
CD33 rs2455069 SNP: Correlation with Alzheimer’s Disease and Hypothesis of Functional Role

 , ,
, ,  , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Genetic Analysis
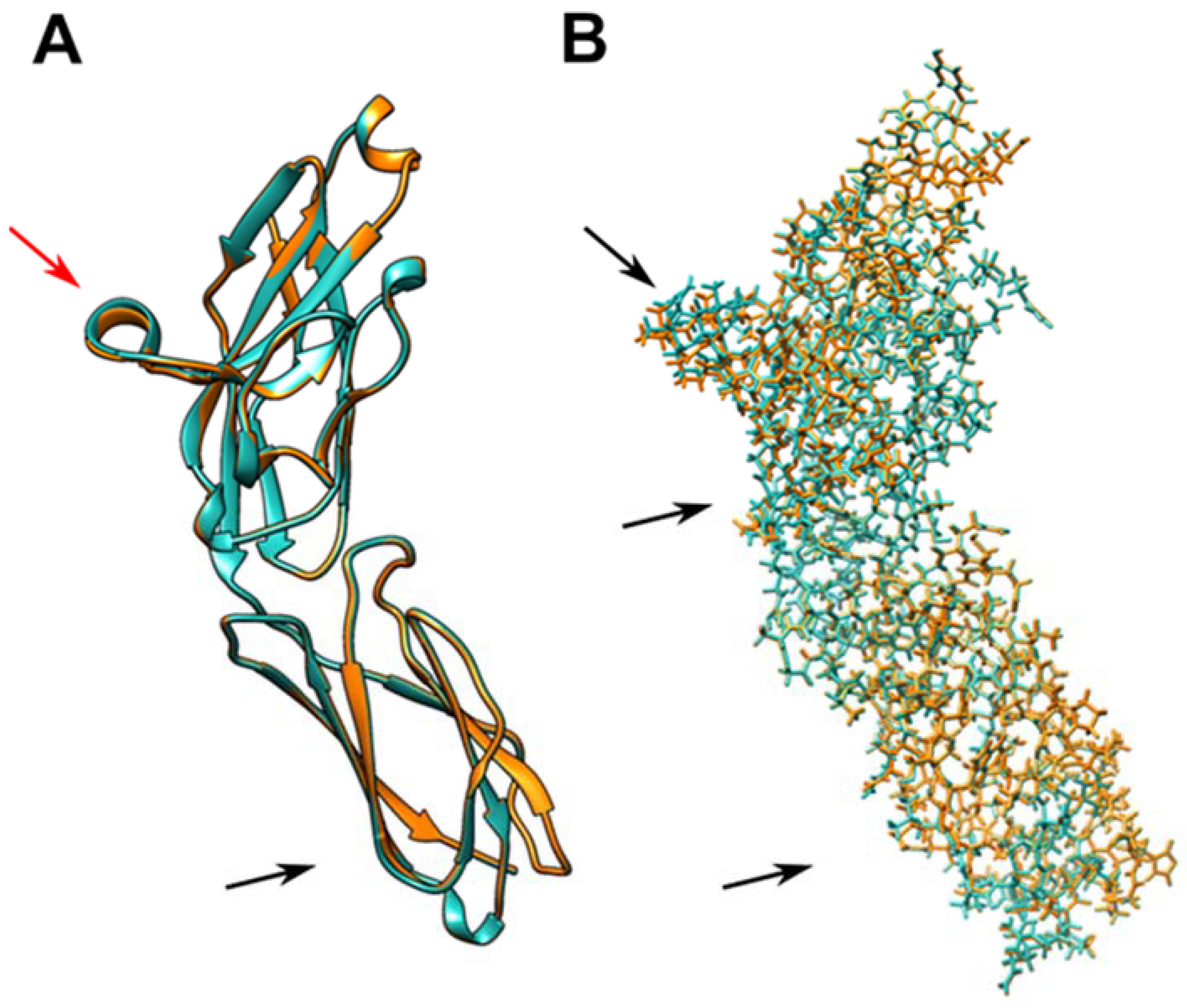
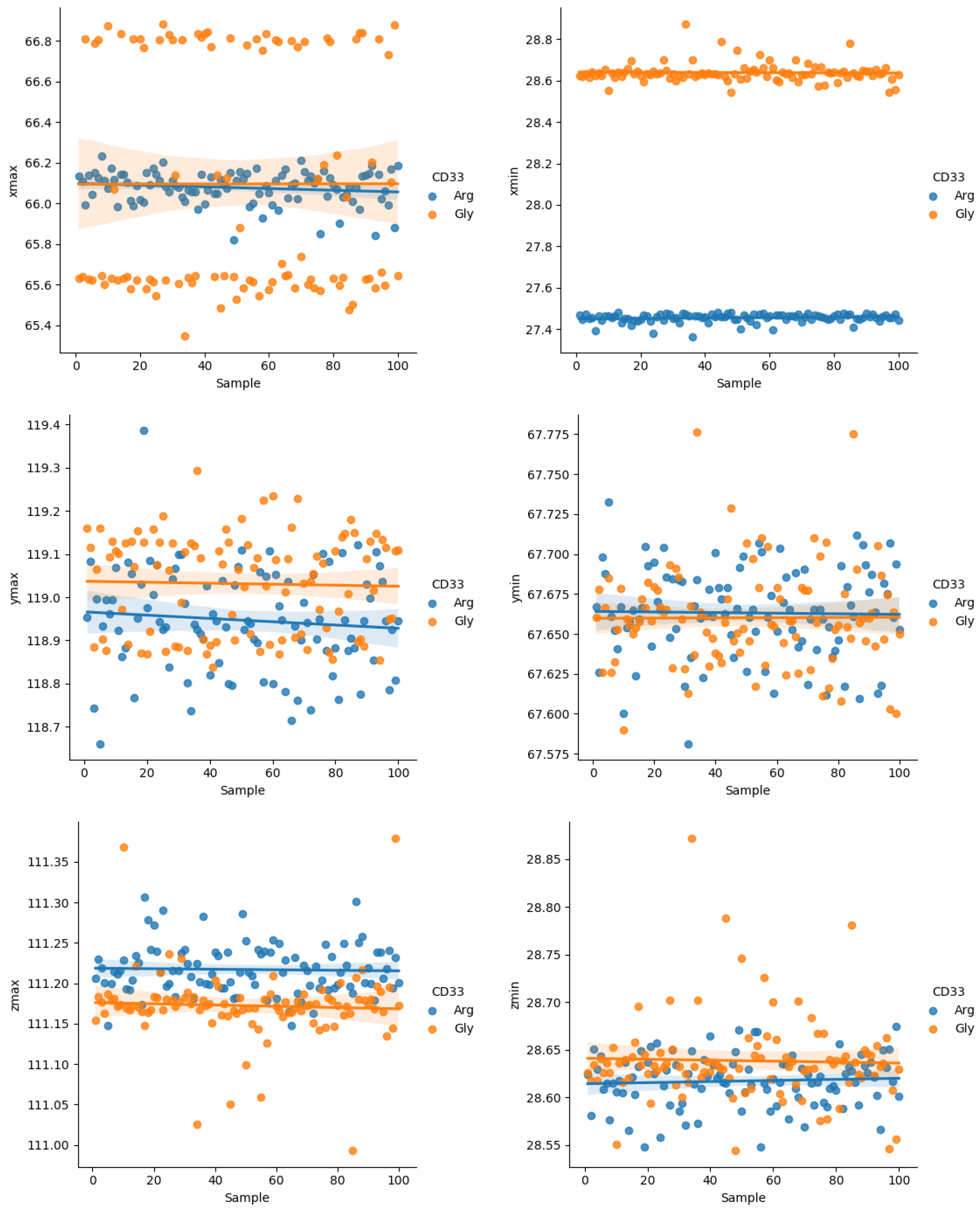
2.2. In Silico Structural Analysis
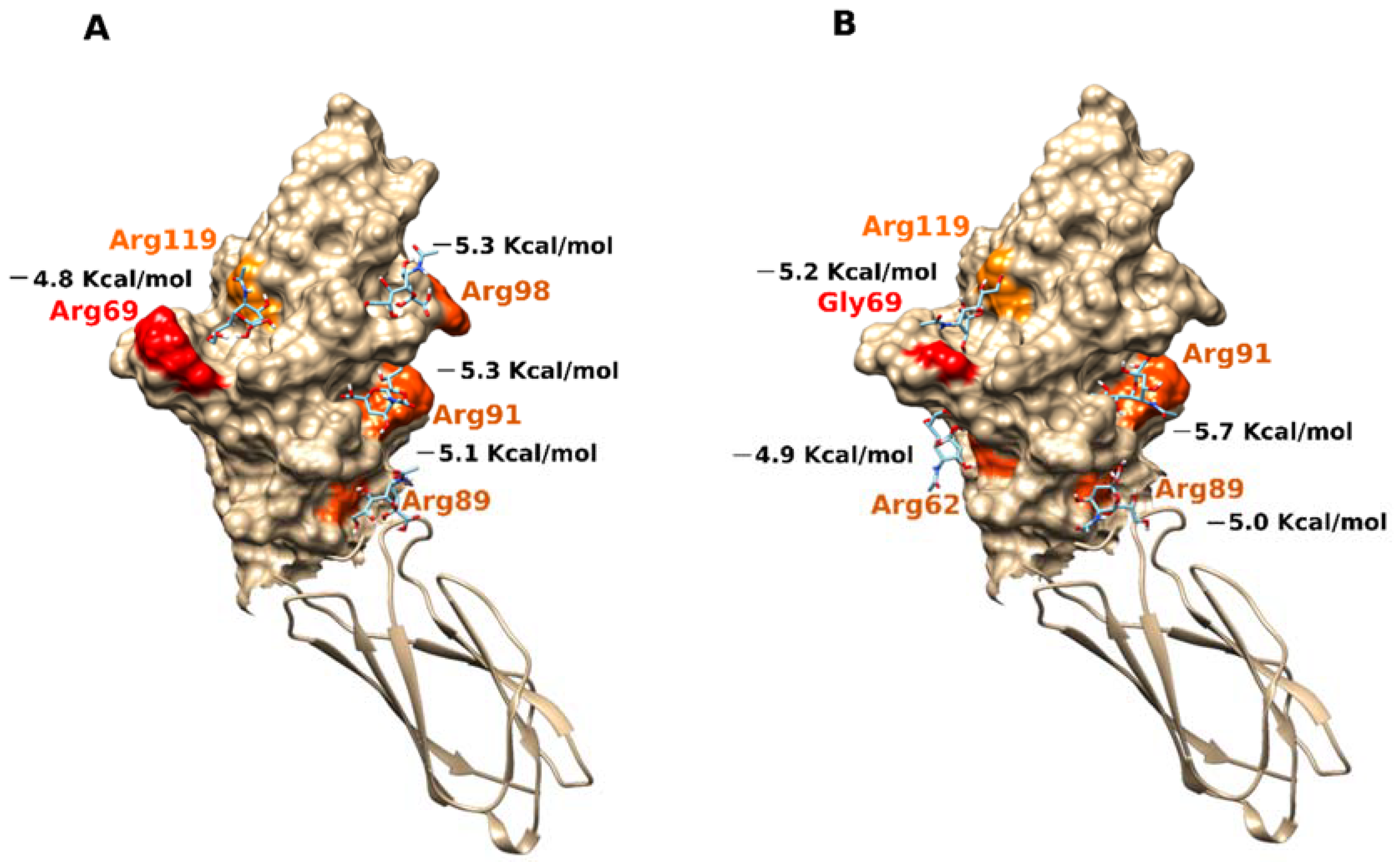
2.3. Docking Analysis
2.4. Virtual Screening Analysis
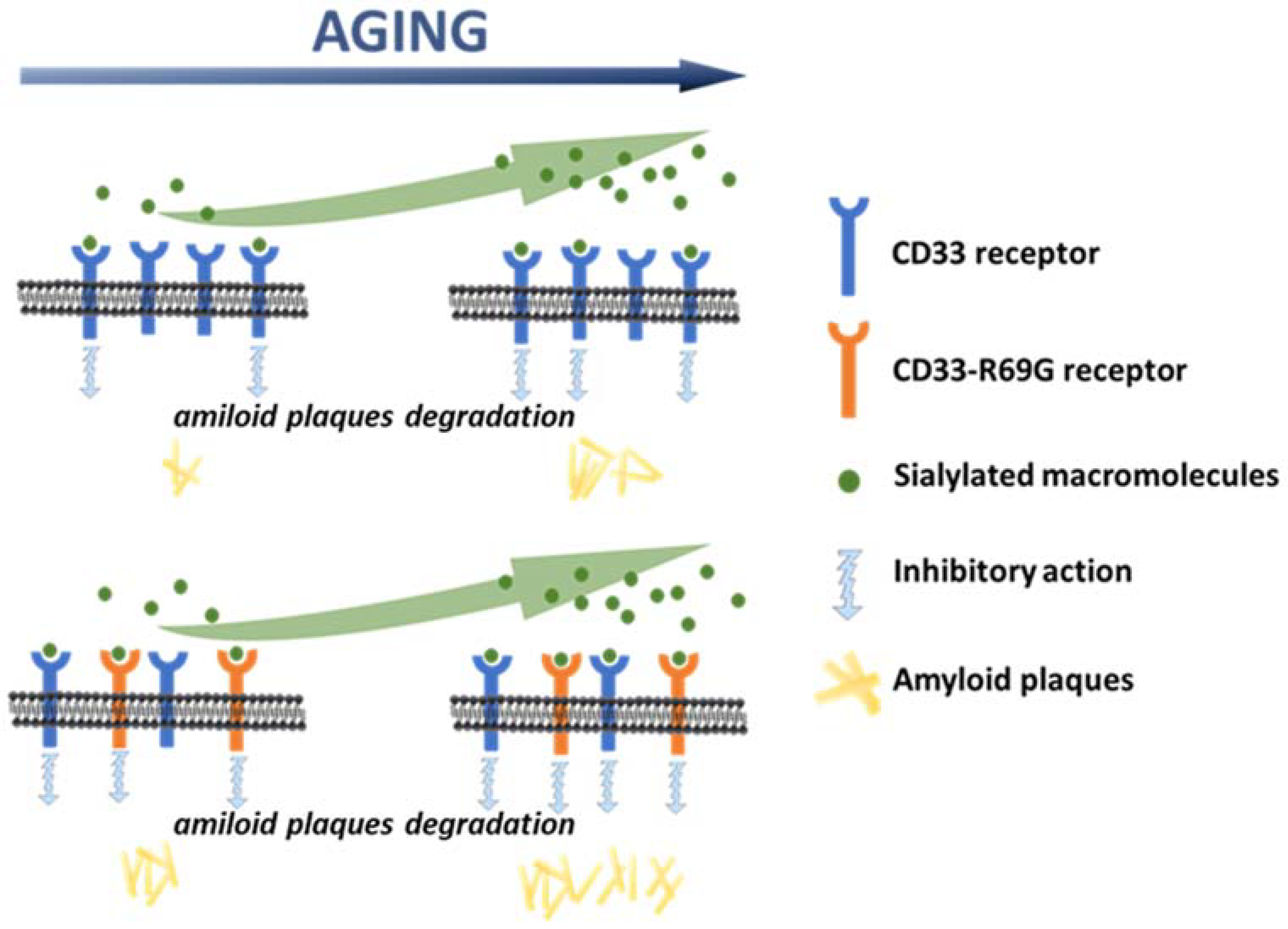
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Participant Recruitment and Analysis
5.2. High-Resolution Melting Analysis (HRM)
5.3. CD33 Structure Editing and Minimization
5.4. Ligand Structure Editing and Minimization
5.5. Docking Analysis
5.6. Virtual Screening Analysis
5.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia. Available online: https://www.alzint.org/u/WorldAlzheimerReport2019.pdf (accessed on 1 September 2019).
- GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022, 7, e105–e125. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-C.; Tan, L.; Wang, H.-F.; Jiang, T.; Cao, L.; Wang, C.; Wang, J.; Tan, C.-C.; Meng, X.-F.; Yu, J.-T. Rate of early onset Alzheimer’s disease: A systematic review and meta-analysis. Ann. Transl. Med. 2015, 3, 38, Erratum in Ann. Transl. Med. 2016, 4, E4. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Kamboh, M.I. Genomics and Functional Genomics of Alzheimer’s Disease. Neurotherapeutics 2021, 1–21. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; Van Der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430, Erratum in Nat. Genet. 2019, 51, 1423–1424. [Google Scholar] [CrossRef] [Green Version]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282, Erratum in Nat. Genet. 2021, 53, 1722. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.; Hooli, B.; DiVito, J.; Ionita, I.; et al. Genome-wide Association Analysis Reveals Putative Alzheimer’s Disease Susceptibility Loci in Addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.-C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Wang, X.-J.; Mao, Z.-F. Associations Between Genetic Variants in 19p13 and 19q13 Regions and Susceptibility to Alzheimer Disease: A Meta-Analysis. Med. Sci. Monit. 2016, 22, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Paulson, J.C. Siglecs as Immune Cell Checkpoints in Disease. Annu. Rev. Immunol. 2020, 38, 365–395. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, J.-T.; Hu, N.; Tan, M.-S.; Zhu, X.-C.; Tan, L. CD33 in Alzheimer’s Disease. Mol. Neurobiol. 2014, 49, 529–535. [Google Scholar] [CrossRef]
- Zhao, L. CD33 in Alzheimer’s Disease—Biology, Pathogenesis, and Therapeutics: A Mini-Review. Gerontology 2019, 65, 323–331. [Google Scholar] [CrossRef]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835.e7. [Google Scholar] [CrossRef]
- Ulland, T.K.; Colonna, M. TREM2—A key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 667–675. [Google Scholar] [CrossRef]
- Neumann, H.; Daly, M.J. Variant TREM2 as Risk Factor for Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 182–184. [Google Scholar] [CrossRef]
- Linnartz, B.; Wang, Y.; Neumann, H. Microglial Immunoreceptor Tyrosine-Based Activation and Inhibition Motif Signaling in Neuroinflammation. Int. J. Alzheimer’s Dis. 2010, 2010, 587463. [Google Scholar] [CrossRef] [Green Version]
- Wißfeld, J.; Nozaki, I.; Mathews, M.; Raschka, T.; Ebeling, C.; Hornung, V.; Brüstle, O.; Neumann, H. Deletion of Alzheimer’s disease-associated CD33 results in an inflammatory human microglia phenotype. Glia 2021, 69, 1393–1412. [Google Scholar] [CrossRef]
- Griciuc, A.; Federico, A.N.; Natasan, J.; Forte, A.M.; McGinty, D.; Nguyen, H.; Volak, A.; LeRoy, S.; Gandhi, S.; Lerner, E.P.; et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum. Mol. Genet. 2020, 29, 2920–2935. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Estus, S.; Shaw, B.C.; Devanney, N.; Katsumata, Y.; Press, E.E.; Fardo, D.W. Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol. 2019, 138, 187–199. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1–42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 19. [Google Scholar] [CrossRef]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 23, 2729–2736. [Google Scholar] [CrossRef] [Green Version]
- Rendina, A.; Drongitis, D.; Donizetti, A.; Fucci, L.; Milan, G.; Tripodi, F.; Giustezza, F.; Postiglione, A.; Pappatà, S.; Ferrari, R.; et al. CD33 and SIGLECL1 Immunoglobulin Superfamily Involved in Dementia. J. Neuropathol. Exp. Neurol. 2020, 79, 891–901. [Google Scholar] [CrossRef]
- Nettiksimmons, J.; Tranah, G.; Evans, D.S.; Yokoyama, J.S.; Yaffe, K. Gene-based aggregate SNP associations between candidate AD genes and cognitive decline. Age 2016, 38, 41. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Hermans, S.J.; Crespi, G.A.; Gooi, J.; Doughty, L.; Nero, T.L.; Markulić, J.; Ebneth, A.; Wroblowski, B.; Oehlrich, D.; et al. Small Molecule Binding to Alzheimer Risk Factor CD33 Promotes Aβ Phagocytosis. iScience 2019, 19, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Floyd, H.; Lopez, F.; Vivier, E.; Crocker, P. The Membrane-Proximal Immunoreceptor Tyrosine-Based Inhibitory Motif Is Critical for the Inhibitory Signaling Mediated by Siglecs-7 and -9, CD33-Related Siglecs Expressed on Human Monocytes and NK Cells. J. Immunol. 2004, 173, 6841–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varki, A.; Angata, T. Siglecs—The major subfamily of I-type lectins. Glycobiology 2006, 16, 1R–27R. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, N.; Yasuda, Y.; Yoshimura, A.; Goshima, A.; Crocker, P.R.; Vergoten, G.; Nishiura, Y.; Takahashi, T.; Hanashima, S.; Matsumoto, K.; et al. Discovery of a new sialic acid binding region that regulates Siglec-7. Sci. Rep. 2020, 10, 8647. [Google Scholar] [CrossRef]
- Olaru, O.G.; Constantin, G.I.; Pena, C.M. Variation of total serum sialic acid concentration in postmenopausal women. Exp. Ther. Med. 2020, 20, 2455–2459. [Google Scholar] [CrossRef]
- Mosconi, L.; Rahman, A.; Diaz, I.; Wu, X.; Scheyer, O.; Hristov, H.W.; Vallabhajosula, S.; Isaacson, R.S.; De Leon, M.J.; Brinton, R.D. Increased Alzheimer’s risk during the menopause transition: A 3-year longitudinal brain imaging study. PLoS ONE 2018, 13, e0207885. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, version 1.8; New York (USA) Schrödinger, LLC: 2015. Available online: Pymol.org/2/support.html (accessed on 15 February 2022).
- Brooks, B.R.; Brooks, C.L., III; MacKerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD33 Genotype | OR a (95% C.I. b) | Chi2 | p |
|---|---|---|---|
| AA | Ref | ||
| AG | 2.149 (1.259–3.668) | 8.02 | p = 0.00463 |
| GG | 3.040 (1.428–6.476) | 8.58 | p = 0.00341 |
| AG + GG | 2.294 (1.363–3.862) | 10.03 | p = 0.00154 |
| A | Ref | ||
| G | 1.555 (1.135–2.130) | 7.61 | p = 0.00582 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tortora, F.; Rendina, A.; Angiolillo, A.; Di Costanzo, A.; Aniello, F.; Donizetti, A.; Febbraio, F.; Vitale, E. CD33 rs2455069 SNP: Correlation with Alzheimer’s Disease and Hypothesis of Functional Role. Int. J. Mol. Sci. 2022, 23, 3629. https://doi.org/10.3390/ijms23073629
Tortora F, Rendina A, Angiolillo A, Di Costanzo A, Aniello F, Donizetti A, Febbraio F, Vitale E. CD33 rs2455069 SNP: Correlation with Alzheimer’s Disease and Hypothesis of Functional Role. International Journal of Molecular Sciences. 2022; 23(7):3629. https://doi.org/10.3390/ijms23073629
Chicago/Turabian StyleTortora, Fabiana, Antonella Rendina, Antonella Angiolillo, Alfonso Di Costanzo, Francesco Aniello, Aldo Donizetti, Ferdinando Febbraio, and Emilia Vitale. 2022. "CD33 rs2455069 SNP: Correlation with Alzheimer’s Disease and Hypothesis of Functional Role" International Journal of Molecular Sciences 23, no. 7: 3629. https://doi.org/10.3390/ijms23073629