 19.47 | 2.37 | 0.989 | 0.55 | |
| 11 | 23.2:1 | 0.78 | 0.56 | 0.41 | 7.00 | 2.43 | 0.964 | 0.52 |
| 46.4:1 | 0.81 | 0.55 | 0.39 | 11.66 | 3.07 | 0.996 | 0.51 | |
| 92.8:1 | 0.77 | 0.53 | 0.38 | 15.83 | 3.42 | 0.991 | 0.49 | |
| 185.6:1 | 0.93 | 0.66 | 0.48 | 24.06 | 3.51 | 0.991 | 0.61 | |
| 371.2:1 | 0.95 | 0.72 | 0.55 | 28.63 | 3.27 | 0.968 | 0.67 | |
| 13 | 23.2:1 | 0.64 | 0.55 | 0.49 | 8.17 | 2.65 | 0.992 | 0.53 |
| 46.4:1 | 0.67 | 0.51 | 0.39 | 14.17 | 4.18 | 0.963 | 0.48 | |
| 92.8:1 | 0.55 | 0.51 | 0.47 | 17.54 | 2.60 | 0.966 | 0.50 | |
| 185.6:1 | 0.71 | 0.68 | 0.65 | 29.89 | 2.54 | 0.997 | 0.67 | |
| 14 | 23.2:1 | 0.85 | 0.73 | 0.63 | 9.04 | 1.78 | 0.994 | 0.70 |
| 46.4:1 | 0.86 | 0.72 | 0.60 | 13.53 | 1.99 | 0.998 | 0.68 | |
| 92.8:1 | 1.00 | 0.86 | 0.74 | 20.47 | 1.99 | 0.996 | 0.82 | |
| 185.6:1 | 1.28 | 1.26 | 1.24 | 30.97 | 1.65 | 0.994 | 1.25 | |
| Compound (Yield) | Column | Flow Rate | Elution | Detection |
|---|---|---|---|---|
| 2 (70.2%) | 24 g silica | 35 mL/min | CH2Cl2:CH3OH (A:B) 12% B (40 min) | 254 nm |
| 3 (78.9%) | 24 g silica | 35 mL/min | CH2Cl2:ethyl-acetate (A:B) 2→4% B (60 min) | 254 nm |
| 4 (6.1%) | (1) biphenyl (2) phenyl-hexyl | 15 mL/min | (1) water:CH3CN (A:B) 26% B 40 min-40% B 40–60 min (2) water:CH3CN (A:B) 44% B (30 min) | 254 nm |
| 5 (19.4 %) | ||||
| 6 (9.4%) | ||||
| 7 (3.4%) | ||||
| 9 (55.8%) | 80 g silica | 60 mL/min | CH2Cl2:CH3OH (A:B) 0→3% B (60 min) | 254 nm |
| 10 (20.3%) | XB-C18 | 15 mL/min | water:CH3CN (A:B) 42% B (40 min) | 254 nm 300 nm |
| 11 (12.0%) | ||||
| 12 (82.0%) | 24 g silica | 35 mL/min | CH2Cl2:CH3OH (A:B) 0→5% B (70 min) | 254 nm |
| 13 (16.6%) | (1) XB-C18 (2) Luna silica | 15 mL/min | (1) water: CH3CN (A:B) 35% B (40 min) (2) cyclohexane:(2-propanol:water 97:3) (A:B) 17% B (40 min) | 245 nm 300 nm |
| 14 (6.1%) | ||||
| 16 (84.6%) | 80 g silica | 60 mL/min | n-hexane:(acetone:2-propanol 8:2) (A:B) 10→15% B (60 min) | 254 nm |
| 17 (6.4%) | (1) Luna silica (2) XB-C18 | 15 mL/min | (1) cyclohexane:2-propanol (A:B) 3% B (40 min) (2) water:CH3CN (A:B) 63% B (40 min) | 254 nm 366 nm |
| 19 (43.9%) | 40 g silica | 40 mL/min | 100% n-hexane (60 min) | 210 nm |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vágvölgyi, M.; Kocsis, E.; Nové, M.; Szemerédi, N.; Spengler, G.; Kele, Z.; Berkecz, R.; Gáti, T.; Tóth, G.; Hunyadi, A. Diversity-Oriented Synthesis Catalyzed by Diethylaminosulfur-Trifluoride—Preparation of New Antitumor Ecdysteroid Derivatives. Int. J. Mol. Sci. 2022, 23, 3447. https://doi.org/10.3390/ijms23073447
Vágvölgyi M, Kocsis E, Nové M, Szemerédi N, Spengler G, Kele Z, Berkecz R, Gáti T, Tóth G, Hunyadi A. Diversity-Oriented Synthesis Catalyzed by Diethylaminosulfur-Trifluoride—Preparation of New Antitumor Ecdysteroid Derivatives. International Journal of Molecular Sciences. 2022; 23(7):3447. https://doi.org/10.3390/ijms23073447
Chicago/Turabian StyleVágvölgyi, Máté, Endre Kocsis, Márta Nové, Nikoletta Szemerédi, Gabriella Spengler, Zoltán Kele, Róbert Berkecz, Tamás Gáti, Gábor Tóth, and Attila Hunyadi. 2022. "Diversity-Oriented Synthesis Catalyzed by Diethylaminosulfur-Trifluoride—Preparation of New Antitumor Ecdysteroid Derivatives" International Journal of Molecular Sciences 23, no. 7: 3447. https://doi.org/10.3390/ijms23073447
APA StyleVágvölgyi, M., Kocsis, E., Nové, M., Szemerédi, N., Spengler, G., Kele, Z., Berkecz, R., Gáti, T., Tóth, G., & Hunyadi, A. (2022). Diversity-Oriented Synthesis Catalyzed by Diethylaminosulfur-Trifluoride—Preparation of New Antitumor Ecdysteroid Derivatives. International Journal of Molecular Sciences, 23(7), 3447. https://doi.org/10.3390/ijms23073447