
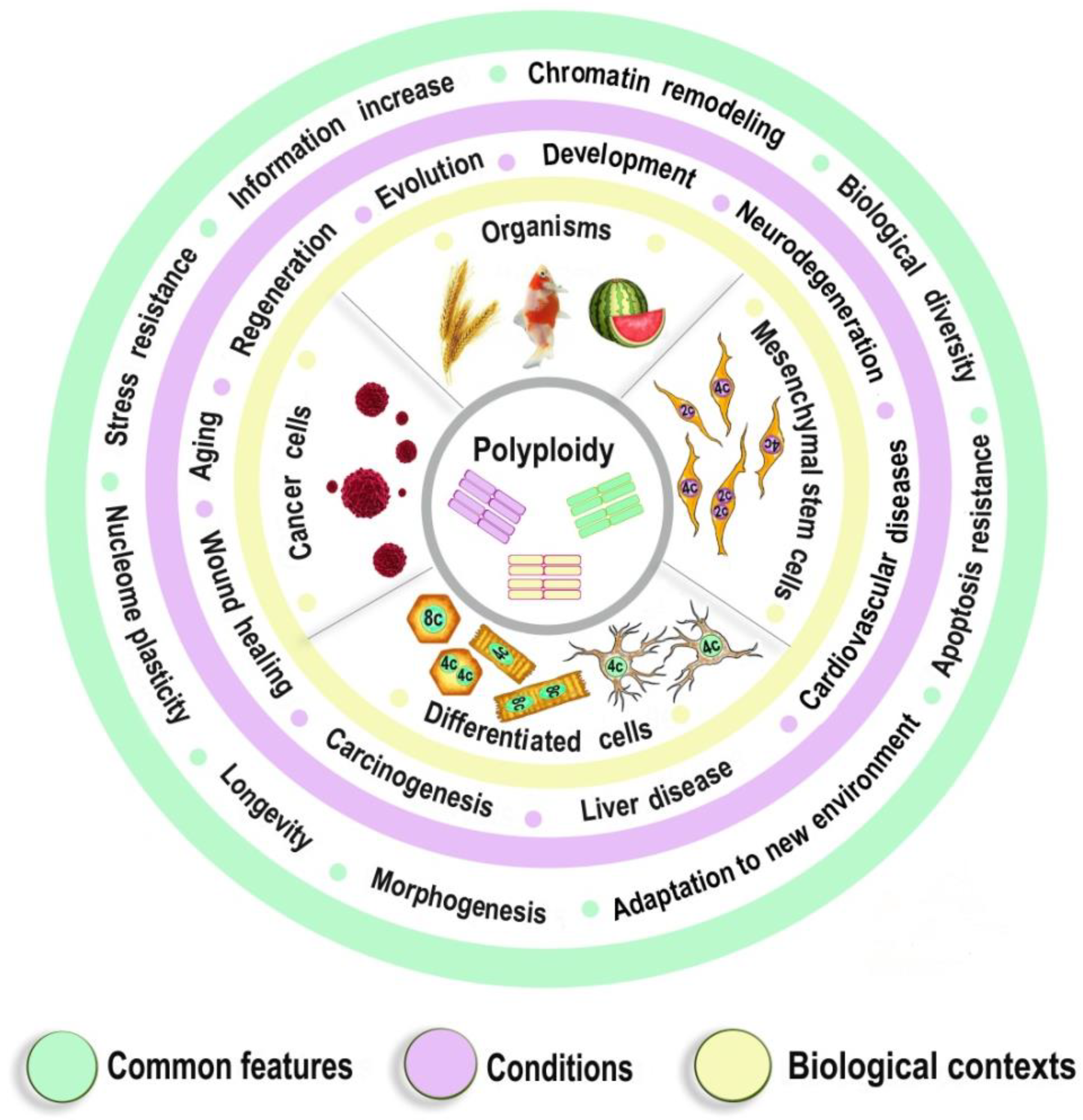
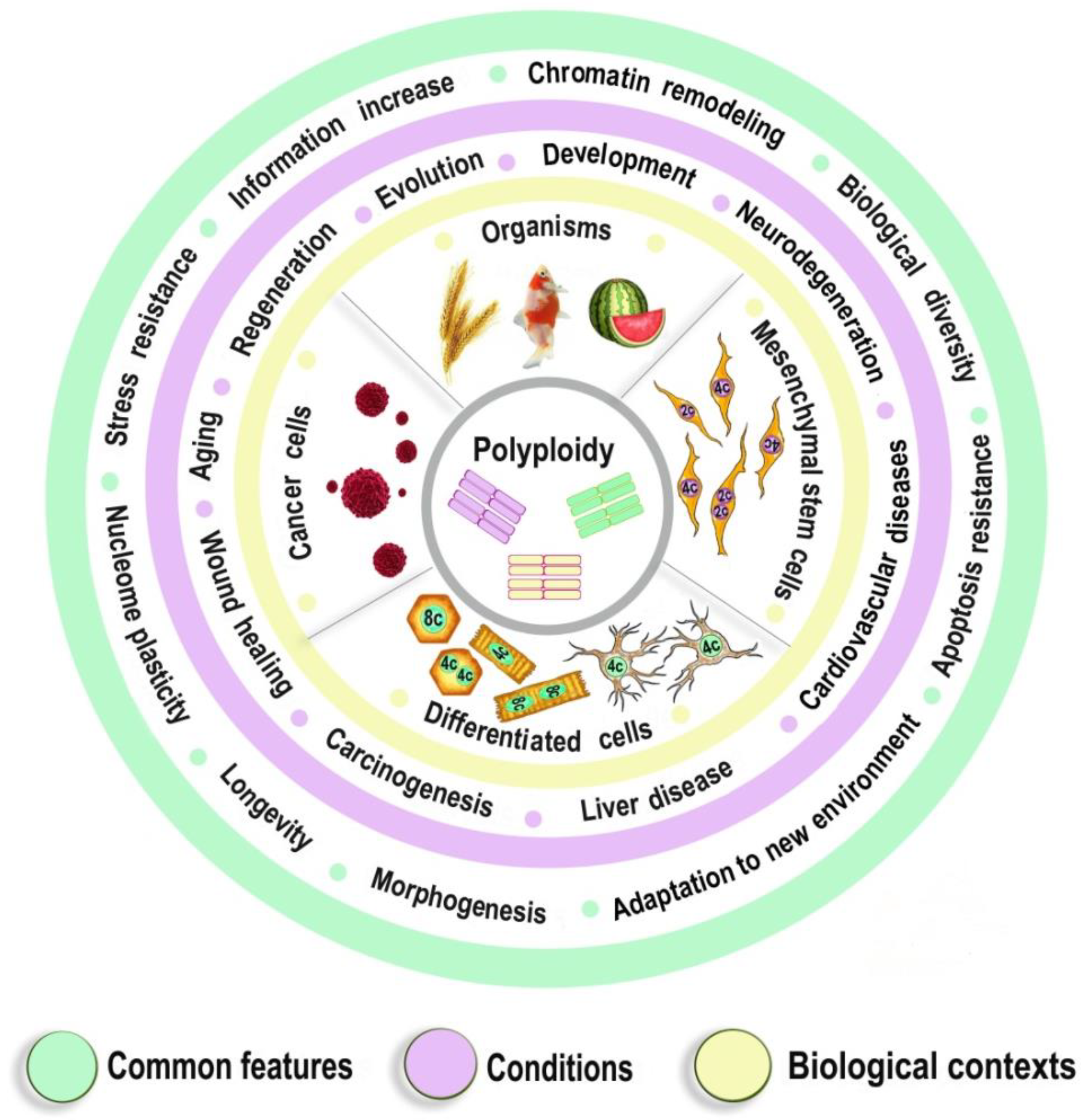
Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Evolution
2.1. General Picture
2.2. Cellular and Molecular Aspects
2.3. Polyploidy in Agriculture and Aquaculture Biotechnology
3. Evolutionary Medicine
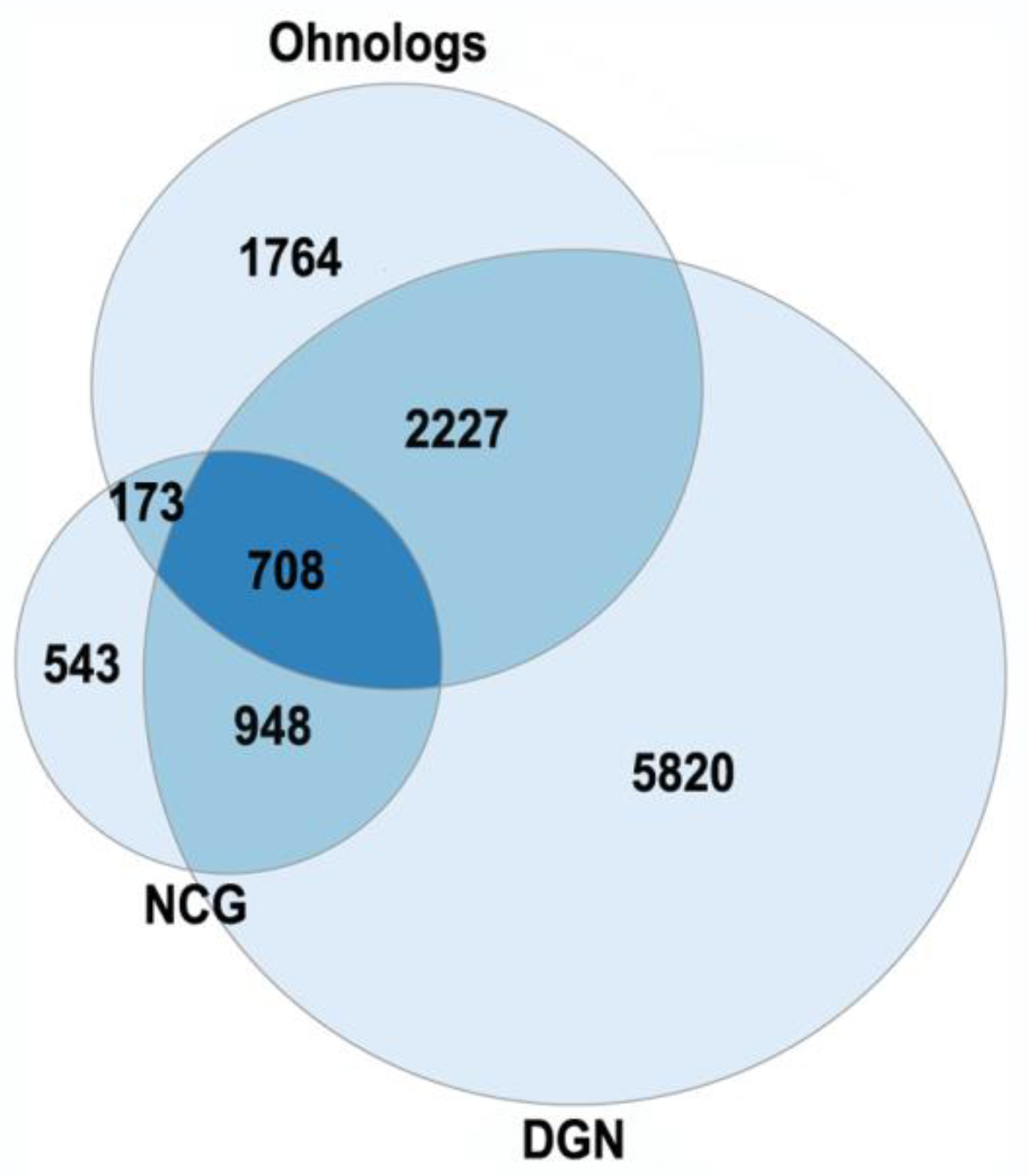
3.1. Ohnologs and Diseases
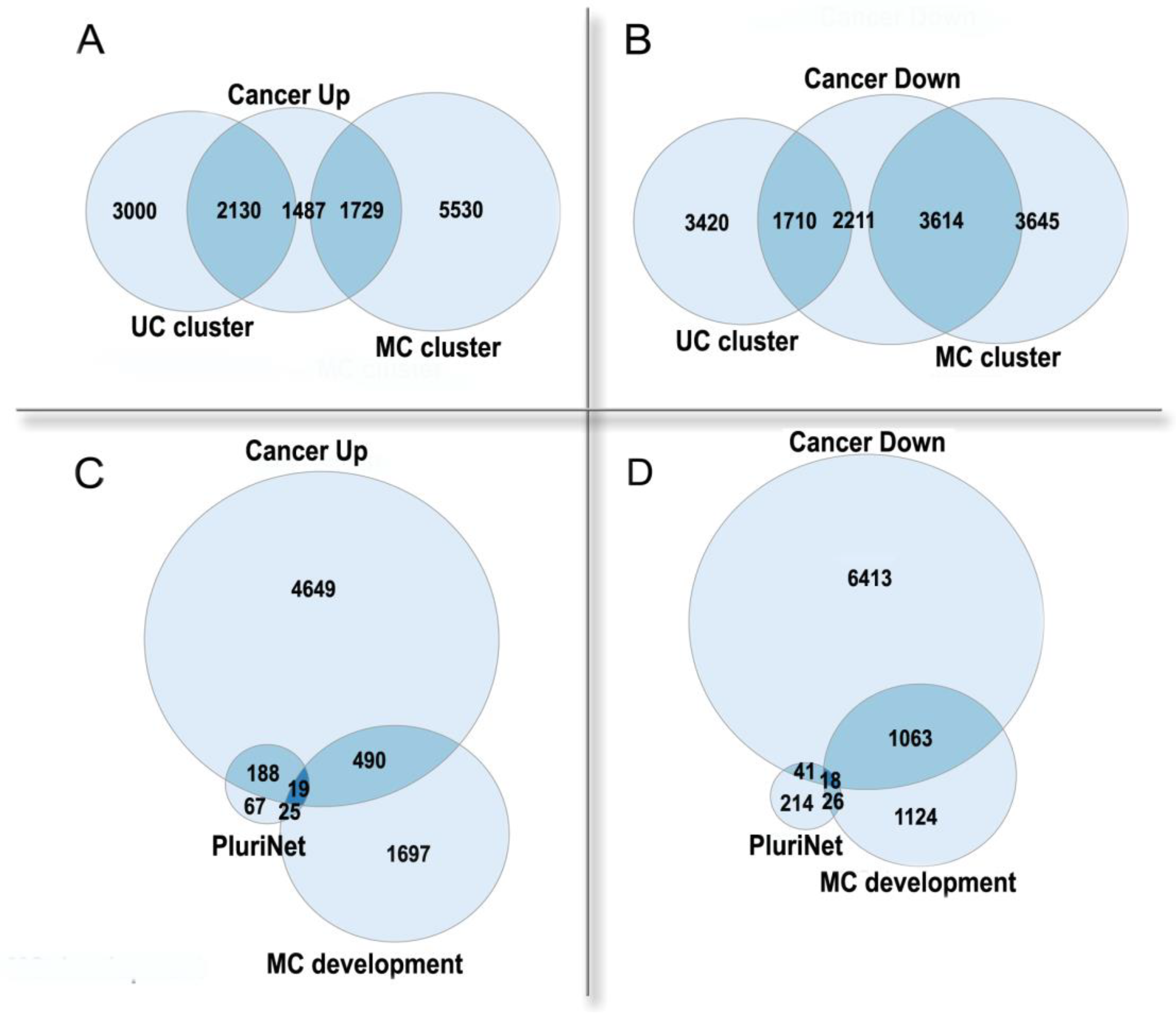
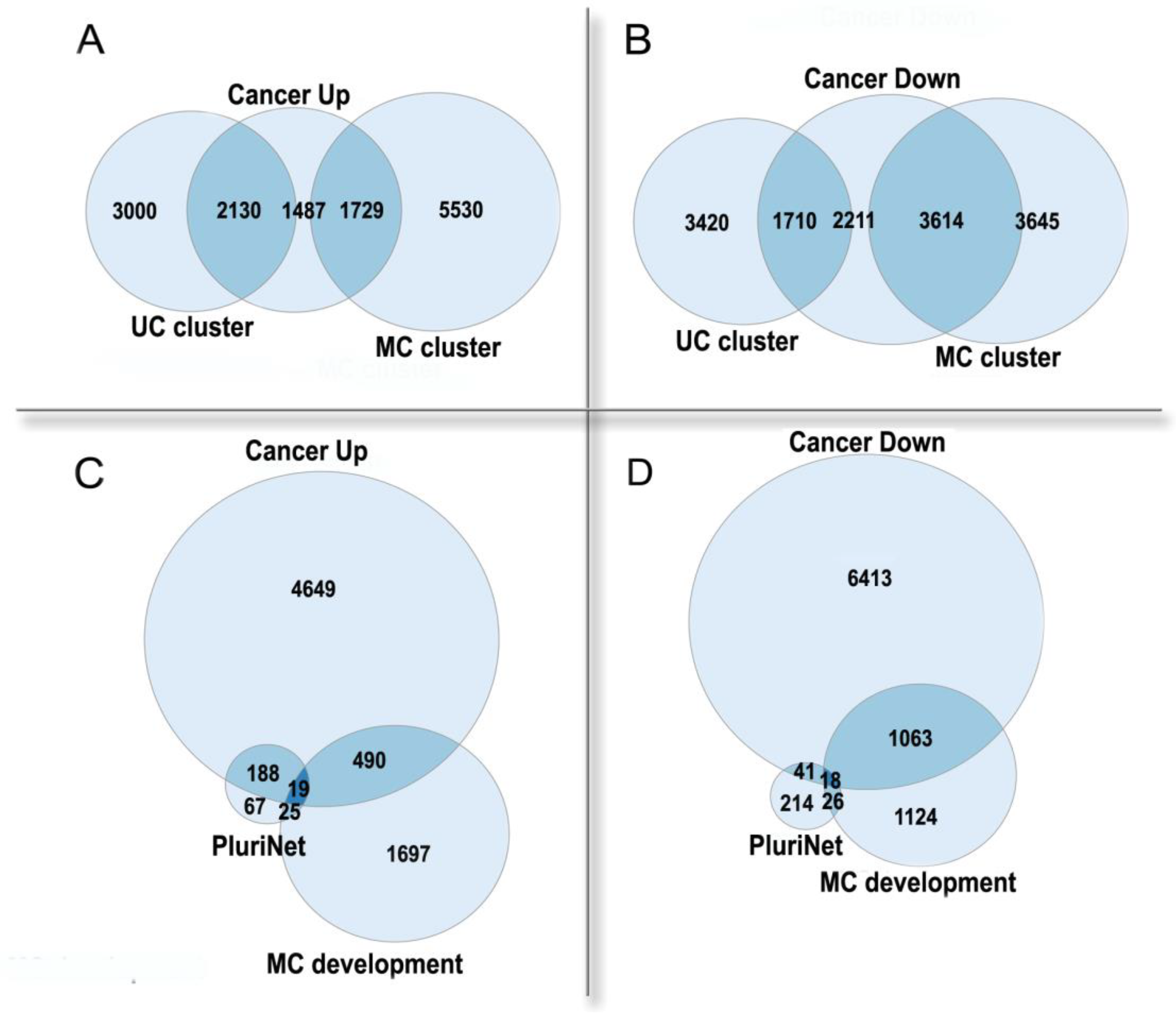
3.2. Polyploidy in Cancer
4. Somatic Polyploidy
4.1. Somatic Polyploidy Is a Way of Adaptation to Stress
4.2. Polyploid Cells Reduce the Functional Capacity of the Organ
4.3. Functional Load Can Control Polyploidization during Postnatal Organogenesis of Heart and Liver
5. Ploidy-Associated Transcriptome Features Are Related to Stress Response, Metabolism, Morphogenesis, and Longevity
5.1. Ploidy-Associated Transcriptomic Features Are Evolutionary Conserved
5.2. The Epigenetics of Ploidy-Associated Transcriptomic Features
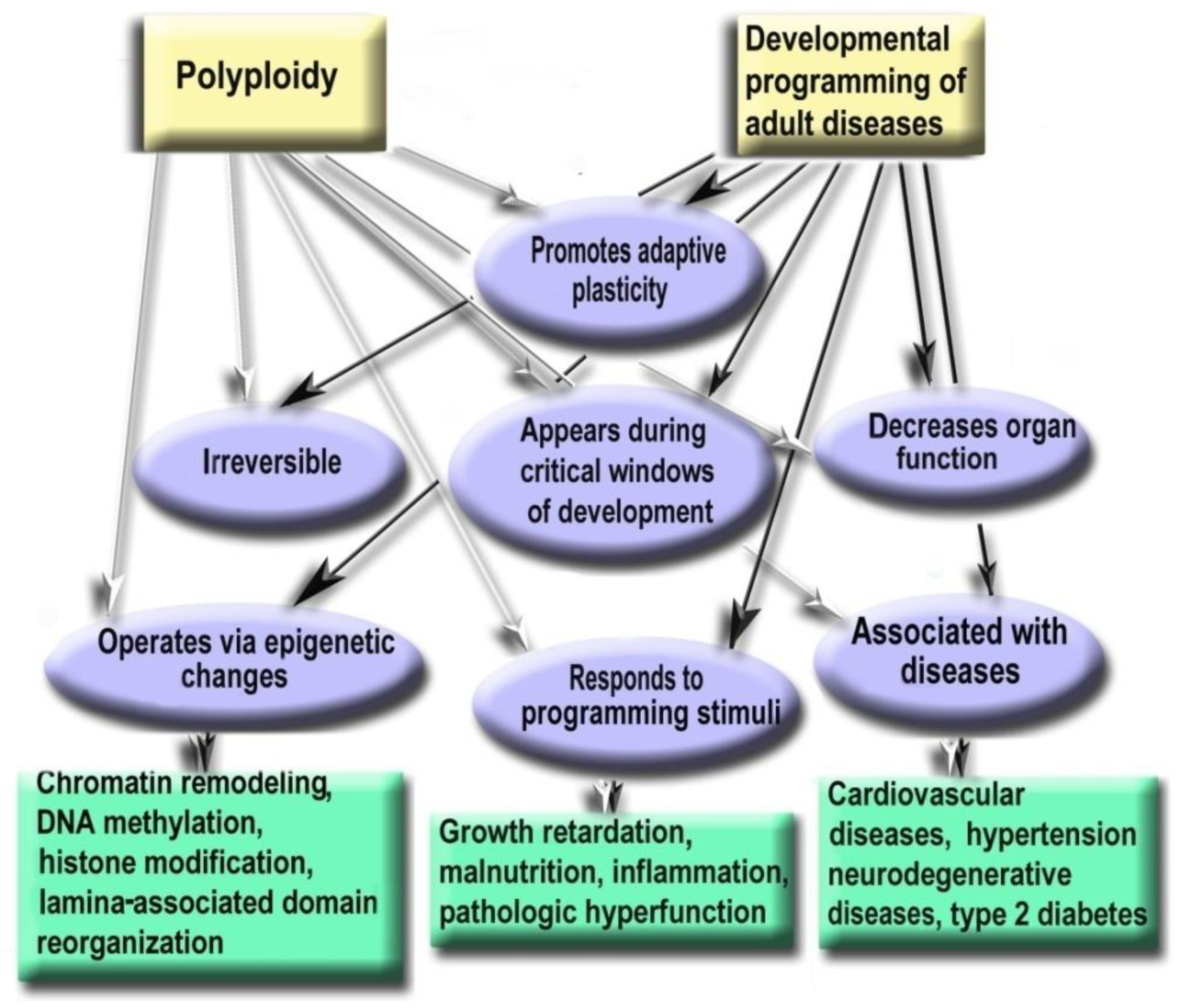
6. Polyploidy Meets the Hallmarks of Developmental Programming of Adult Diseases in Slowly Renewing or Terminally Differentiated Organs
- Polyploidy helps to cope with the adverse environments via the augmentation of stress resistance and adaptation through epigenetic mechanisms [3,78,112]. Furthermore, it is one of the most variable characteristics of somatic cells. The degree of polyploidization in homologous organs shows large across-species diversity. The percentage of cardiomyocytes with polyploid nuclei varies several folds in mammals of similar weight. For example, about 50% of human cardiomyocytes contain nuclei with 4, 8, 16, or even 32 genomes, whereas cardiomyocytes of the grey wolf or reindeer show only about 1% of cells with polyploid nuclei [71,82]. Accordingly, cardiomyocyte ploidy also varies between individuals of the same species. The mean ploidy in the normal human heart varies from about 4× to 10× [77,100,108]. Thus, polyploidy is characterized by the degree of biologic plasticity similar to the renowned factors of ontogenetic programming.
- Polyploid cells (e.g., cardiomyocytes, megakaryocytes, hepatocytes, pancreacytes, vascular epithelial cells, retina epithelium) appeared in the perinatal and early postnatal ontogenesis [9]. These periods are characterized by high biological plasticity and coincide in time with the critical periods of development [131,132].
- Cells of slowly renewing organs, including neurons of neocortex and cerebellum, cardiomyocytes, and hepatocytes, which accumulate additional genomes in infancy, childhood, and pre-pubertant period, retain the increased genome amount throughout their lives, regardless of environmental conditions [9,72,77,100,145].
- Polyploidization is associated with a decrease in organ functional potential [71,82,104,109]. This decrease probably originates from the involvement of polyploidy in the trade-off between proliferation and function that is also a sign of the developmental programming of adult diseases factor [71,107,131].
- The level of ploidy, particularly in cardiomyocytes, responds to the well-established stimuli of developmental programming (including adverse growth conditions, increased functional load, inflammation, and malnutrition) similarly in the various species and various cells [9,71,132,141]. For example, in mammal hepatocytes, cardiomyocytes, retinocytes, and drosophila somatic cells, polyploidy is associated with the increased response to stress, activated pathways of morphogenesis and glycolytic metabolism, and the weakened aerobic metabolism and apoptosis [3,11,112,146].
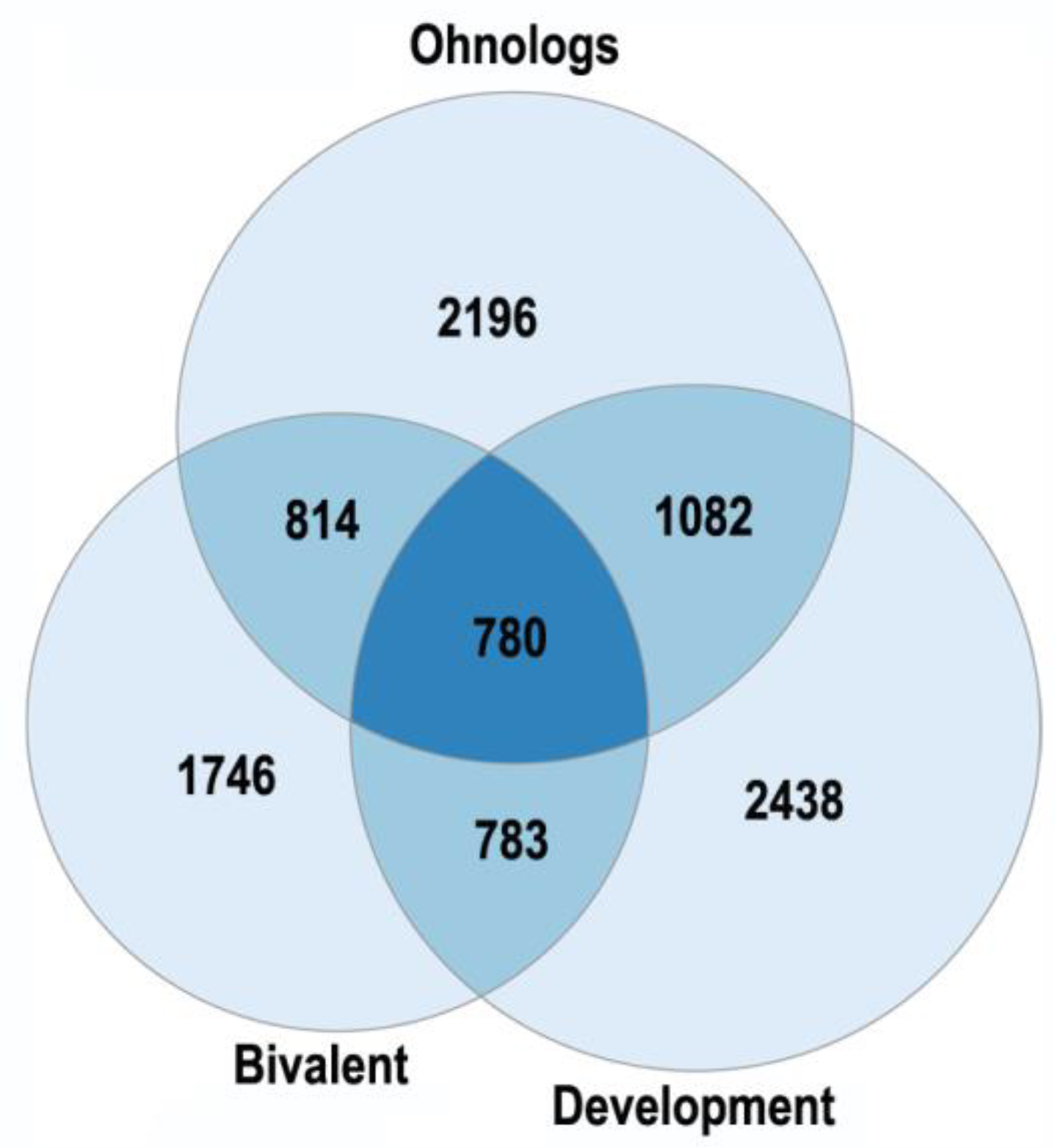
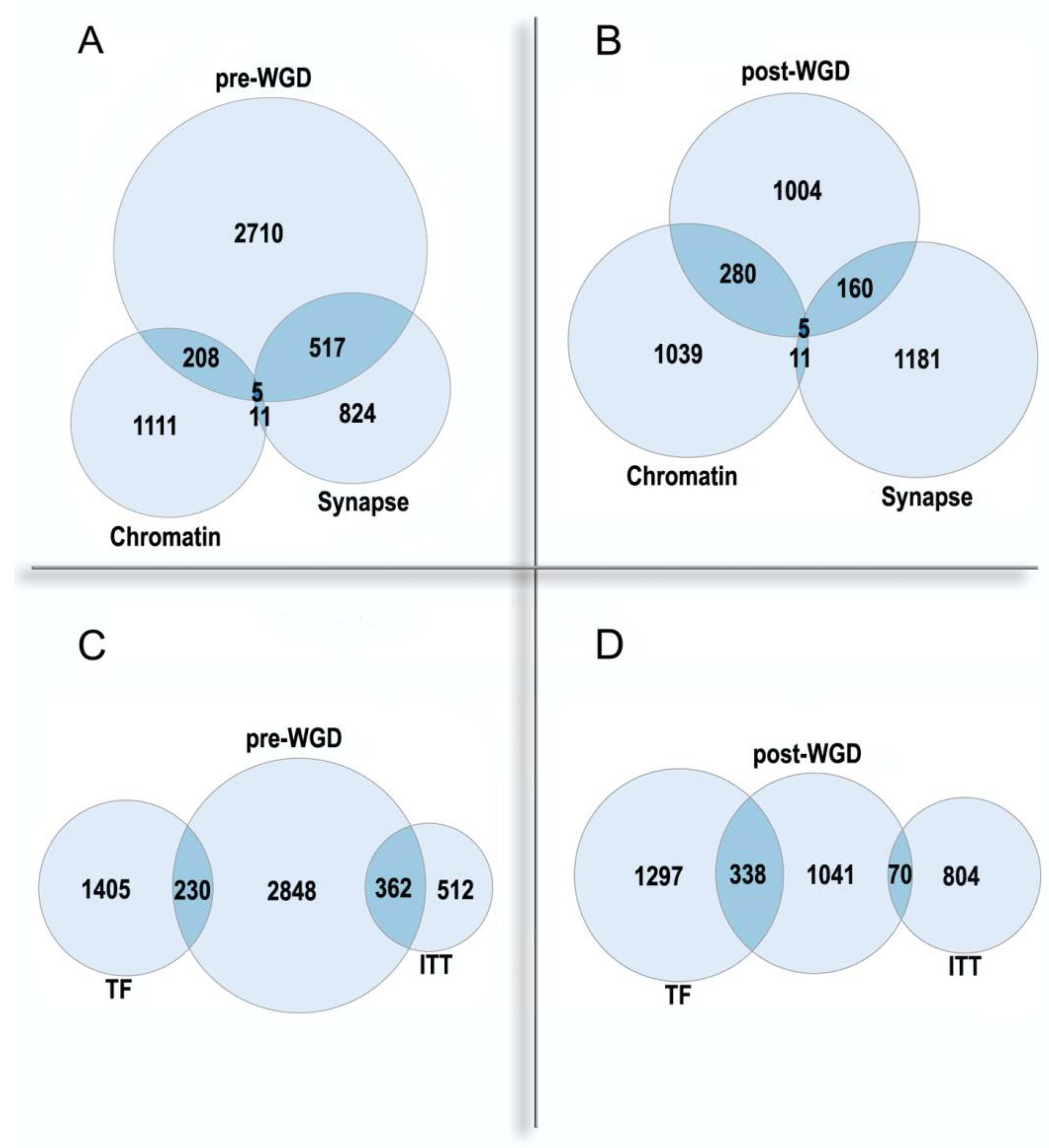
- Polyploidy is associated with epigenetic changes at various levels of genome organization leading to chromatin remodeling and genome instability [95,123,124]. The association between polyploidy and chromatin decompactization under stress was well documented for cardiomyocytes and hepatocytes [76,128]. Polyploidy can alter global patterns of DNA methylation, microRNA expression, and histone modification in mammalian, insect, and plant cells [1,2,78,95,112,127,129]. Polyploid cells show higher expression of bivalent genes, which harbor both activating (H3K4me3) and repressive (H3K27me3) chromatin domains, allowing rapid switching between cellular programs [112]. Overall, ploidy-associated transcriptomic changes occur through the same epigenetic mechanisms as in the developmental programming of health and disease, including chromatin remodeling, DNA methylation, histone modification, and others.
Experimental Studies Confirm the Role of Polyploidy in the Developmental Programming of Health and Disease
7. Genome Duplication in Regeneration and Aging
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gjelsvik, K.J.; Besen-McNally, R.; Losick, V.P. Solving the Polyploid Mystery in Health and Disease. Trends Genet. 2019, 35, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in Liver Development, Homeostasis and Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.T.; Soltis, D.E.; Soltis, P.S.; Ashman, T.-L.; Van de Peer, Y. Polyploidy: A Biological Force From Cells to Ecosystems. Trends Cell Biol. 2020, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Dörnen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811. [Google Scholar] [CrossRef] [Green Version]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The Evolutionary Significance of Polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Van de Peer, Y.; Ashman, T.-L.; Soltis, P.S.; Soltis, D.E. Polyploidy: An Evolutionary and Ecological Force in Stressful Times. Plant Cell 2021, 33, 11–26. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Genome Multiplication as Adaptation to Tissue Survival: Evidence from Gene Expression in Mammalian Heart and Liver. Genomics 2007, 89, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E. Somatic Polyploidy Promotes Cell Function under Stress and Energy Depletion: Evidence from Tissue-Specific Mammal Transcriptome. Funct. Integr. Genom. 2010, 10, 433–446. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Sidorenko, N.V.; Beyer, T.V.; Vinogradov, A.E. Neonatal Cardiomyocyte Ploidy Reveals Critical Windows of Heart Development. Int. J. Cardiol. 2010, 141, 81–91. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Erenpreisa, J.A.; Salmina, K.A.; Vazquez-Martin, A.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Polyploidy Activates Biological Pathways Related to Morphogenesis in Mammalian Tissues. MOJ Immunol. 2018, 6, 90–93. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Anatskaya, O.V.; Giuliani, A.; Erenpreisa, J.; Huang, S.; Salmina, K.; Inashkina, I.; Huna, A.; Nikolsky, N.N.; Vinogradov, A.E. Somatic Polyploidy Is Associated with the Upregulation of C-MYC Interacting Genes and EMT-like Signature. Oncotarget 2016, 7, 75235–75260. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Evolutionary Framework of the Human Interactome: Unicellular and Multicellular Giant Clusters. Biosystems 2019, 181, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-86661-6. [Google Scholar]
- Singh, P.P.; Isambert, H. OHNOLOGS v2: A Comprehensive Resource for the Genes Retained from Whole Genome Duplication in Vertebrates. Nucleic Acids Res. 2020, 48, D724–D730. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.; Sato, Y.; Sinclair, R.; Tsukamoto, K.; Nishida, M. Rapid Genome Reshaping by Multiple-Gene Loss after Whole-Genome Duplication in Teleost Fish Suggested by Mathematical Modeling. Proc. Natl. Acad. Sci. USA 2015, 112, 14918–14923. [Google Scholar] [CrossRef] [Green Version]
- Robertson, F.M.; Gundappa, M.K.; Grammes, F.; Hvidsten, T.R.; Redmond, A.K.; Lien, S.; Martin, S.A.M.; Holland, P.W.H.; Sandve, S.R.; Macqueen, D.J. Lineage-Specific Rediploidization Is a Mechanism to Explain Time-Lags between Genome Duplication and Evolutionary Diversification. Genome Biol. 2017, 18, 111. [Google Scholar] [CrossRef] [Green Version]
- Soppa, J. Polyploidy and Community Structure. Nat. Microbiol. 2017, 2, 16261. [Google Scholar] [CrossRef]
- Hu, G.; Wendel, J.F. Cis-Trans Controls and Regulatory Novelty Accompanying Allopolyploidization. New Phytol. 2019, 221, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.J.; Carter, T.F.; Greenbaum, E.; Gvoždík, V.; Kelley, D.B.; McLaughlin, P.J.; Pauwels, O.S.G.; Portik, D.M.; Stanley, E.L.; Tinsley, R.C.; et al. Genetics, Morphology, Advertisement Calls, and Historical Records Distinguish Six New Polyploid Species of African Clawed Frog (Xenopus, Pipidae) from West and Central Africa. PLoS ONE 2015, 10, e0142823. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Stöck, M.; Kneitz, S.; Klopp, C.; Woltering, J.M.; Adolfi, M.C.; Feron, R.; Prokopov, D.; Makunin, A.; Kichigin, I.; et al. The Sterlet Sturgeon Genome Sequence and the Mechanisms of Segmental Rediploidization. Nat. Ecol. Evol. 2020, 4, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Moritz, C.; Bi, K. Spontaneous Speciation by Ploidy Elevation: Laboratory Synthesis of a New Clonal Vertebrate. Proc. Natl. Acad. Sci. USA 2011, 108, 9733–9734. [Google Scholar] [CrossRef] [Green Version]
- Otto, S.P. The Evolutionary Consequences of Polyploidy. Cell 2007, 131, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Evans, B.J.; Upham, N.S.; Golding, G.B.; Ojeda, R.A.; Ojeda, A.A. Evolution of the Largest Mammalian Genome. Genome Biol. Evol. 2017, 9, 1711–1724. [Google Scholar] [CrossRef]
- Barker, M.S.; Arrigo, N.; Baniaga, A.E.; Li, Z.; Levin, D.A. On the Relative Abundance of Autopolyploids and Allopolyploids. New Phytol. 2016, 210, 391–398. [Google Scholar] [CrossRef]
- Venkatachalam, A.B.; Parmar, M.B.; Wright, J.M. Evolution of the Duplicated Intracellular Lipid-Binding Protein Genes of Teleost Fishes. Mol. Genet. Genom. MGG 2017, 292, 699–727. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Cai, X.; Liang, J.; Freeling, M.; Wang, X. Gene Retention, Fractionation and Subgenome Differences in Polyploid Plants. Nat. Plants 2018, 4, 258–268. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Global versus Local Centrality in Evolution of Yeast Protein Network. J. Mol. Evol. 2009, 68, 192–196. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Anatskaya, O.V. Loss of Protein Interactions and Regulatory Divergence in Yeast Whole-Genome Duplicates. Genomics 2009, 93, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, L.M.; Kuo, A.; Marcet-Houben, M.; Polaino, S.; Salamov, A.; Villalobos-Escobedo, J.M.; Grimwood, J.; Álvarez, M.I.; Avalos, J.; Bauer, D.; et al. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016, 26, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Court, F.; Arnaud, P. An Annotated List of Bivalent Chromatin Regions in Human ES Cells: A New Tool for Cancer Epigenetic Research. Oncotarget 2017, 8, 4110–4124. [Google Scholar] [CrossRef] [Green Version]
- Jeon, A.-J.; Tucker-Kellogg, G. Bivalent Genes That Undergo Transcriptional Switching Identify Networks of Key Regulators of Embryonic Stem Cell Differentiation. BMC 2020, 21, 614. [Google Scholar] [CrossRef]
- Vinogradov, A.E. “Genome Design” Model and Multicellular Complexity: Golden Middle. Nucleic Acids Res. 2006, 34, 5906–5914. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Growth of Biological Complexity from Prokaryotes to Hominids Reflected in the Human Genome. Int. J. Mol. Sci. 2021, 22, 11640. [Google Scholar] [CrossRef]
- Hu, H.; Miao, Y.-R.; Jia, L.-H.; Yu, Q.-Y.; Zhang, Q.; Guo, A.-Y. AnimalTFDB 3.0: A Comprehensive Resource for Annotation and Prediction of Animal Transcription Factors. Nucleic Acids Res. 2019, 47, D33–D38. [Google Scholar] [CrossRef]
- Mattenberger, F.; Sabater-Muñoz, B.; Toft, C.; Fares, M.A. The Phenotypic Plasticity of Duplicated Genes in Saccharomyces Cerevisiae and the Origin of Adaptations. G3: Genes Genomes Genet. 2017, 7, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Wang, X.; Cheng, F. Plant Polyploidy: Origin, Evolution, and Its Influence on Crop Domestication. Hortic. Plant J. 2019, 5, 231–239. [Google Scholar] [CrossRef]
- Ruiz, M.; Quiñones, A.; Martínez-Cuenca, M.R.; Aleza, P.; Morillon, R.; Navarro, L.; Primo-Millo, E.; Martínez-Alcántara, B. Tetraploidy Enhances the Ability to Exclude Chloride from Leaves in Carrizo Citrange Seedlings. J. Plant Physiol. 2016, 205, 1–10. [Google Scholar] [CrossRef]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Wegulo, S.N.; Dababat, A.A.; Erginbas-Orakci, G.; Bouhssini, M.E.; Gautam, P.; Poland, J.; Akci, N.; et al. Genome-Wide Association Study for Multiple Biotic Stress Resistance in Synthetic Hexaploid Wheat. Int. J. Mol. Sci. 2019, 20, 3667. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Carretero-Paulet, L.; Van de Peer, Y. Using Digital Organisms to Study the Evolutionary Consequences of Whole Genome Duplication and Polyploidy. PLoS ONE 2019, 14, e0220257. [Google Scholar] [CrossRef] [Green Version]
- Salman-Minkov, A.; Sabath, N.; Mayrose, I. Whole-Genome Duplication as a Key Factor in Crop Domestication. Nat. Plants 2016, 2, 16115. [Google Scholar] [CrossRef]
- Glombik, M.; Bačovský, V.; Hobza, R.; Kopecký, D. Competition of Parental Genomes in Plant Hybrids. Front. Plant Sci. 2020, 11, 200. [Google Scholar] [CrossRef]
- Guo, H.; Mendrikahy, J.N.; Xie, L.; Deng, J.; Lu, Z.; Wu, J.; Li, X.; Shahid, M.Q.; Liu, X. Transcriptome Analysis of Neo-Tetraploid Rice Reveals Specific Differential Gene Expressions Associated with Fertility and Heterosis. Sci. Rep. 2017, 7, 40139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julião, S.A.; do Ribeiro, C.V.; Lopes, J.M.L.; de Matos, E.M.; Reis, A.C.; Peixoto, P.H.P.; Machado, M.A.; Azevedo, A.L.S.; Grazul, R.M.; de Campos, J.M.S.; et al. Induction of Synthetic Polyploids and Assessment of Genomic Stability in Lippia Alba. Front. Plant Sci. 2020, 11, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Gui, J. Natural and Artificial Polyploids in Aquaculture. Aquac. Fish. 2017, 2, 103–111. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Borkin, L.J.; Günther, R.; Rosanov, J.M. Two Germ Cell Lineages with Genomes of Different Species in One and the Same Animal. Hereditas 1991, 114, 245–251. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Borkin, L.J.; Günther, R.; Rosanov, J.M. Genome Elimination in Diploid and Triploid Rana Esculenta Males: Cytological Evidence from DNA Flow Cytometry. Genome 1990, 33, 619–627. [Google Scholar] [CrossRef]
- Voskarides, K. Editorial: A New Bright Era for Evolutionary Medicine. J. Mol. Evol. 2020, 88, 1–2. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “Virgin Birth”, Polyploidy, and the Origin of Cancer. Oncoscience 2014, 2, 3–14. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered Interactions between Unicellular and Multicellular Genes Drive Hallmarks of Transformation in a Diverse Range of Solid Tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, V.F. ACLS Cancers: Genomic and Epigenetic Changes Transform the Cell of Origin of Cancer into a Tumorigenic Pathogen of Unicellular Organization and Lifestyle. Gene 2020, 726, 144174. [Google Scholar] [CrossRef]
- Fotiou, E.; Williams, S.; Martin-Geary, A.; Robertson, D.L.; Tenin, G.; Hentges, K.E.; Keavney, B. Integration of Large-Scale Genomic Data Sources With Evolutionary History Reveals Novel Genetic Loci for Congenital Heart Disease. Circ. Genom. Precis. Med. 2019, 12, 442–451. [Google Scholar] [CrossRef]
- Arbabian, A.; Iftinca, M.; Altier, C.; Singh, P.P.; Isambert, H.; Coscoy, S. Mutations in Calmodulin-Binding Domains of TRPV4/6 Channels Confer Invasive Properties to Colon Adenocarcinoma Cells. Channels 2020, 14, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Makino, T.; Khor, S.-S.; Toyoda, H.; Miyagawa, T.; Liu, X.; Kuwabara, H.; Kano, Y.; Shimada, T.; Sugiyama, T.; et al. Sensitivity to Gene Dosage and Gene Expression Affects Genes with Copy Number Variants Observed among Neuropsychiatric Diseases. BMC Med. Genom. 2020, 13, 55. [Google Scholar] [CrossRef]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A Comprehensive Catalogue of Known and Candidate Cancer Genes from Cancer Sequencing Screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Dujon, A.M.; Aktipis, A.; Alix-Panabières, C.; Amend, S.R.; Boddy, A.M.; Brown, J.S.; Capp, J.-P.; DeGregori, J.; Ewald, P.; Gatenby, R.; et al. Identifying Key Questions in the Ecology and Evolution of Cancer. Evol. Appl. 2021, 14, 877–892. [Google Scholar] [CrossRef]
- Pienta, K.J.; Hammarlund, E.U.; Brown, J.S.; Amend, S.R.; Axelrod, R.M. Cancer Recurrence and Lethality Are Enabled by Enhanced Survival and Reversible Cell Cycle Arrest of Polyaneuploid Cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2020838118. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Human Transcriptome Nexuses: Basic-Eukaryotic and Metazoan. Genomics 2010, 95, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V. Cell-Cycle Dependence of Transcriptome Gene Modules: Comparison of Regression Lines. FEBS J. 2020, 287, 4427–4439. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. How the Evolution of Multicellularity Set the Stage for Cancer. Br. J. Cancer 2018, 118, 145–152. [Google Scholar] [CrossRef]
- Liu, J. The Dualistic Origin of Human Tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Brock, A.; Huang, S. Precision Oncology: Between Vaguely Right and Precisely Wrong. Cancer Res. 2017, 77, 6473–6479. [Google Scholar] [CrossRef] [Green Version]
- Shabo, I.; Svanvik, J.; Lindström, A.; Lechertier, T.; Trabulo, S.; Hulit, J.; Sparey, T.; Pawelek, J. Roles of Cell Fusion, Hybridization and Polyploid Cell Formation in Cancer Metastasis. World J. Clin. Oncol. 2020, 11, 121–135. [Google Scholar] [CrossRef]
- Vinogradov, A.E.; Ezhevsky, S.A.; Rosanov, J.M.; Kazhdan, I.A.; Zweibach, A.S. Loosening of Cell Cycle Controls of Human Lymphocytes under the Action of Tumour Promoter TPA. Cell Prolif. 1991, 24, 493–505. [Google Scholar] [CrossRef]
- Miles, D.M.; Desdouets, C.; Géli, V. Histone Stress: An Unexplored Source of Chromosomal Instability in Cancer? Curr. Genet. 2019, 65, 1081–1088. [Google Scholar] [CrossRef]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Kim, Y.K. Cancer Stem Cells as a Potential Target to Overcome Multidrug Resistance. Front. Oncol. 2020, 10, 764. [Google Scholar] [CrossRef]
- Shu, Z.; Row, S.; Deng, W.-M. Endoreplication: The Good, the Bad, and the Ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-Genome Doubling Confers Unique Genetic Vulnerabilities on Tumour Cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E. Heart and Liver as Developmental Bottlenecks of Mammal Design: Evidence from Cell Polyploidization. Biol. J. Linn. Soc. 2004, 83, 175–186. [Google Scholar] [CrossRef]
- Lazzeri, E.; Angelotti, M.L.; Conte, C.; Anders, H.-J.; Romagnani, P. Surviving Acute Organ Failure: Cell Polyploidization and Progenitor Proliferation. Trends Mol. Med. 2019, 25, 366–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandakumar, S.; Rozich, E.; Buttitta, L. Cell Cycle Re-Entry in the Nervous System: From Polyploidy to Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 698661. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.C.; Kobielski, S.; Park, J.; Losick, V.P. Polyploidy in Tissue Repair and Regeneration. Cold Spring Harb. Perspect. Biol. 2021, 13, a040881. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Matveev, I.V.; Sidorenko, N.V.; Kharchenko, M.V.; Kropotov, A.V.; Vinogradov, A.E. Changes in the Heart of Neonatal Rats after Cryptosporidial Gastroenteritis of Different Degrees of Severity. J. Evol. Biochem. Physiol. 2013, 49, 509–518. [Google Scholar] [CrossRef]
- Silva, I.S.; Ghiraldini, F.G.; Veronezi, G.M.B.; Mello, M.L.S. Polyploidy and Nuclear Phenotype Characteristics of Cardiomyocytes from Diabetic Adult and Normoglycemic Aged Mice. Acta Histochem. 2018, 120, 84–94. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Anatskaya, O.; Cragg, M.S. Paradoxes of Cancer: Survival at the Brink. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Kirillova, A.; Han, L.; Liu, H.; Kühn, B. Polyploid Cardiomyocytes: Implications for Heart Regeneration. Dev. Camb. Engl. 2021, 148, dev199401. [Google Scholar] [CrossRef]
- Neiman, M.; Beaton, M.J.; Hessen, D.O.; Jeyasingh, P.D.; Weider, L.J. Endopolyploidy as a Potential Driver of Animal Ecology and Evolution. Biol. Rev. Camb. Philos. Soc. 2017, 92, 234–247. [Google Scholar] [CrossRef] [Green Version]
- Pandit, S.K.; Westendorp, B.; de Bruin, A. Physiological Significance of Polyploidization in Mammalian Cells. Trends Cell Biol. 2013, 23, 556–566. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E. Paradoxical Relationship between Protein Content and Nucleolar Activity in Mammalian Cardiomyocytes. Genome 2004, 47, 565–578. [Google Scholar] [CrossRef]
- Lin, H.; Huang, Y.-S.; Fustin, J.-M.; Doi, M.; Chen, H.; Lai, H.-H.; Lin, S.-H.; Lee, Y.-L.; King, P.-C.; Hou, H.-S.; et al. Hyperpolyploidization of Hepatocyte Initiates Preneoplastic Lesion Formation in the Liver. Nat. Commun. 2021, 12, 645. [Google Scholar] [CrossRef]
- Zheng, L.; Dai, H.; Zhou, M.; Li, X.; Liu, C.; Guo, Z.; Wu, X.; Wu, J.; Wang, C.; Zhong, J.; et al. Polyploid Cells Rewire DNA Damage Response Networks to Overcome Replication Stress-Induced Barriers for Tumour Progression. Nat. Commun. 2012, 3, 815. [Google Scholar] [CrossRef]
- Patterson, M.; Swift, S.K. Residual Diploidy in Polyploid Tissues: A Cellular State with Enhanced Proliferative Capacity for Tissue Regeneration? Stem Cells Dev. 2019, 28, 1527–1539. [Google Scholar] [CrossRef]
- Müller, M.; May, S.; Bird, T.G. Ploidy Dynamics Increase the Risk of Liver Cancer Initiation. Nat. Commun. 2021, 12, 1896. [Google Scholar] [CrossRef]
- Clay, D.E.; Fox, D.T. DNA Damage Responses during the Cell Cycle: Insights from Model Organisms and Beyond. Genes 2021, 12, 1882. [Google Scholar] [CrossRef]
- Matsumoto, T.; Wakefield, L.; Peters, A.; Peto, M.; Spellman, P.; Grompe, M. Proliferative Polyploid Cells Give Rise to Tumors via Ploidy Reduction. Nat. Commun. 2021, 12, 646. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, K.; Luo, X.; Li, L.; Tu, H.-C.; Sehgal, A.; Nguyen, L.H.; Zhang, Y.; Gopal, P.; Tarlow, B.D.; et al. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Dev. Cell 2018, 44, 447–459.e5. [Google Scholar] [CrossRef] [Green Version]
- Newcomb, R.; Dean, E.; McKinney, B.J.; Alvarez, J.V. Context-Dependent Effects of Whole-Genome Duplication during Mammary Tumor Recurrence. Sci. Rep. 2021, 11, 14932. [Google Scholar] [CrossRef]
- Windner, S.E.; Manhart, A.; Brown, A.; Mogilner, A.; Baylies, M.K. Nuclear Scaling Is Coordinated among Individual Nuclei in Multinucleated Muscle Fibers. Dev. Cell 2019, 49, 48–62.e3. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of Mononuclear Diploid Cardiomyocytes Underlies Natural Variation in Heart Regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
- Porrello, E.R.; Olson, E.N. A Neonatal Blueprint for Cardiac Regeneration. Stem Cell Res. 2014, 13, 556–570. [Google Scholar] [CrossRef] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Choudhury, S.; Mich-Basso, J.D.; Ammanamanchi, N.; Ganapathy, B.; Suresh, S.; Khaladkar, M.; Singh, J.; Maehr, R.; Zuppo, D.A.; et al. Lamin B2 Levels Regulate Polyploidization of Cardiomyocyte Nuclei and Myocardial Regeneration. Dev. Cell 2020, 53, 42–59.e11. [Google Scholar] [CrossRef]
- Herrtwich, L.; Nanda, I.; Evangelou, K.; Nikolova, T.; Horn, V.; Erny, D.; Stefanowski, J.; Rogell, L.; Klein, C.; Gharun, K.; et al. DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell 2016, 167, 1264–1280.e18. [Google Scholar] [CrossRef]
- Zybina, T.G.; Stein, G.I.; Pozharisski, K.M.; Zybina, E.V. Invasion and Genome Reproduction of the Trophoblast Cells of Placenta Junctional Zone in the Field Vole, Microtus Rossiaemeridionalis. Cell Biol. Int. 2014, 38, 136–143. [Google Scholar] [CrossRef]
- Zybina, T.G.; Zybina, E.V. Role of Cell Cycling and Polyploidy in Placental Trophoblast of Different Mammalian Species. Reprod. Domest. Anim. Zuchthyg. 2020, 55, 895–904. [Google Scholar] [CrossRef]
- Zybina, T.G. Genome Modifications Involved in Developmental Programs of the Placental Trophoblast. In Cytogenetics-Classical and Molecular Strategies for Analysing Heredity Material; Larramendy, M., Soloneski, S., Eds.; IntechOpen: London, UK, 2021; ISBN 978-1-83968-941-3. [Google Scholar]
- Brodsky, V.Y.; Sarkisov, D.S.; Arefyeva, A.M.; Panova, N.W.; Gvasava, I.G. Polyploidy in Cardiac Myocytes of Normal and Hypertrophic Human Hearts; Range of Values. Virchows Arch. Int. J. Pathol. 1994, 424, 429–435. [Google Scholar] [CrossRef]
- Herget, G.W.; Neuburger, M.; Plagwitz, R.; Adler, C.P. DNA Content, Ploidy Level and Number of Nuclei in the Human Heart after Myocardial Infarction. Cardiovasc. Res. 1997, 36, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krane, M.; Dreßen, M.; Santamaria, G.; My, I.; Schneider, C.M.; Dorn, T.; Laue, S.; Mastantuono, E.; Berutti, R.; Rawat, H.; et al. Sequential Defects in Cardiac Lineage Commitment and Maturation Cause Hypoplastic Left Heart Syndrome. Circulation 2021, 144, 1409–1428. [Google Scholar] [CrossRef] [PubMed]
- Besen-McNally, R.; Gjelsvik, K.J.; Losick, V.P. Wound-Induced Polyploidization Is Dependent on Integrin-Yki Signaling. Biol. Open 2021, 10, bio055996. [Google Scholar] [CrossRef] [PubMed]
- Anatskaya, O.V.; Vinogradov, A.E. Myocyte Ploidy in Heart Chambers of Birds with Different Locomotor Activity. J. Exp. Zool. 2002, 293, 427–441. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Vinogradov, A.E.; Kudryavtsev, B.N. Cardiomyocyte Ploidy Levels in Birds with Different Growth Rates. J. Exp. Zool. 2001, 289, 48–58. [Google Scholar] [CrossRef]
- Drigo, A.E.R.; Lev-Ram, V.; Tyagi, S.; Ramachandra, R.; Deerinck, T.; Bushong, E.; Phan, S.; Orphan, V.; Lechene, C.; Ellisman, M.H.; et al. Age Mosaicism across Multiple Scales in Adult Tissues. Cell Metab. 2019, 30, 343–351.e3. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, W.Y.; Uryvaeva, I.V. Cell Polyploidy: Its Relation to Tissue Growth and Function. Int. Rev. Cytol. 1977, 50, 275–332. [Google Scholar] [CrossRef]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.-Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte Proliferation Contributes to Heart Growth in Young Humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [Green Version]
- Vinogradov, A.E.; Anatskaya, O.V.; Kudryavtsev, B.N. Relationship of Hepatocyte Ploidy Levels with Body Size and Growth Rate in Mammals. Genome 2001, 44, 350–360. [Google Scholar] [CrossRef]
- Potapova, T.A.; Seidel, C.W.; Box, A.C.; Rancati, G.; Li, R. Transcriptome Analysis of Tetraploid Cells Identifies Cyclin D2 as a Facilitator of Adaptation to Genome Doubling in the Presence of P53. Mol. Biol. Cell 2016, 27, 3065–3084. [Google Scholar] [CrossRef]
- Katsuda, T.; Hosaka, K.; Matsuzaki, J.; Usuba, W.; Prieto-Vila, M.; Yamaguchi, T.; Tsuchiya, A.; Terai, S.; Ochiya, T. Transcriptomic Dissection of Hepatocyte Heterogeneity: Linking Ploidy, Zonation, and Stem/Progenitor Cell Characteristics. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 161–183. [Google Scholar] [CrossRef] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E.; Vainshelbaum, N.M.; Giuliani, A.; Erenpreisa, J. Phylostratic Shift of Whole-Genome Duplications in Normal Mammalian Tissues towards Unicellularity Is Driven by Developmental Bivalent Genes and Reveals a Link to Cancer. Int. J. Mol. Sci. 2020, 21, 8759. [Google Scholar] [CrossRef]
- Øvrebø, J.I.; Edgar, B.A. Polyploidy in Tissue Homeostasis and Regeneration. Dev. Camb. Engl. 2018, 145, dev156034. [Google Scholar] [CrossRef] [Green Version]
- Sikora, E.; Czarnecka-Herok, J.; Bojko, A.; Sunderland, P. Therapy-Induced Polyploidization and Senescence: Coincidence or Interconnection? Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Walen, K.H. Cell Cycle Stress in Normal Human Cells: A Route to “First Cells” (with/without Fitness Gain) and Cancer-like Cell-Shape Changes. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar] [CrossRef]
- Broughton, K.M.; Khieu, T.; Nguyen, N.; Rosa, M.; Mohsin, S.; Quijada, P.; Wang, B.J.; Echeagaray, O.H.; Kubli, D.A.; Kim, T.; et al. Cardiac Interstitial Tetraploid Cells Can Escape Replicative Senescence in Rodents but Not Large Mammals. Commun. Biol. 2019, 2, 205. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Sidorenko, N.V.; Vinogradov, A.E.; Beyer, T.V. Impact of Neonatal Cryptosporidial Gastroenteritis on Epigenetic Programming of Rat Hepatocytes. Cell Biol. Int. 2007, 31, 420–427. [Google Scholar] [CrossRef]
- Malik, A.; Korol, A.; Weber, M.; Hankeln, T.; Avivi, A.; Band, M. Transcriptome Analysis of the Spalax Hypoxia Survival Response Includes Suppression of Apoptosis and Tight Control of Angiogenesis. BMC Genom. 2012, 13, 615. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Gladyshev, V.N. Molecular Signatures of Longevity: Insights from Cross-Species Comparative Studies. Semin. Cell Dev. Biol. 2017, 70, 190–203. [Google Scholar] [CrossRef]
- Mayfield-Jones, D.; Washburn, J.D.; Arias, T.; Edger, P.P.; Pires, J.C.; Conant, G.C. Watching the Grin Fade: Tracing the Effects of Polyploidy on Different Evolutionary Time Scales. Semin. Cell Dev. Biol. 2013, 24, 320–331. [Google Scholar] [CrossRef]
- Michiue, T.; Yamamoto, T.; Yasuoka, Y.; Goto, T.; Ikeda, T.; Nagura, K.; Nakayama, T.; Taira, M.; Kinoshita, T. High Variability of Expression Profiles of Homeologous Genes for Wnt, Hh, Notch, and Hippo Signaling Pathways in Xenopus Laevis. Dev. Biol. 2017, 426, 270–290. [Google Scholar] [CrossRef]
- Blanc, G.; Wolfe, K.H. Widespread Paleopolyploidy in Model Plant Species Inferred from Age Distributions of Duplicate Genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [Green Version]
- Kind, J.; Pagie, L.; de Vries, S.S.; Nahidiazar, L.; Dey, S.S.; Bienko, M.; Zhan, Y.; Lajoie, B.; de Graaf, C.A.; Amendola, M.; et al. Genome-Wide Maps of Nuclear Lamina Interactions in Single Human Cells. Cell 2015, 163, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D Structures of Individual Mammalian Genomes Studied by Single-Cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Malashicheva, A.; Perepelina, K. Diversity of Nuclear Lamin A/C Action as a Key to Tissue-Specific Regulation of Cellular Identity in Health and Disease. Front. Cell Dev. Biol. 2021, 9, 761469. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lozano, M.; Natarajan, P.; Levi, A.; Katam, R.; Lopez-Ortiz, C.; Nimmakayala, P.; Reddy, U.K. Altered Chromatin Conformation and Transcriptional Regulation in Watermelon Following Genome Doubling. Plant J. Cell Mol. Biol. 2021, 106, 588–600. [Google Scholar] [CrossRef]
- Kuga, T.; Nie, H.; Kazami, T.; Satoh, M.; Matsushita, K.; Nomura, F.; Maeshima, K.; Nakayama, Y.; Tomonaga, T. Lamin B2 Prevents Chromosome Instability by Ensuring Proper Mitotic Chromosome Segregation. Oncogenesis 2014, 3, e94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghiraldini, F.G.; Silva, I.S.; Mello, M.L.S. Polyploidy and Chromatin Remodeling in Hepatocytes from Insulin-Dependent Diabetic and Normoglycemic Aged Mice. Cytom. Part J. Int. Soc. Anal. Cytol. 2012, 81, 755–764. [Google Scholar] [CrossRef]
- Gilsbach, R.; Preissl, S.; Grüning, B.A.; Schnick, T.; Burger, L.; Benes, V.; Würch, A.; Bönisch, U.; Günther, S.; Backofen, R.; et al. Dynamic DNA Methylation Orchestrates Cardiomyocyte Development, Maturation and Disease. Nat. Commun. 2014, 5, 5288. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Y.; Xia, E.-H.; Yao, Q.-Y.; Liu, X.-D.; Gao, L.-Z. Autotetraploid Rice Methylome Analysis Reveals Methylation Variation of Transposable Elements and Their Effects on Gene Expression. Proc. Natl. Acad. Sci. USA 2015, 112, E7022–E7029. [Google Scholar] [CrossRef] [Green Version]
- Bateson, P. Robustness and Plasticity in Development. Wiley Interdiscip. Rev. Cogn. Sci. 2017, 8, e1386. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Low, F.M. Evolutionary and Developmental Mismatches Are Consequences of Adaptive Developmental Plasticity in Humans and Have Implications for Later Disease Risk. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2019, 374, 20180109. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Gutierrez, A.M.; Antonelli, M.C.; Frasch, M.G. Microglial Memory of Early Life Stress and Inflammation: Susceptibility to Neurodegeneration in Adulthood. Neurosci. Biobehav. Rev. 2020, 117, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, Z.; Ghahramani Seno, M.M.; Fereidoni, M. A Neonatal Mild Defect in Brain Insulin Signaling Predisposes a Subclinical Model of Sporadic Alzheimer’s to Develop the Disease. J. Mol. Neurosci. MN 2021, 71, 1473–1484. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Infant Mortality, Childhood Nutrition, and Ischaemic Heart Disease in England and Wales. Lancet Lond. Engl. 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Barker, D.J.P. Coronary Heart Disease: A Disorder of Growth. Horm. Res. 2003, 59 (Suppl. 1), 35–41. [Google Scholar] [CrossRef]
- Golubnitschaja, O.; Costigliola, V. Predictive, Preventive and Personalised Medicine as the Medicine of the Future: Anticipatory Scientific Innovation and Advanced Medical Services. In Anticipation and Medicine; Nadin, M., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 69–85. ISBN 978-3-319-45140-4. [Google Scholar]
- Aagaard-Tillery, K.M.; Grove, K.; Bishop, J.; Ke, X.; Fu, Q.; McKnight, R.; Lane, R.H. Developmental Origins of Disease and Determinants of Chromatin Structure: Maternal Diet Modifies the Primate Fetal Epigenome. J. Mol. Endocrinol. 2008, 41, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Simmons, R.A. Developmental Origins of Adult Disease. Pediatr. Clin. N. Am. 2009, 56, 449–466. [Google Scholar] [CrossRef]
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic Responses and the Developmental Origins of Health and Disease. J. Endocrinol. 2019, 242, T105–T119. [Google Scholar] [CrossRef] [Green Version]
- Ryznar, R.J.; Phibbs, L.; Van Winkle, L.J. Epigenetic Modifications at the Center of the Barker Hypothesis and Their Transgenerational Implications. Int. J. Environ. Res. Public. Health 2021, 18, 12728. [Google Scholar] [CrossRef]
- Lurbe, E.; Ingelfinger, J. Developmental and Early Life Origins of Cardiometabolic Risk Factors: Novel Findings and Implications. Hypertension 2021, 77, 308–318. [Google Scholar] [CrossRef]
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625. [Google Scholar] [CrossRef]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.-C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child Health, Developmental Plasticity, and Epigenetic Programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef]
- Bensley, J.G.; Stacy, V.K.; De Matteo, R.; Harding, R.; Black, M.J. Cardiac Remodelling as a Result of Pre-Term Birth: Implications for Future Cardiovascular Disease. Eur. Heart J. 2010, 31, 2058–2066. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Erenpreisa, J.A.; Nikolsky, N.N.; Vinogradov, A.E. Pairwise Comparison of Mammalian Transcriptomes Associated with the Effect of Polyploidy on the Expression Activity of Developmental Gene Modules. Cell Tissue Biol. 2016, 10, 122–132. [Google Scholar] [CrossRef]
- Gan, P.; Patterson, M.; Velasquez, A.; Wang, K.; Tian, D.; Windle, J.J.; Tao, G.; Judge, D.P.; Makita, T.; Park, T.J.; et al. Tnni3k Alleles Influence Ventricular Mononuclear Diploid Cardiomyocyte Frequency. PLoS Genet. 2019, 15, e1008354. [Google Scholar] [CrossRef]
- Bergmann, O. Cardiomyocytes in Congenital Heart Disease: Overcoming Cytokinesis Failure in Tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 2021, 161, 1587–1590. [Google Scholar] [CrossRef]
- Finch, C.E. Evolution in Health and Medicine Sackler Colloquium: Evolution of the Human Lifespan and Diseases of Aging: Roles of Infection, Inflammation, and Nutrition. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. 1), 1718–1724. [Google Scholar] [CrossRef] [Green Version]
- Curione, M.; Aratari, A.; Amato, S.; Colotto, M.; Barbato, M.; Leone, S.; Tego, A.; Panetti, D.; Parlapiano, C. A Study on QT Interval in Patients Affected with Inflammatory Bowel Disease without Cardiac Involvement. Intern. Emerg. Med. 2010, 5, 307–310. [Google Scholar] [CrossRef]
- Filatova, N.A.; Knyazev, N.A.; Skarlato, S.O.; Anatskaya, O.V.; Vinogradov, A.E. Natural Killer Cell Activity Irreversibly Decreases after Cryptosporidium Gastroenteritis in Neonatal Mice. Parasite Immunol. 2018, 40, e12524. [Google Scholar] [CrossRef]
- Kimmel, G.J.; Dane, M.; Heiser, L.M.; Altrock, P.M.; Andor, N. Integrating Mathematical Modeling with High-Throughput Imaging Explains How Polyploid Populations Behave in Nutrient-Sparse Environments. Cancer Res. 2020, 80, 5109–5120. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Matveev, I.V.; Sidorenko, N.V.; Kharchenko, M.V.; Kropotov, A.V.; Vinogradov, A.E. Remodeling of Rat Cardiomyocytes after Neonatal Cryptosporidiosis. I. Change of Ratio of Isoforms of Myosin Heavy Chains. Cell Tissue Biol. 2012, 6, 40–51. [Google Scholar] [CrossRef]
- Han, P.; Li, W.; Yang, J.; Shang, C.; Lin, C.-H.; Cheng, W.; Hang, C.T.; Cheng, H.-L.; Chen, C.-H.; Wong, J.; et al. Epigenetic Response to Environmental Stress: Assembly of BRG1-G9a/GLP-DNMT3 Repressive Chromatin Complex on Myh6 Promoter in Pathologically Stressed Hearts. Biochim. Biophys. Acta 2016, 1863, 1772–1781. [Google Scholar] [CrossRef]
- Petruseva, I.O.; Evdokimov, A.N.; Lavrik, O.I. Genome Stability Maintenance in Naked Mole-Rat. Acta Nat. 2017, 9, 31–41. [Google Scholar]
- Lucchetta, E.M.; Ohlstein, B. Amitosis of Polyploid Cells Regenerates Functional Stem Cells in the Drosophila Intestine. Cell Stem Cell 2017, 20, 609–620.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alié, A.; Hayashi, T.; Sugimura, I.; Manuel, M.; Sugano, W.; Mano, A.; Satoh, N.; Agata, K.; Funayama, N. The Ancestral Gene Repertoire of Animal Stem Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7093–E7100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz-Trillo, I.; de Mendoza, A. Towards Understanding the Origin of Animal Development. Dev. Camb. Engl. 2020, 147, dev192575. [Google Scholar] [CrossRef]
- Vinogradov, A.E. Accelerated Pathway Evolution in Mouse-like Rodents Involves Cell Cycle Control. Mamm. Genome 2015, 26, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.E.; Anatskaya, O.V. Gene Golden Age Paradox and Its Partial Solution. Genomics 2019, 111, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Sikora, E.; Bielak-Zmijewska, A.; Mosieniak, G. Cellular Senescence in Ageing, Age-Related Disease and Longevity. Curr. Vasc. Pharmacol. 2014, 12, 698–706. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anatskaya, O.V.; Vinogradov, A.E. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. Int. J. Mol. Sci. 2022, 23, 3542. https://doi.org/10.3390/ijms23073542
Anatskaya OV, Vinogradov AE. Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. International Journal of Molecular Sciences. 2022; 23(7):3542. https://doi.org/10.3390/ijms23073542
Chicago/Turabian StyleAnatskaya, Olga V., and Alexander E. Vinogradov. 2022. "Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases" International Journal of Molecular Sciences 23, no. 7: 3542. https://doi.org/10.3390/ijms23073542
APA StyleAnatskaya, O. V., & Vinogradov, A. E. (2022). Polyploidy as a Fundamental Phenomenon in Evolution, Development, Adaptation and Diseases. International Journal of Molecular Sciences, 23(7), 3542. https://doi.org/10.3390/ijms23073542