
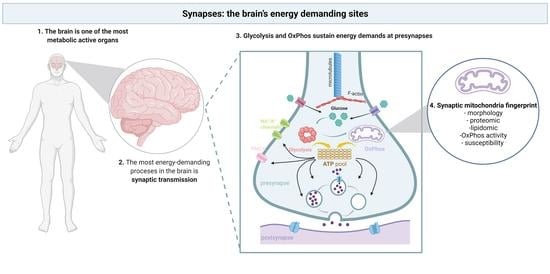
Synapses: The Brain’s Energy-Demanding Sites


Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Brain: The Energy Demanding Organ
2.1. Brain: The “Sugar” Monster
2.2. Synaptic Department Claims a Large Chunk of the Brain’s Energy Budget
3. Energy Production at Presynapses
3.1. At Resting Conditions

3.2. Upon Synaptic Stimulation
3.3. Factors That Shift Synaptic Dependence on Glycolysis or OxPhos
4. Other Mitochondrial Functions at Synapses
4.1. Calcium Uptake
4.2. ROS Regulation
5. To Be or Not to Be a Synaptic Mitochondria?
6. When Synaptic Energy Production Fails: Neurodegenerative Diseases
6.1. Glycolytic Dysfunctions
6.2. Mitochondrial Dysfunctions
7. Conclusions and Open Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The Energetics of CNS White Matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Howarth, C.; Peppiatt-Wildman, C.; Attwell, D. The Energy Use Associated with Neural Computation in the Cerebellum. J. Cereb. Blood Flow Metab. 2009, 30, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. J. Cereb. Blood Flow Metab. 2012, 32, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Hirabayashi, Y.; Kwon, S.-K.; Lewis, T.L.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, E.; Musich, P.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef]
- Clarke, D.; Sokoloff, L. Circulation and energy metabolism in the brain. Basic Neurochem. Mol. Cell. Med. Asp. 1999, 81, 10019861449. [Google Scholar]
- O’Malley, G.; Lazzer, S.; Vermorel, M. Metabolic and Mechanical Cost of Sedentary and Physical Activities in Obese Children and Adolescents; ECOG: Philadelphia, PA, USA, 2015. [Google Scholar]
- Raichle, M.E. Two views of brain function. Trends Cogn. Sci. 2010, 14, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Shulman, R.G.; Hyder, F.; Rothman, D.L. Baseline brain energy supports the state of consciousness. Proc. Natl. Acad. Sci. USA 2009, 106, 11096–11101. [Google Scholar] [CrossRef]
- Sokoloff, L. Local Cerebral Energy Metabolism: Its Relationships to Local Functional Activity and Blood Flow. In Cerebral Vascular Smooth Muscle and Its Control; Elliott, K., O’Connor, M., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2009; Volume 27, pp. 11096–11101. [Google Scholar] [CrossRef]
- Sokoloff, L.; Mangold, R.; Wechsler, R.L.; Kennedy, C.; Kety, S.S. The effect of mental arithmetic on cerebral circulation and metabolism. J. Clin. Investig. 1955, 34, 1101–1108. [Google Scholar] [CrossRef]
- Lin, A.-L.; Fox, P.T.; Hardies, J.; Duong, T.Q.; Gao, J.-H. Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8446–8451. [Google Scholar] [CrossRef]
- Aanerud, J.; Borghammer, P.; Rodell, A.; Jónsdottir, K.Y.; Gjedde, A. Sex differences of human cortical blood flow and energy metabolism. J. Cereb. Blood Flow Metab. 2017, 37, 2433–2440. [Google Scholar] [CrossRef]
- KETY, S.S. The general metabolism of the brain in vivo. In Metabolism of the Nervous System; Richter, D., Ed.; Pergamon Press: London, UK; New York, NY, USA, 1957; pp. 221–237. [Google Scholar]
- Hyder, F.; Fulbright, R.K.; Shulman, R.G.; Rothman, D.L. Glutamatergic Function in the Resting Awake Human Brain is Supported by Uniformly High Oxidative Energy. J. Cereb. Blood Flow Metab. 2013, 33, 339–347. [Google Scholar] [CrossRef]
- Blazey, T.; Snyder, A.Z.; Goyal, M.S.; Vlassenko, A.G.; Raichle, M.E. A systematic meta-analysis of oxygen-to-glucose and oxygen-to-carbohydrate ratios in the resting human brain. PLoS ONE 2018, 13, e0204242. [Google Scholar] [CrossRef]
- Bueschke, N.; Amaral-Silva, L.D.; Adams, S.; Santin, J.M. Transforming a neural circuit to function without oxygen and glucose delivery. Curr. Biol. 2021, 31, R1564–R1565. [Google Scholar] [CrossRef]
- Lucas, S.J.; Michel, C.B.; Marra, V.; Smalley, J.L.; Hennig, M.; Graham, B.P.; Forsythe, I.D. Glucose and lactate as metabolic constraints on presynaptic transmission at an excitatory synapse. J. Physiol. 2018, 596, 1699–1721. [Google Scholar] [CrossRef]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, S.V.; Waseem, T.V. Metabolic regulation of synaptic activity. Rev. Neurosci. 2018, 29, 825–835. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, D.; Giménez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wilson, G.S. A Temporary Local Energy Pool Coupled to Neuronal Activity: Fluctuations of Extracellular Lactate Levels in Rat Brain Monitored with Rapid-Response Enzyme-Based Sensor. J. Neurochem. 1997, 69, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Prichard, J.; Rothman, D.; Novotny, E.; Petroff, O.; Kuwabara, T.; Avison, M.; Howseman, A.; Hanstock, C.; Shulman, R. Lactate rise detected by 1H NMR in human visual cortex during physiologic stimulation. Proc. Natl. Acad. Sci. USA 1991, 88, 5829–5831. [Google Scholar] [CrossRef]
- Schaller, B.; Mekle, R.; Xin, L.; Kunz, N.; Gruetter, R. Net increase of lactate and glutamate concentration in activated human visual cortex detected with magnetic resonance spectroscopy at 7 tesla. J. Neurosci. Res. 2013, 91, 1076–1083. [Google Scholar] [CrossRef]
- Vaishnavi, S.N.; Vlassenko, A.G.; Rundle, M.M.; Snyder, A.Z.; Mintun, M.A.; Raichle, M.E. Regional aerobic glycolysis in the human brain. Proc. Natl. Acad. Sci. USA 2010, 107, 17757–17762. [Google Scholar] [CrossRef]
- Alle, H.; Roth, A.; Geiger, J.R.P. Energy-Efficient Action Potentials in Hippocampal Mossy Fibers. Science 2009, 325, 1405–1408. [Google Scholar] [CrossRef]
- Sengupta, B.; Stemmler, M.; Laughlin, S.; Niven, J.E. Action Potential Energy Efficiency Varies Among Neuron Types in Vertebrates and Invertebrates. PLoS Comput. Biol. 2010, 6, e1000840. [Google Scholar] [CrossRef]
- Vida, I.; Frotscher, M. A hippocampal interneuron associated with the mossy fiber system. Proc. Natl. Acad. Sci. USA 2000, 97, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Williams, J.; Nathans, J. Complete morphologies of basal forebrain cholinergic neurons in the mouse. Elife 2014, 3, e02444. [Google Scholar] [CrossRef] [PubMed]
- Hubley, M.J.; Locke, B.; Moerland, T.S. The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1996, 1291, 115–121. [Google Scholar] [CrossRef]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Pathak, D.; Shields, L.Y.; Mendelsohn, B.A.; Haddad, D.; Lin, W.; Gerencser, A.A.; Kim, H.; Brand, M.; Edwards, R.H.; Nakamura, K. The Role of Mitochondrially Derived ATP in Synaptic Vesicle Recycling. J. Biol. Chem. 2015, 290, 22325–22336. [Google Scholar] [CrossRef] [PubMed]
- Pulido, C.; Ryan, T.A. Synaptic vesicle pools are a major hidden resting metabolic burden of nerve terminals. Sci. Adv. 2021, 7, 9027. [Google Scholar] [CrossRef]
- Choi, S.W.; Gerencser, A.A.; Nicholls, D.G. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: Spare respiratory capacity and stochastic mitochondrial failure. J. Neurochem. 2009, 109, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; Choi, S.W.; Day, N.U.; Gerencser, A.A.; Hubbard, A.; Melov, S. Impaired spare respiratory capacity in cortical synaptosomes from Sod2 null mice. Free Radic. Biol. Med. 2011, 50, 866–873. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Silver, I.; Erecinska, M. Extracellular glucose concentration in mammalian brain: Continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J. Neurosci. 1994, 14, 5068–5076. [Google Scholar] [CrossRef]
- Gruetter, R.; Novotny, E.J.; Boulware, S.D.; Rothman, D.L.; Mason, G.F.; I Shulman, G.; Shulman, R.G.; Tamborlane, W.V. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc. Natl. Acad. Sci. USA 1992, 89, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Schwarcz, A.D.; Dénes, Á. Mitochondrial Ultrastructure Is Coupled to Synaptic Performance at Axonal Release Sites. Eneuro 2018, 5, ENEURO.0390–17. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, R.; Fornasiero, E.F.; Cyganek, L.; Montoya, J.; Jakobs, S.; O Rizzoli, S.; Rehling, P.; Pacheu-Grau, D. Monitoring mitochondrial translation in living cells. EMBO Rep. 2021, 22, e51635. [Google Scholar] [CrossRef] [PubMed]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xiong, G.-J.; Huang, N.; Sheng, Z.-H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef] [PubMed]
- Kasischke, K.A.; Vishwasrao, H.D.; Fisher, P.J.; Zipfel, W.R.; Webb, W.W. Neural Activity Triggers Neuronal Oxidative Metabolism Followed by Astrocytic Glycolysis. Science 2004, 305, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Windels, F.; Bruet, N.; Poupard, A.; Urbain, N.; Chouvet, G.; Feuerstein, C.; Savasta, M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur. J. Neurosci. 2000, 12, 4141–4146. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Voutsinos-Porche, B.; Bonvento, G.; Tanaka, K.; Steiner, P.; Welker, E.; Chatton, J.-Y.; Magistretti, P.J.; Pellerin, L. Glial Glutamate Transporters Mediate a Functional Metabolic Crosstalk between Neurons and Astrocytes in the Mouse Developing Cortex. Neuron 2003, 37, 275–286. [Google Scholar] [CrossRef]
- Yellen, G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018, 217, 2235–2246. [Google Scholar] [CrossRef]
- Hall, C.; Klein-Flugge, M.; Howarth, C.; Attwell, D. Oxidative Phosphorylation, Not Glycolysis, Powers Presynaptic and Postsynaptic Mechanisms Underlying Brain Information Processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef]
- Díaz-García, C.M.; Mongeon, R.; Lahmann, C.; Koveal, D.; Zucker, H.; Yellen, G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017, 26, 361–374.e4. [Google Scholar] [CrossRef]
- Segarra-Mondejar, M.; Casellas-Díaz, S.; Ramiro-Pareta, M.; Sánchez, C.A.M.; Martorell-Riera, A.; Hermelo, I.; Reina, M.; Aragonés, J.; Estrada, O.M.M.; Soriano, F.X. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018, 37, e97368. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Wu, Z.; Farrell, R.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e3. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Nelson, J.C.; Bend, E.G.; Rodríguez-Laureano, L.; Tueros, F.G.; Cartagenova, L.; Underwood, K.; Jorgensen, E.M.; Colón-Ramos, D.A. Glycolytic Enzymes Localize to Synapses under Energy Stress to Support Synaptic Function. Neuron 2016, 90, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Knull, H. Compartmentation of glycolytic enzymes in nerve endings as determined by glutaraldehyde fixation. J. Biol. Chem. 1980, 255, 6439–6444. [Google Scholar] [CrossRef]
- Zala, D.; Hinckelmann, M.-V.; Yu, H.; Lyra Da Cunha, M.M.; Liot, G.; Cordelières, F.P.; Marco, S.; Saudou, F. Vesicular Glycolysis Provides On-Board Energy for Fast Axonal Transport. Cell 2013, 152, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.W.; Dunham, P.B. Membrane-bound ATP fuels the Na/K pump. Studies on membrane-bound glycolytic enzymes on inside-out vesicles from human red cell membranes. J. Gen. Physiol. 1981, 78, 547–568. [Google Scholar] [CrossRef] [PubMed]
- Knull, H.R. Association of glycolytic enzymes with particulate fractions from nerve endings. Biochim. Biophys. Acta (BBA)-Enzym. 1978, 522, 1–9. [Google Scholar] [CrossRef]
- Ashrafi, G.; A Ryan, T. Glucose metabolism in nerve terminals. Curr. Opin. Neurobiol. 2017, 45, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Maeno-Hikichi, Y.; Polo-Parada, L.; Kastanenka, K.V.; Landmesser, L.T. Frequency-Dependent Modes of Synaptic Vesicle Endocytosis and Exocytosis at Adult Mouse Neuromuscular Junctions. J. Neurosci. 2011, 31, 1093–1105. [Google Scholar] [CrossRef]
- Harata, N.C.; Choi, S.; Pyle, J.L.; Aravanis, A.M.; Tsien, R.W. Frequency-Dependent Kinetics and Prevalence of Kiss-and-Run and Reuse at Hippocampal Synapses Studied with Novel Quenching Methods. Neuron 2006, 49, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Sobieski, C.; Fitzpatrick, M.J.; Mennerick, S.J. Differential Presynaptic ATP Supply for Basal and High-Demand Transmission. J. Neurosci. 2017, 37, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, R.; Fornasiero, E.F.; Cyganek, L.; Jakobs, S.; Rizzoli, O.; Rehling, P.; Pacheu-Grau, D. Local translation in synaptic mitochondria influences synaptic transmission. bioRxiv 2020, 215194. [Google Scholar] [CrossRef]
- Williams, J.M.; Thompson, V.L.; Mason-Parker, S.E.; Abraham, W.C.; Tate, W.P. Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Mol. Brain Res. 1998, 60, 50–56. [Google Scholar] [CrossRef]
- Krishna, S.; e Drigo, R.A.; Capitanio, J.S.; Ramachandra, R.; Ellisman, M.; Hetzer, M.W. Identification of long-lived proteins in the mitochondria reveals increased stability of the electron transport chain. Dev. Cell 2021, 56, 2952–2965. [Google Scholar] [CrossRef]
- Bomba-Warczak, E.; Edassery, S.L.; Hark, T.J.; Savas, J.N. Long-lived mitochondrial cristae proteins in mouse heart and brain. J. Cell Biol. 2021, 220, e202005193. [Google Scholar] [CrossRef]
- Fornasiero, E.F.; Mandad, S.; Wildhagen, H.; Alevra, M.; Rammner, B.; Keihani, S.; Opazo, F.; Urban, I.; Ischebeck, T.; Sakib, M.S.; et al. Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 2018, 9, 4230. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Soustek, M.S.; Thomas, A.; Cogliati, S.; Poyatos, C.G.; Martín-García, E.; Jedrychowski, M.; Gygi, S.P.; Enríquez, J.A.; Puigserver, P. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Mol. Cell 2019, 74, 877–890.e6. [Google Scholar] [CrossRef]
- Gonzalez-Franquesa, A.; Stocks, B.; Chubanava, S.; Hattel, H.B.; Moreno-Justicia, R.; Peijs, L.; Treebak, J.T.; Zierath, J.R.; Deshmukh, A.S. Mass-spectrometry-based proteomics reveals mitochondrial supercomplexome plasticity. Cell Rep. 2021, 35, 109180. [Google Scholar] [CrossRef]
- Nesterov, S.; Chesnokov, Y.; Kamyshinsky, R.; Panteleeva, A.; Lyamzaev, K.; Vasilov, R.; Yaguzhinsky, L. Ordered Clusters of the Complete Oxidative Phosphorylation System in Cardiac Mitochondria. Int. J. Mol. Sci. 2021, 22, 1462. [Google Scholar] [CrossRef]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic Glycolysis in the Human Brain Is Associated with Development and Neotenous Gene Expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Tindale, L.; Lone, A.; Singh, O.; Macauley, S.L.; Stanley, M.; Holtzman, D.M.; Bartha, R.; Cumming, R.C. Aerobic Glycolysis in the Frontal Cortex Correlates with Memory Performance in Wild-Type Mice But Not the APP/PS1 Mouse Model of Cerebral Amyloidosis. J. Neurosci. 2016, 36, 1871–1878. [Google Scholar] [CrossRef]
- Almeida, A.; Brooks, K.; Sammut, I.; Keelan, J.; Davey, G.; Clark, J.; Bates, T. Postnatal Development of the Complexes of the Electron Transport Chain in Synaptic Mitochondria from Rat Brain. Dev. Neurosci. 1995, 17, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.I.; Keine, C.; Okayama, S.; Satterfield, R.; Musgrove, M.; Guerrero-Given, D.; Kamasawa, N.; Young, S.M. Presynaptic Mitochondria Volume and Abundance Increase during Development of a High-Fidelity Synapse. J. Neurosci. 2019, 39, 7994–8012. [Google Scholar] [CrossRef]
- Agostini, M.; Romeo, F.; Inoue, S.; Niklison-Chirou, M.V.; Elia, A.J.; Dinsdale, D.; Morone, N.; Knight, R.A.; Mak, T.W.; Melino, G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016, 23, 1502–1514. [Google Scholar] [CrossRef]
- Bardy, C.; van den Hurk, M.; Eames, T.; Marchand, C.; Hernandez, R.V.; Kellogg, M.; Gorris, M.; Galet, B.; Palomares, V.; Brown, J.; et al. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc. Natl. Acad. Sci. USA 2015, 112, E2725–E2734. [Google Scholar] [CrossRef]
- Satir, T.M.; Nazir, F.H.; Vizlin-Hodzic, D.; Hardselius, E.; Blennow, K.; Wray, S.; Zetterberg, H.; Agholme, L.; Bergström, P. Accelerated neuronal and synaptic maturation by BrainPhys medium increases Aβ secretion and alters Aβ peptide ratios from iPSC-derived cortical neurons. Sci. Rep. 2020, 10, 601. [Google Scholar] [CrossRef]
- Jackson, T.C.; Kotermanski, S.E.; Jackson, E.K.; Kochanek, P.M. BrainPhys® increases neurofilament levels in CNS cultures, and facilitates investigation of axonal damage after a mechanical stretch-injury in vitro. Exp. Neurol. 2018, 300, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, V.; Carter, J.R. Metabolic effects of 2-deoxy-D-glucose in isolated fat cells. Biochim. Biophys. Acta (BBA)-Gen. Subj. 1977, 496, 278–291. [Google Scholar] [CrossRef]
- Lujan, B.; Kushmerick, C.; Das Banerjee, T.; Dagda, R.K.; Renden, R. Glycolysis selectively shapes the presynaptic action potential waveform. J. Neurophysiol. 2016, 116, 2523–2540. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Aoki, C.; Elste, A.; Rogalski-Wilk, A.A.; Siekevitz, P. The synthesis of ATP by glycolytic enzymes in the postsynaptic density and the effect of endogenously generated nitric oxide. Proc. Natl. Acad. Sci. USA 1997, 94, 13273–13278. [Google Scholar] [CrossRef]
- Laschet, J.J.; Minier, F.; Kurcewicz, I.; Bureau, M.H.; Trottier, S.; Jeanneteau, F.; Griffon, N.; Samyn, B.; Van Beeumen, J.; Louvel, J.; et al. Glyceraldehyde-3-Phosphate Dehydrogenase Is a GABAA Receptor Kinase Linking Glycolysis to Neuronal Inhibition. J. Neurosci. 2004, 24, 7614–7622. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef]
- E Wernette, M.; Ochs, R.S.; A Lardy, H. Ca2+ stimulation of rat liver mitochondrial glycerophosphate dehydrogenase. J. Biol. Chem. 1981, 256, 12767–12771. [Google Scholar] [CrossRef]
- Turkan, A.; Hiromasa, A.Y.; Roche, T.E. Formation of a Complex of the Catalytic Subunit of Pyruvate Dehydrogenase Phosphatase Isoform 1 (PDP1c) and the L2 Domain Forms a Ca2+ Binding Site and Captures PDP1c as a Monomer. Biochemistry 2004, 43, 15073–15085. [Google Scholar] [CrossRef]
- Rutter, G.; Denton, R.M. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochem. J. 1989, 263, 453–462. [Google Scholar] [CrossRef]
- Hubbard, M.J.; McHugh, N.J. Mitochondrial ATP synthase F1-β-subunit is a calcium-binding protein. FEBS Lett. 1996, 391, 323–329. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Csordás, G.; Szanda, G.; Golenár, T.; Debattisti, V.; Bartok, A.; Wang, N.; Moffat, C.; Seifert, E.L.; Spät, A.; et al. Tissue-Specific Mitochondrial Decoding of Cytoplasmic Ca2+ Signals Is Controlled by the Stoichiometry of MICU1/2 and MCU. Cell Rep. 2017, 18, 2291–2300. [Google Scholar] [CrossRef]
- Chouhan, A.; Ivannikov, M.V.; Lu, Z.; Sugimori, M.; Llinas, R.R.; MacLeod, G.T. Cytosolic Calcium Coordinates Mitochondrial Energy Metabolism with Presynaptic Activity. J. Neurosci. 2012, 32, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-G.; Zucker, R.S. Mitochondrial Involvement in Post-Tetanic Potentiation of Synaptic Transmission. Neuron 1997, 18, 483–491. [Google Scholar] [CrossRef]
- Wong, H.-T.C.; Zhang, Q.; Beirl, A.J.; Petralia, R.S.; Wang, Y.-X.; Kindt, K. Synaptic mitochondria regulate hair-cell synapse size and function. Elife 2019, 8, e48914. [Google Scholar] [CrossRef]
- Datta, S.; Jaiswal, M. Mitochondrial calcium at the synapse. Mitochondrion 2021, 59, 135–153. [Google Scholar] [CrossRef]
- Tufi, R.; Gleeson, T.P.; von Stockum, S.; Hewitt, V.L.; Lee, J.J.; Terriente-Felix, A.; Sanchez-Martinez, A.; Ziviani, E.; Whitworth, A.J. Comprehensive Genetic Characterization of Mitochondrial Ca2+ Uniporter Components Reveals Their Different Physiological Requirements In Vivo. Cell Rep. 2019, 27, 1541–1550.e5. [Google Scholar] [CrossRef]
- Marland, J.; Hasel, P.; Bonnycastle, K.; Cousin, M.A. Mitochondrial Calcium Uptake Modulates Synaptic Vesicle Endocytosis in Central Nerve Terminals. J. Biol. Chem. 2016, 291, 2080–2086. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef]
- Niescier, R.F.; Hong, K.; Park, D.; Min, K.-T. MCU Interacts with Miro1 to Modulate Mitochondrial Functions in Neurons. J. Neurosci. 2018, 38, 4666–4677. [Google Scholar] [CrossRef] [PubMed]
- Cardanho-Ramos, C.; Faria-Pereira, A.; Morais, V.A. Orchestrating mitochondria in neurons: Cytoskeleton as the conductor. Cytoskeleton 2019, 77, 65–75. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 Is a Calcium Sensor for Glutamate Receptor-Dependent Localization of Mitochondria at Synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+-Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Hernansanz-Agustín, P.; Enríquez, J. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Tsatmali, M.; Walcott, E.C.; Crossin, K.L. Newborn neurons acquire high levels of reactive oxygen species and increased mitochondrial proteins upon differentiation from progenitors. Brain Res. 2005, 1040, 137–150. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Accardi, M.V.; Daniels, B.A.; Brown, P.; Fritschy, J.-M.; Tyagarajan, S.; Bowie, D. Mitochondrial reactive oxygen species regulate the strength of inhibitory GABA-mediated synaptic transmission. Nat. Commun. 2014, 5, 3168. [Google Scholar] [CrossRef]
- Jia, Q.; Sieburth, D. Mitochondrial hydrogen peroxide positively regulates neuropeptide secretion during diet-induced activation of the oxidative stress response. Nat. Commun. 2021, 12, 2304. [Google Scholar] [CrossRef]
- Bao, L.; Avshalumov, M.V.; Patel, J.C.; Lee, C.R.; Miller, E.W.; Chang, C.J.; Rice, M.E. Mitochondria Are the Source of Hydrogen Peroxide for Dynamic Brain-Cell Signaling. J. Neurosci. 2009, 29, 9002–9010. [Google Scholar] [CrossRef]
- Sidlauskaite, E.; Gibson, J.W.; Megson, I.L.; Whitfield, P.D.; Tovmasyan, A.; Batinic-Haberle, I.; Murphy, M.; Moult, P.R.; Cobley, J.N. Mitochondrial ROS cause motor deficits induced by synaptic inactivity: Implications for synapse pruning. Redox Biol. 2018, 16, 344–351. [Google Scholar] [CrossRef]
- Chavan, V.; Willis, J.; Walker, S.K.; Clark, H.R.; Liu, X.; Fox, M.A.; Srivastava, S.; Mukherjee, K. Central Presynaptic Terminals Are Enriched in ATP but the Majority Lack Mitochondria. PLoS ONE 2015, 10, e0125185. [Google Scholar] [CrossRef] [PubMed]
- Freeman, D.W.; Petralia, R.S.; Wang, Y.-X.; Mattson, M.P.; Yao, P.J. Mitochondria in hippocampal presynaptic and postsynaptic compartments differ in size as well as intensity. Matters 2017, 3. [Google Scholar] [CrossRef]
- Lees, R.M.; Johnson, J.D.; Ashby, M.C. Presynaptic Boutons That Contain Mitochondria Are More Stable. Front. Synaptic Neurosci. 2020, 11, 37. [Google Scholar] [CrossRef]
- Delgado, T.; Petralia, R.S.; Freeman, D.W.; Sedlacek, M.; Wang, Y.-X.; Brenowitz, S.D.; Sheu, S.-H.; Gu, J.W.; Kapogiannis, D.; Mattson, M.P.; et al. Comparing 3D ultrastructure of presynaptic and postsynaptic mitochondria. Biol. Open 2019, 8, bio044834. [Google Scholar] [CrossRef]
- Rybka, V.; Suzuki, Y.J.; Gavrish, A.S.; Dibrova, V.A.; Gychka, S.G.; Shults, N.V. Transmission Electron Microscopy Study of Mitochondria in Aging Brain Synapses. Antioxidants 2019, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.T.W.; Honick, A.S.; Reynolds, I.J. Mitochondrial Trafficking to Synapses in Cultured Primary Cortical Neurons. J. Neurosci. 2006, 26, 7035–7045. [Google Scholar] [CrossRef] [PubMed]
- Smit-Rigter, L.; Rajendran, R.; Silva, C.A.; Spierenburg, L.; Groeneweg, F.; Ruimschotel, E.M.; van Versendaal, D.; van der Togt, C.; Eysel, U.; Heimel, J.A.; et al. Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Curr. Biol. 2016, 26, 2609–2616. [Google Scholar] [CrossRef]
- Vaccaro, V.; Devine, M.; Higgs, N.F.J.; Kittler, J.T. Miro1-dependent mitochondrial positioning drives the rescaling of presynaptic Ca2+ signals during homeostatic plasticity. EMBO Rep. 2017, 18, 231–240. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Yan, S.; Sosunov, A.A.; McKhann, G.M.; Yan, S.S.D. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 18670–18675. [Google Scholar] [CrossRef]
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic Mitochondria Are More Susceptible to Ca2+ Overload than Nonsynaptic Mitochondria. J. Biol. Chem. 2006, 281, 11658–11668. [Google Scholar] [CrossRef] [PubMed]
- Naga, K.K.; Sullivan, P.G.; Geddes, J.W. High Cyclophilin D Content of Synaptic Mitochondria Results in Increased Vulnerability to Permeability Transition. J. Neurosci. 2007, 27, 7469–7475. [Google Scholar] [CrossRef]
- Kiebish, M.A.; Han, X.; Cheng, H.; Lunceford, A.; Clarke, C.F.; Moon, H.; Chuang, J.H.; Seyfried, T.N. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 2008, 106, 299–312. [Google Scholar] [CrossRef]
- Stauch, K.L.; Purnell, P.; Fox, H.S. Quantitative Proteomics of Synaptic and Nonsynaptic Mitochondria: Insights for Synaptic Mitochondrial Vulnerability. J. Proteome Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef]
- Völgyi, K.; Gulyássy, P.; Háden, K.; Kis, V.; Badics, K.; Kékesi, K.A.; Simor, A.; Gyorffy, B.; Tóth, E.A.; Lubec, G.; et al. Synaptic mitochondria: A brain mitochondria cluster with a specific proteome. J. Proteom. 2015, 120, 142–157. [Google Scholar] [CrossRef]
- Graham, L.C.; Eaton, S.L.; Brunton, P.; Atrih, A.; Smith, C.; Lamont, D.J.; Gillingwater, T.; Pennetta, G.; Skehel, P.A.; Wishart, T.M. Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture. Mol. Neurodegener. 2017, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.F.; Lai, J.C.K.; Lim, L.; Clark, J.B. The Activities of Some Energy-Metabolising Enzymes in Nonsynaptic (Free) and Synaptic Mitochondria Derived from Selected Brain Regions. J. Neurochem. 1984, 42, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.T. Cytochrome oxidase: An endogenous metabolic marker for neuronal activity. Trends Neurosci. 1989, 12, 94–101. [Google Scholar] [CrossRef]
- Villa, R.F.; Gorini, A.; Hoyer, S. Differentiated effect of ageing on the enzymes of Krebs’ cycle, electron transfer complexes and glutamate metabolism of non-synaptic and intra-synaptic mitochondria from cerebral cortex. J. Neural Transm. 2006, 113, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Davey, G.P.; Peuchen, S.; Clark, J.B. Energy Thresholds in Brain Mitochondria: Potential involvement in neurodegeneration *. J. Biol. Chem. 1998, 273, 12753–12757. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.A.; Torres, A.K.; Jara, C.; Murphy, M.P.; Tapia-Rojas, C. Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 2020, 34, 101558. [Google Scholar] [CrossRef]
- Perkins, G.A.; Tjong, J.; Brown, J.M.; Poquiz, P.H.; Scott, R.T.; Kolson, D.R.; Ellisman, M.H.; Spirou, G.A. The Micro-Architecture of Mitochondria at Active Zones: Electron Tomography Reveals Novel Anchoring Scaffolds and Cristae Structured for High-Rate Metabolism. J. Neurosci. 2010, 30, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, S.; Enríquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Kim, Y.; Katti, P.; Willingham, T.B. The Functional Impact of Mitochondrial Structure Across Subcellular Scales. Front. Physiol. 2020, 11, 1462. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, S.V.; Waseem, T.V.; Puchkova, L.V. Biogenetic and morphofunctional heterogeneity of mitochondria: The case of synaptic mitochondria. Rev. Neurosci. 2017, 28, 363–373. [Google Scholar] [CrossRef]
- Cai, Q.; Zakaria, H.M.; Simone, A.; Sheng, Z.-H. Spatial Parkin Translocation and Degradation of Damaged Mitochondria via Mitophagy in Live Cortical Neurons. Curr. Biol. 2012, 22, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Wong, H.-T.C.; Pinter, K.; Mosqueda, N.; Beirl, A.; Lomash, R.M.; Won, S.; Kindt, K.S.; Drerup, C.M. Retrograde Mitochondrial Transport Is Essential for Organelle Distribution and Health in Zebrafish Neurons. J. Neurosci. 2021, 41, 1371–1392. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Su, X.; Cai, Q. Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 2020, 21, e49801. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Arnold, B.; Howlett, E.H.; Calderon, M.J.; Croix, C.M.S.; Greenamyre, J.T.; Sanders, L.H.; Berman, S.B. Evidence for Compartmentalized Axonal Mitochondrial Biogenesis: Mitochondrial DNA Replication Increases in Distal Axons As an Early Response to Parkinson’s Disease-Relevant Stress. J. Neurosci. 2018, 38, 7505–7515. [Google Scholar] [CrossRef] [PubMed]
- Cardanho-Ramos, C.; Morais, V.A. Mitochondrial Biogenesis in Neurons: How and Where. Int. J. Mol. Sci. 2021, 22, 13059. [Google Scholar] [CrossRef]
- Li, S.; Sheng, Z.-H. Energy matters: Presynaptic metabolism and the maintenance of synaptic transmission. Nat. Rev. Neurosci. 2021, 23, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Rose, J.; McCrory, C.; McKenzie, C.-A.; King, D.; Smith, C.; Gillingwater, T.; Henstridge, C.; Spires-Jones, T.L. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol. 2018, 136, 747–757. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Raichle, M.E. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 27–37. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Diabetes Mellitus and Risk of Alzheimer Disease and Decline in Cognitive Function. Arch. Neurol. 2004, 61, 661–666. [Google Scholar] [CrossRef]
- Wang, K.-C.; Woung, L.-C.; Tsai, M.-T.; Liu, C.-C.; Su, Y.-H.; Li, C.-Y. Risk of Alzheimer’s Disease in Relation to Diabetes: A Population-Based Cohort Study. Neuroepidemiology 2012, 38, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: The Hisayama Study. Neurology 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Chakravarty, M.; Jonsdottir, K.Y.; Sato, N.; Matsuda, H.; Ito, K.; Arahata, Y.; Kato, T.; Gjedde, A. Cortical hypometabolism and hypoperfusion in Parkinson’s disease is extensive: Probably even at early disease stages. Brain Struct. Funct. 2010, 214, 303–317. [Google Scholar] [CrossRef]
- Yang, S.-Q.; Tian, Q.; Li, D.; He, S.-Q.; Hu, M.; Liu, S.-Y.; Zou, W.; Chen, Y.-J.; Zhang, P.; Tang, X.-Q. Leptin mediates protection of hydrogen sulfide against 6-hydroxydopamine-induced Parkinson’s disease: Involving enhancement in Warburg effect. Neurochem. Int. 2020, 135, 104692. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Carasa, I.F.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.; Atassi, N. Positron Emission Tomography Molecular Imaging Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Lederer, C.W.; Torrisi, A.; Pantelidou, M.; Santama, N.; Cavallaro, S. Pathways and genes differentially expressed in the motor cortex of patients with sporadic amyotrophic lateral sclerosis. BMC Genom. 2007, 8, 26. [Google Scholar] [CrossRef]
- Han, R.; Liang, J.; Zhou, B. Glucose Metabolic Dysfunction in Neurodegenerative Diseases—New Mechanistic Insights and the Potential of Hypoxia as a Prospective Therapy Targeting Metabolic Reprogramming. Int. J. Mol. Sci. 2021, 22, 5887. [Google Scholar] [CrossRef] [PubMed]
- McFarland, R.; Taylor, R.W.; Turnbull, D. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010, 9, 829–840. [Google Scholar] [CrossRef]
- Kilbride, S.M.; Telford, J.E.; Tipton, K.F.; Davey, G.P.; Kilbride, S.M. Partial inhibition of complex I activity increases Ca2+-independent glutamate release rates from depolarized synaptosomes. J. Neurochem. 2008, 106, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Maglioni, S. Neuroligin-mediated neurodevelopmental defects are induced by mitochondrial dysfunction and prevented by lutein in C. elegans. bioRxiv 2021, 957225. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Lee, H.-G.; Li, X.; Perry, G.; Smith, M.A.; Zhu, X. Impaired Balance of Mitochondrial Fission and Fusion in Alzheimer’s Disease. J. Neurosci. 2009, 29, 9090–9103. [Google Scholar] [CrossRef] [PubMed]
- DuBoff, B.; Götz, J.; Feany, M.B. Tau Promotes Neurodegeneration via DRP1 Mislocalization In Vivo. Neuron 2012, 75, 618–632. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2011, 21, 406–420. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.-F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Amyloid-β-Derived Diffusible Ligands Cause Impaired Axonal Transport of Mitochondria in Neurons. Neurodegener. Dis. 2010, 7, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Sun, X.; Starovoytov, V.; Cai, Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 2015, 24, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-L.; Wang, W.-A.; Tan, J.-X.; Huang, J.-K.; Zhang, X.; Zhang, B.-Z.; Wang, Y.; Yangcheng, H.-Y.; Zhu, H.-L.; Sun, X.-J.; et al. Expression of β-Amyloid Induced Age-Dependent Presynaptic and Axonal Changes in Drosophila. J. Neurosci. 2010, 30, 1512–1522. [Google Scholar] [CrossRef]
- Kim-Han, J.S.; Antenor-Dorsey, J.A.; O’Malley, K.L. The Parkinsonian Mimetic, MPP+, Specifically Impairs Mitochondrial Transport in Dopamine Axons. J. Neurosci. 2011, 31, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Shahpasand, K.; Uemura, I.; Saito, T.; Asano, T.; Hata, K.; Shibata, K.; Toyoshima, Y.; Hasegawa, M.; Hisanaga, S.-I. Regulation of Mitochondrial Transport and Inter-Microtubule Spacing by Tau Phosphorylation at the Sites Hyperphosphorylated in Alzheimer’s Disease. J. Neurosci. 2012, 32, 2430–2441. [Google Scholar] [CrossRef]
- Li, L.; Nadanaciva, S.; Berger, Z.; Shen, W.; Paumier, K.; Schwartz, J.; Mou, K.; Loos, P.; Milici, A.J.; Dunlop, J.; et al. Human A53T α-Synuclein Causes Reversible Deficits in Mitochondrial Function and Dynamics in Primary Mouse Cortical Neurons. PLoS ONE 2013, 8, e85815. [Google Scholar] [CrossRef]
- Dukes, A.A.; Bai, Q.; Van Laar, V.S.; Zhou, Y.; Ilin, V.; David, C.N.; Agim, Z.S.; Bonkowsky, J.L.; Cannon, J.R.; Watkins, S.C.; et al. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP+ exposure. Neurobiol. Dis. 2016, 95, 238–249. [Google Scholar] [CrossRef]
- Gonçalves, F.; Morais, V. PINK1: A Bridge between Mitochondria and Parkinson’s Disease. Life 2021, 11, 371. [Google Scholar] [CrossRef] [PubMed]
- Kamienieva, I.; Duszyński, J.; Szczepanowska, J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson’s disease. Transl. Neurodegener. 2021, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Aidt, F.H.; Nielsen, S.M.B.; Kanters, J.; Pesta, D.; Nielsen, T.T.; Nørremølle, A.; Hasholt, L.; Christiansen, M.; Hagen, C.M. Dysfunctional mitochondrial respiration in the striatum of the Huntington’s disease transgenic R6/2 mouse model. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Characterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Cookson, M.R.; Taylor, R.W.; Turnbull, U.M.; Chrzanowska-Lightowlers, Z.M.A.; Dong, L.; Figlewicz, D.A.; Shaw, P. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain 2002, 125, 1522–1533. [Google Scholar] [CrossRef]
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal mitochondrial dysfunction in sporadic amyotrophic lateral sclerosis is developmentally regulated. Sci. Rep. 2021, 11, 18916. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Mallik, B.; Frank, C.A. Roles for Mitochondrial Complex I subunits in regulating synaptic transmission and growth. bioRxiv 2022. [Google Scholar] [CrossRef]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Adami, P.V.M.; Quijano, C.; Magnani, N.; Galeano, P.; Evelson, P.; Cassina, A.; Carmo, S.D.; Leal, M.C.; Castaño, E.M.; Cuello, A.C.; et al. Synaptosomal bioenergetic defects are associated with cognitive impairment in a transgenic rat model of early Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2016, 37, 69–84. [Google Scholar] [CrossRef]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.; Ottemann, B.M.; Emanuel, K.; Fox, H.S. Loss of Pink1 modulates synaptic mitochondrial bioenergetics in the rat striatum prior to motor symptoms: Concomitant complex I respiratory defects and increased complex II-mediated respiration. Proteom.-Clin. Appl. 2016, 10, 1205–1217. [Google Scholar] [CrossRef]
- Monteiro-Cardoso, V.F.; Oliveira, M.M.; Melo, T.; Domingues, M.R.; Moreira, P.I.; Ferreiro, E.; Peixoto, F.; Videira, R.A. Cardiolipin Profile Changes are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2013, 179, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.K.; Grady, J.; Cosgrave, E.M.; Bennison, E.; Chen, C.; Hepplewhite, P.D.; Morris, C.M. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Park. Dis. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Ultrastructural study of synapses in the anterior horn neurons of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1996, 204, 53–56. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; de la Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Dromard, Y.; Arango-Lievano, M.; Fontanaud, P.; Tricaud, N.; Jeanneteau, F. Dual imaging of dendritic spines and mitochondria in vivo reveals hotspots of plasticity and metabolic adaptation to stress. Neurobiol. Stress 2021, 15, 100402. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faria-Pereira, A.; Morais, V.A. Synapses: The Brain’s Energy-Demanding Sites. Int. J. Mol. Sci. 2022, 23, 3627. https://doi.org/10.3390/ijms23073627
Faria-Pereira A, Morais VA. Synapses: The Brain’s Energy-Demanding Sites. International Journal of Molecular Sciences. 2022; 23(7):3627. https://doi.org/10.3390/ijms23073627
Chicago/Turabian StyleFaria-Pereira, Andreia, and Vanessa A. Morais. 2022. "Synapses: The Brain’s Energy-Demanding Sites" International Journal of Molecular Sciences 23, no. 7: 3627. https://doi.org/10.3390/ijms23073627
APA StyleFaria-Pereira, A., & Morais, V. A. (2022). Synapses: The Brain’s Energy-Demanding Sites. International Journal of Molecular Sciences, 23(7), 3627. https://doi.org/10.3390/ijms23073627