
Role of ERK Pathway in the Pathogenesis of Atopic Dermatitis and Its Potential as a Therapeutic Target

 and
and
Abstract
:1. Introduction
2. Results
2.1. p-ERK Is Highly Expressed in Mouse and Human AD Skin
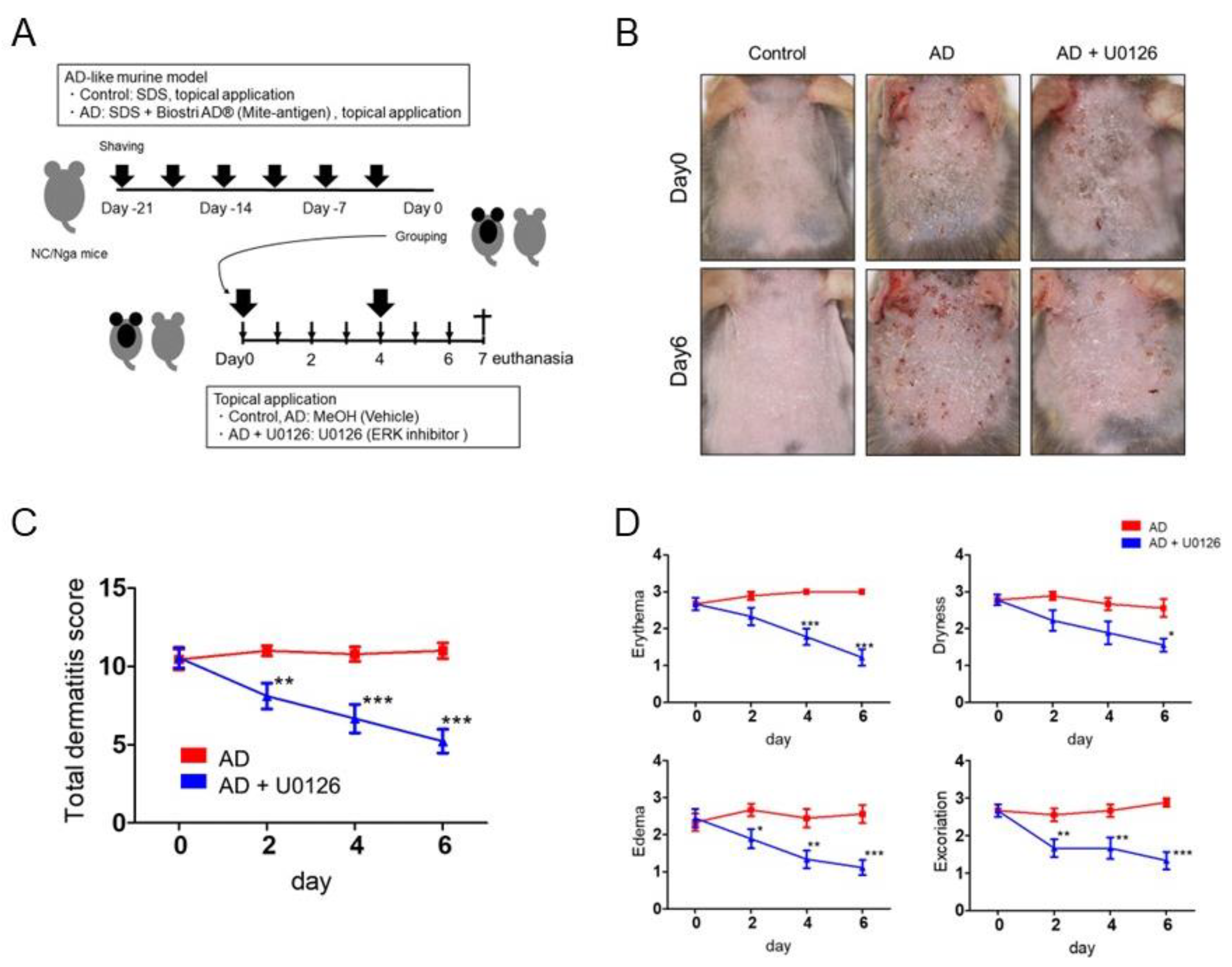
2.2. Topical Application of ERK Inhibitor Alleviates the Clinical Symptoms of Mite Antigen-Induced AD in Mice
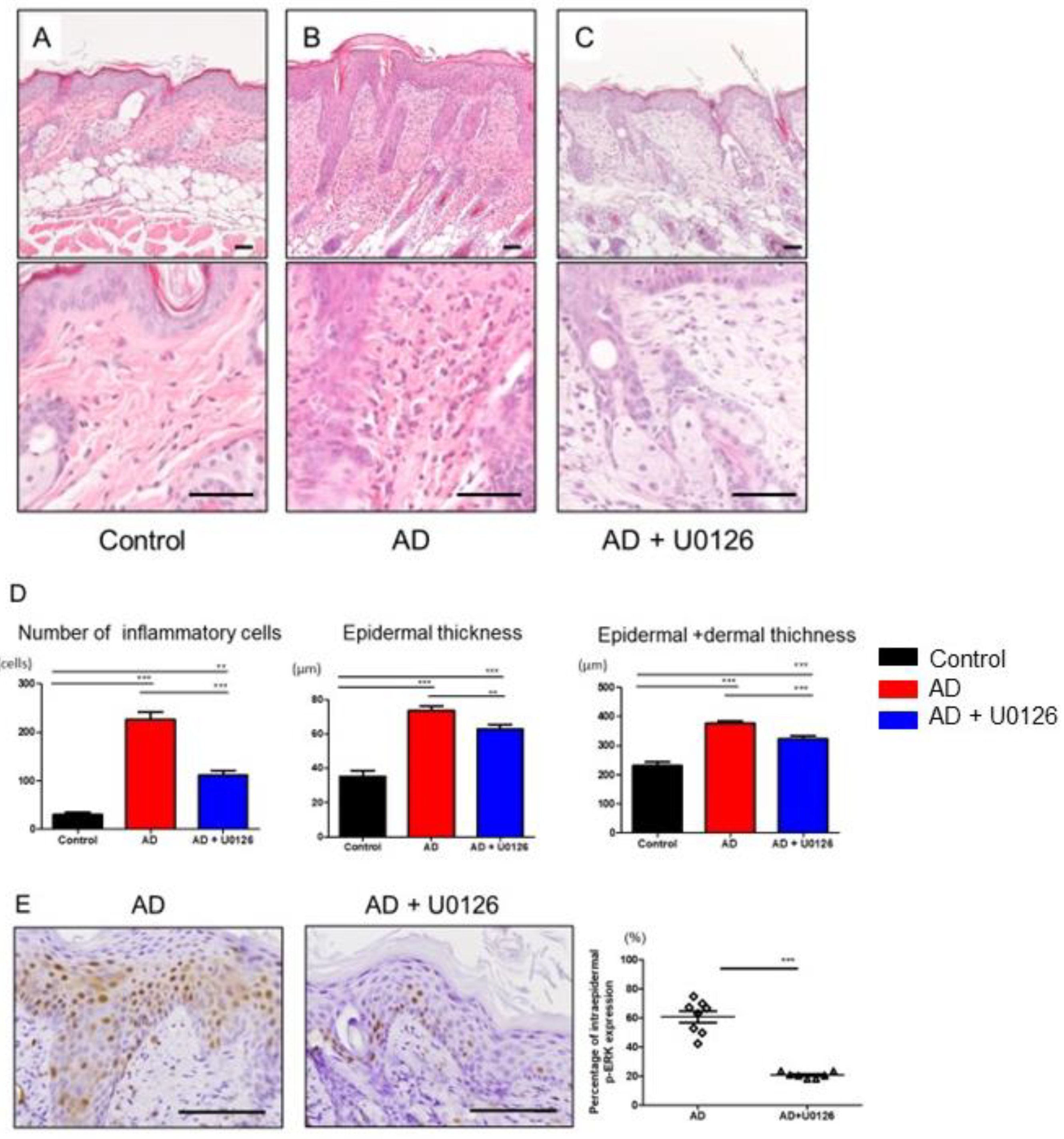
2.3. Topical Application of ERK Inhibitor Improved the Histological Changes of Mite Antigen-Induced AD in Mice
2.4. Topical Application of ERK Inhibitor Improved TEWL and Restored FLG Expression in Mite Antigen-Induced AD in Mice
2.5. ERK Inhibitor Restored the Reduction in the Expression of Filaggrin in NHEK
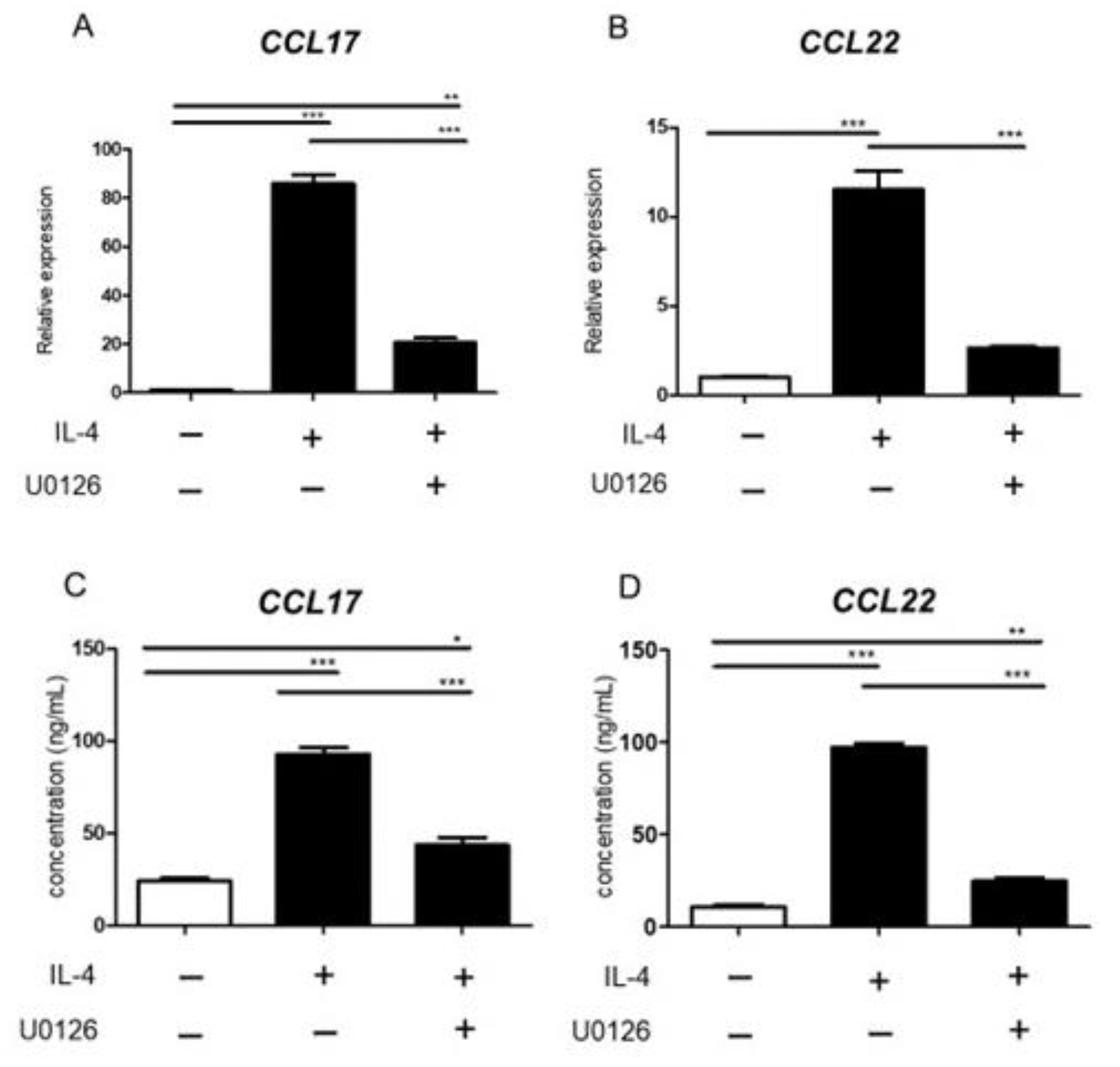
2.6. ERK Inhibitor Inhibited Chemokine Production from BMDC Induced by IL-4
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Mice
4.3. Induction of Mite Antigen-Induced AD-like Murine Model
4.4. Evaluation of Skin Lesions
4.5. Transepidermal Water Loss (TEWL)
4.6. Histological Examination and Immunohistochemical Staining
4.7. Cell Culture
4.8. Generation of Bone Marrow-Derived DCs (BMDCs)
4.9. Real-Time Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
4.10. Western Blotting
4.11. ELISA
4.12. Tissue Sample
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| IFN-γ | interferon gamma |
| IL- | interleukin |
| MAPK | mitogen-activated protein kinases |
| STAT | signal transducer and activator of transcription |
| JAK | Janus kinase |
| ERK | extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinases |
| ET | endothelin |
| TARC | thymus and activation-regulated chemokine |
| NHEK | normal human epidermal keratinocytes |
| BMDC | bone marrow-derived dendritic cells |
| FLG | filaggrin |
| LOR | loricrin |
| IVL | involucrin |
| Th | T helper cell |
| TEWL | transepidermal water loss |
| CCL | C-C motif chemokine |
| MDC | macrophage-derived chemokine |
| CLDN | claudin |
| TJs | tight junctions |
References
- Brunner, P.M.; Leung, D.Y.M.; Guttman-Yassky, E. Immunologic, microbial, and epithelial interactions in atopic dermatitis. Ann. Allergy Asthma Immunol. 2018, 120, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, M.; Chiba, T.; Tsuji, G.; Ulzii, D.; Kido-Nakahara, M.; Nakahara, T.; Kadono, T. Atopic dermatitis: Immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol. Int. 2017, 66, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Ulzii, D.; Vu, Y.H.; Tsuji, G.; Kido-Nakahara, M.; Nakahara, T. Pathogenesis of Atopic Dermatitis: Current Paradigm. Iran. J. Immunol. 2019, 16, 97–107. [Google Scholar] [PubMed]
- Guttman-Yassky, E.; Krueger, J.G. Atopic dermatitis and psoriasis: Two different immune diseases or one spectrum? Curr. Opin. Immunol. 2017, 48, 68–73. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Tsuji, G.; Furue, M. Basics and recent advances in the pathophysiology of atopic dermatitis. J. Dermatol. 2021, 48, 130–139. [Google Scholar] [CrossRef]
- Kabashima, K. New concept of the pathogenesis of atopic dermatitis: Interplay among the barrier, allergy, and pruritus as a trinity. J. Dermatol. Sci. 2013, 70, 3–11. [Google Scholar] [CrossRef]
- McKenzie, C.; Silverberg, J.I. The prevalence and persistence of atopic dermatitis in urban United States children. Ann. Allergy Asthma Immunol. 2019, 123, 173–178.e1. [Google Scholar] [CrossRef]
- Nakahara, T.; Fujita, H.; Arima, K.; Taguchi, Y.; Motoyama, S.; Furue, M. Treatment satisfaction in atopic dermatitis relates to patient-reported severity: A cross-sectional study. Allergy 2019, 74, 1179–1181. [Google Scholar] [CrossRef]
- Drislane, C.; Irvine, A.D. The role of filaggrin in atopic dermatitis and allergic disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef] [Green Version]
- van den Bogaard, E.H.; Bergboer, J.G.; Vonk-Bergers, M.; van Vlijmen-Willems, I.M.; Hato, S.V.; van der Valk, P.G.; Schröder, J.M.; Joosten, I.; Zeeuwen, P.L.; Schalkwijk, J. Coal tar induces AHR-dependent skin barrier repair in atopic dermatitis. J. Clin. Investig. 2013, 123, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, G.; Ito, T.; Chiba, T.; Mitoma, C.; Nakahara, T.; Uchi, H.; Furue, M. The role of the OVOL1-OVOL2 axis in normal and diseased human skin. J. Dermatol. Sci. 2018, 90, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Roan, F.; Ziegler, S.F. The atopic march: Current insights into skin barrier dysfunction and epithelial cell-derived cytokines. Immunol. Rev. 2017, 278, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.L.; Bieber, T.; Guttman-Yassky, E.; Beck, L.A.; Blauvelt, A.; Cork, M.J.; Silverberg, J.I.; Deleuran, M.; Kataoka, Y.; Lacour, J.P.; et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med. 2016, 375, 2335–2348. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Meephansan, J.; Komine, M.; Tsuda, H.; Karakawa, M.; Tominaga, S.; Ohtsuki, M. Expression of IL-33 in the epidermis: The mechanism of induction by IL-17. J. Dermatol. Sci. 2013, 71, 107–114. [Google Scholar] [CrossRef]
- Lejeune, D.; Dumoutier, L.; Constantinescu, S.; Kruijer, W.; Schuringa, J.J.; Renauld, J.-C. Interleukin-22 (IL-22) Activates the JAK/STAT, ERK, JNK, and p38 MAP Kinase Pathways in a Rat Hepatoma Cell Line. J. Biol. Chem. 2002, 277, 33676–33682. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Semba, T.; Sammons, R.; Wang, X.; Xie, X.; Dalby, K.N.; Ueno, N.T. JNK Signaling in Stem Cell Self-Renewal and Differentiation. Int. J. Mol. Sci. 2020, 21, 2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and Obesity-Related Cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Principe, D.R.; Singh, S.K.; Viswakarma, N.; Sondarva, G.; Rana, B.; Rana, A. Mitogen-Activated Protein Kinase Inhibitors and T-Cell-Dependent Immunotherapy in Cancer. Pharmaceuticals 2020, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Aktar, M.K.; Kido-Nakahara, M.; Furue, M.; Nakahara, T. Mutual upregulation of endothelin-1 and IL-25 in atopic dermatitis. Allergy 2015, 70, 846–854. [Google Scholar] [CrossRef]
- Nakahara, T.; Kido-Nakahara, M.; Ohno, F.; Ulzii, D.; Chiba, T.; Tsuji, G.; Furue, M. The pruritogenic mediator endothelin-1 shifts the dendritic cell-T-cell response toward Th17/Th1 polarization. Allergy 2018, 73, 511–515. [Google Scholar] [CrossRef]
- Moroi, Y.; Yu, B.; Urabe, K.; Koga, T.; Nakahara, T.; Dainichi, T.; Uchi, H.; Furue, M. Effects of MAPK inhibitors on CCR4-mediated chemotaxis against thymus and activation-regulated chemokine (TARC/CCL17). J. Dermatol. Sci. 2004, 36, 186–188. [Google Scholar] [CrossRef]
- Ryu, W.I.; Lee, H.; Bae, H.C.; Jeon, J.; Ryu, H.J.; Kim, J.; Imai, Y.; Yamanishi, K. IL-33 down-regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J. Dermatol. Sci. 2018, 90, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Kido-Nakahara, M.; Wang, B.; Ohno, F.; Tsuji, G.; Ulzii, D.; Takemura, M.; Furue, M.; Nakahara, T. Inhibition of mite-induced dermatitis, pruritus, and nerve sprouting in mice by the endothelin receptor antagonist bosentan. Allergy 2021, 76, 291–301. [Google Scholar] [CrossRef]
- Alexander, H.; Brown, S.; Danby, S.; Flohr, C. Research Techniques Made Simple: Transepidermal Water Loss Measurement as a Research Tool. J. Investig. Dermatol. 2018, 138, 2295–2300. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Izuhara, K.; Onozuka, D.; Nunomura, S.; Tamagawa-Mineoka, R.; Masuda, K.; Ichiyama, S.; Saeki, H.; Kabata, Y.; Abe, R.; et al. Exploration of biomarkers to predict clinical improvement of atopic dermatitis in patients treated with dupilumab: A study protocol. Medicine 2020, 99, e22043. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Silverberg, J.I.; Nemoto, O.; Forman, S.B.; Wilke, A.; Prescilla, R.; de la Peña, A.; Nunes, F.P.; Janes, J.; Gamalo, M.; et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 2019, 80, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Kido-Nakahara, M.; Buddenkotte, J.; Kempkes, C.; Ikoma, A.; Cevikbas, F.; Akiyama, T.; Nunes, F.; Seeliger, S.; Hasdemir, B.; Mess, C.; et al. Neural peptidase endothelin-converting enzyme 1 regulates endothelin 1–induced pruritus. J. Clin. Investig. 2014, 124, 2683–2695. [Google Scholar] [CrossRef] [Green Version]
- Kakinuma, T.; Nakamura, K.; Wakugawa, M.; Mitsui, H.; Tada, Y.; Saeki, H.; Torii, H.; Asahina, A.; Onai, N.; Matsushima, K.; et al. Thymus and activation-regulated chemokine in atopic dermatitis: Serum thymus and activation-regulated chemokine level is closely related with disease activity. J. Allergy Clin. Immunol. 2001, 107, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y. Thymus and activation-regulated chemokine as a clinical biomarker in atopic dermatitis. J. Dermatol. 2014, 41, 221–229. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Qiu, L.; Song, H.; Dang, N. MAPK Pathway Involved in Epidermal Terminal Differentiation of Normal Human Epidermal Keratinocytes. Open Med. 2018, 13, 189–195. [Google Scholar] [CrossRef]
- Cursons, J.; Gao, J.; Hurley, D.G.; Print, C.G.; Dunbar, P.R.; Jacobs, M.D.; Crampin, E.J. Regulation of ERK-MAPK signaling in human epidermis. BMC Syst. Biol. 2015, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Ryu, W.-I.; Lee, H.; Kim, J.H.; Bae, H.C.; Ryu, H.J.; Son, S.W. IL-33 induces Egr-1-dependent TSLP expression via the MAPK pathways in human keratinocytes. Exp. Dermatol. 2015, 24, 857–863. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, T.; Oba, J.; Shimomura, C.; Kido-Nakahara, M.; Furue, M. Early Tumor-Infiltrating Dendritic Cells Change their Characteristics Drastically in Association with Murine Melanoma Progression. J. Investig. Dermatol. 2016, 136, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamcheu, J.C.; Esnault, S.; Adhami, V.M.; Noll, A.L.; Banang-Mbeumi, S.; Roy, T.; Singh, S.S.; Huang, S.; Kousoulas, K.G.; Mukhtar, H. Fisetin, a 3,7,3’,4’-Tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK Pathways and Ameliorates Psoriasis Pathology in 2D and 3D Organotypic Human Inflammatory Skin Models. Cells 2019, 8, 1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′to 3′) | ||
|---|---|---|---|
| human | β-actin | forward | ATTGCCGACAGGATGCAGA |
| reverse | GAGTACTTGCGCTCAGGAGGA | ||
| human | Filaggrin | forward | CATGGCAGCTATGGTAGTGCAGA |
| reverse | ACCAAACGCACTTGCTTTACAGA | ||
| human | Involucrin | forward | TAACCACCCGCAGTGTCCAG |
| reverse | ACAGATGAGACGGGCCACCTA | ||
| mouse | β-actin | forward | GGCTGTATTCCCCTCCATCG |
| reverse | CCAGTTGGTAACAATGCCATGT | ||
| mouse | CCL-17 | forward | AGGTCACTTCAGATGCTGCTC |
| reverse | ACTCTCGGCCTACATTGGTG | ||
| mouse | CCL-22 | forward | GACACCTGACGAGGACACA |
| reverse | GCAGAGGGTGACGGATGTAG | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeze, N.; Kido-Nakahara, M.; Tsuji, G.; Maehara, E.; Sato, Y.; Sakai, S.; Fujishima, K.; Hashimoto-Hachiya, A.; Furue, M.; Nakahara, T. Role of ERK Pathway in the Pathogenesis of Atopic Dermatitis and Its Potential as a Therapeutic Target. Int. J. Mol. Sci. 2022, 23, 3467. https://doi.org/10.3390/ijms23073467
Zeze N, Kido-Nakahara M, Tsuji G, Maehara E, Sato Y, Sakai S, Fujishima K, Hashimoto-Hachiya A, Furue M, Nakahara T. Role of ERK Pathway in the Pathogenesis of Atopic Dermatitis and Its Potential as a Therapeutic Target. International Journal of Molecular Sciences. 2022; 23(7):3467. https://doi.org/10.3390/ijms23073467
Chicago/Turabian StyleZeze, Nahoko, Makiko Kido-Nakahara, Gaku Tsuji, Eriko Maehara, Yuki Sato, Sawako Sakai, Kei Fujishima, Akiko Hashimoto-Hachiya, Masutaka Furue, and Takeshi Nakahara. 2022. "Role of ERK Pathway in the Pathogenesis of Atopic Dermatitis and Its Potential as a Therapeutic Target" International Journal of Molecular Sciences 23, no. 7: 3467. https://doi.org/10.3390/ijms23073467
APA StyleZeze, N., Kido-Nakahara, M., Tsuji, G., Maehara, E., Sato, Y., Sakai, S., Fujishima, K., Hashimoto-Hachiya, A., Furue, M., & Nakahara, T. (2022). Role of ERK Pathway in the Pathogenesis of Atopic Dermatitis and Its Potential as a Therapeutic Target. International Journal of Molecular Sciences, 23(7), 3467. https://doi.org/10.3390/ijms23073467