
Wwox Binding to the Murine Brca1-BRCT Domain Regulates Timing of Brip1 and CtIP Phospho-Protein Interactions with This Domain at DNA Double-Strand Breaks, and Repair Pathway Choice

 , and
, and
Abstract
:1. Introduction
2. Results
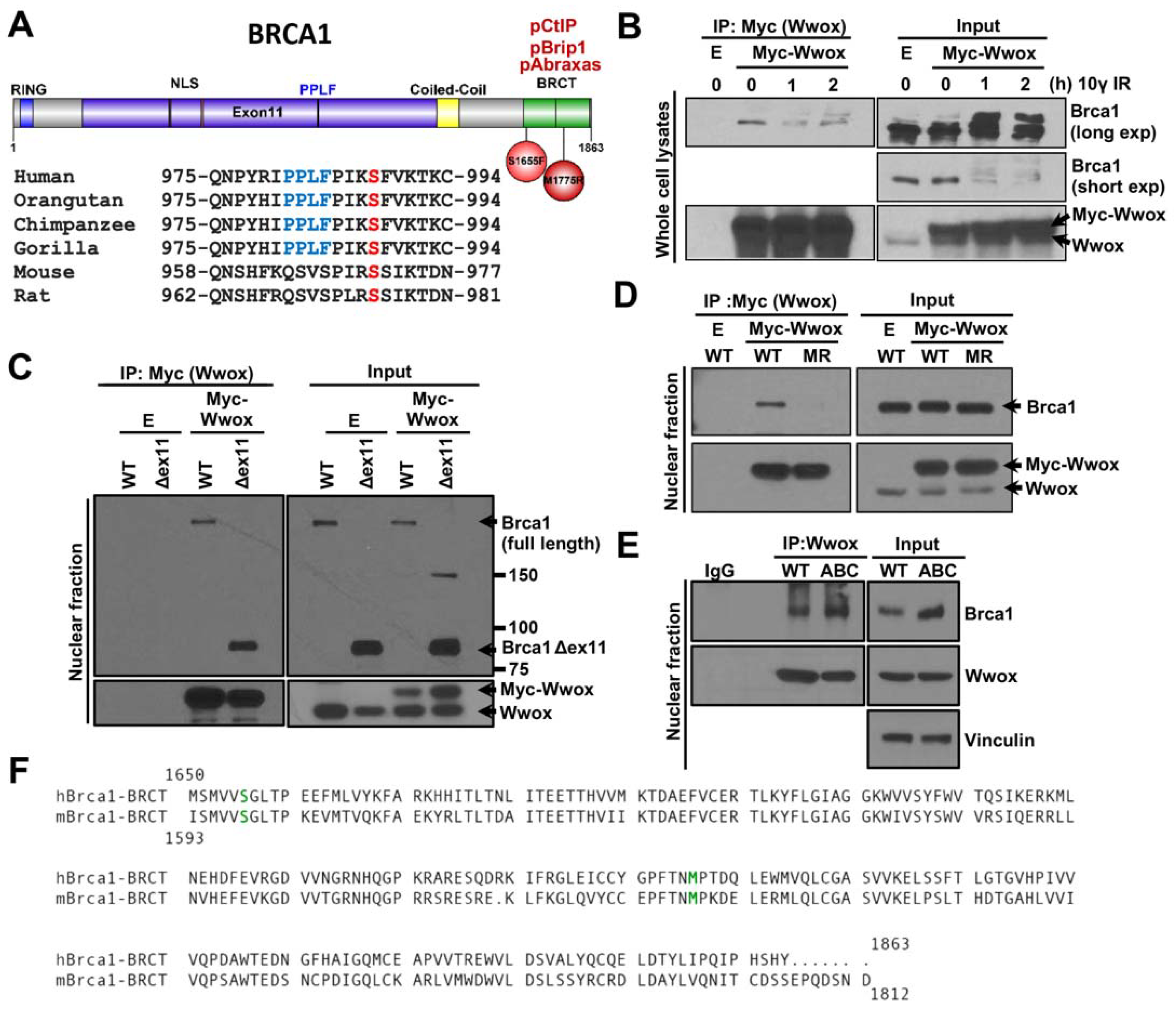
2.1. Wwox Binds to Brca1 through the BRCT Domain
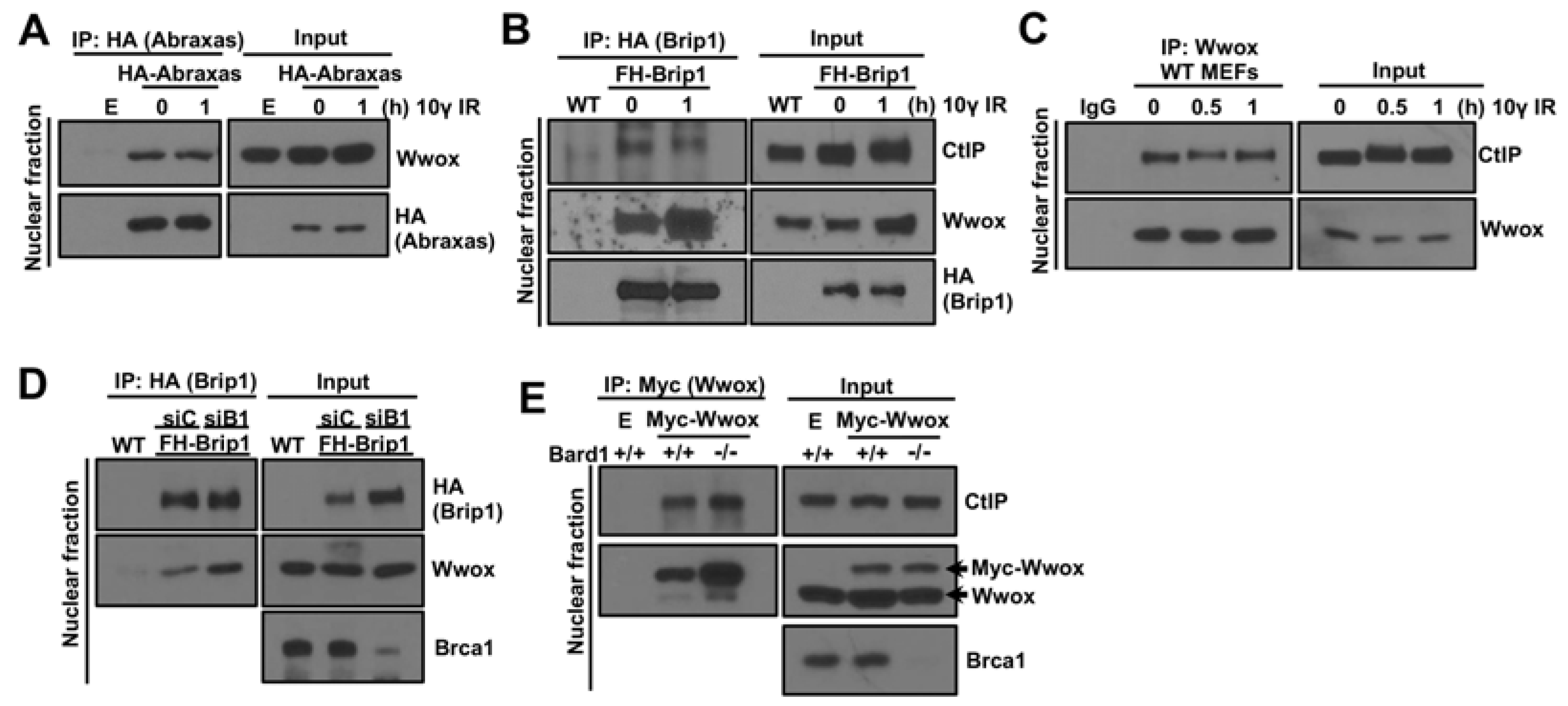
2.2. Wwox Interaction with Brca1-BRCT Is Independent of Binding of Abraxas, Brip1, and CtIP
2.3. Brca1 Independent Interaction of Wwox with Abraxas, Brip1 and CtIP
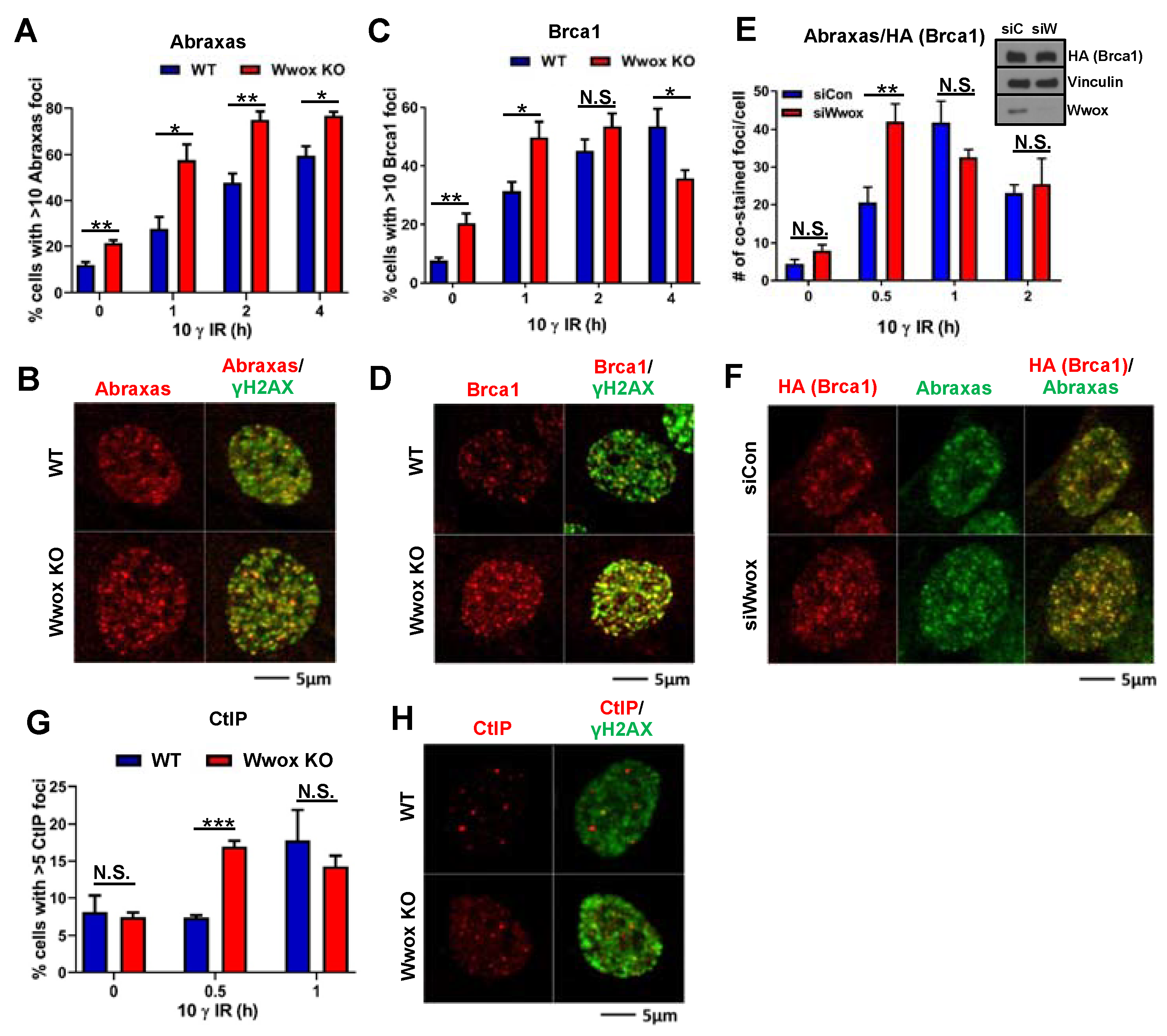
2.4. Effect of Wwox Loss on Timing of Appearance of Abraxas, Brca1 and CtIP Proteins at IR-Induced DSB Foci
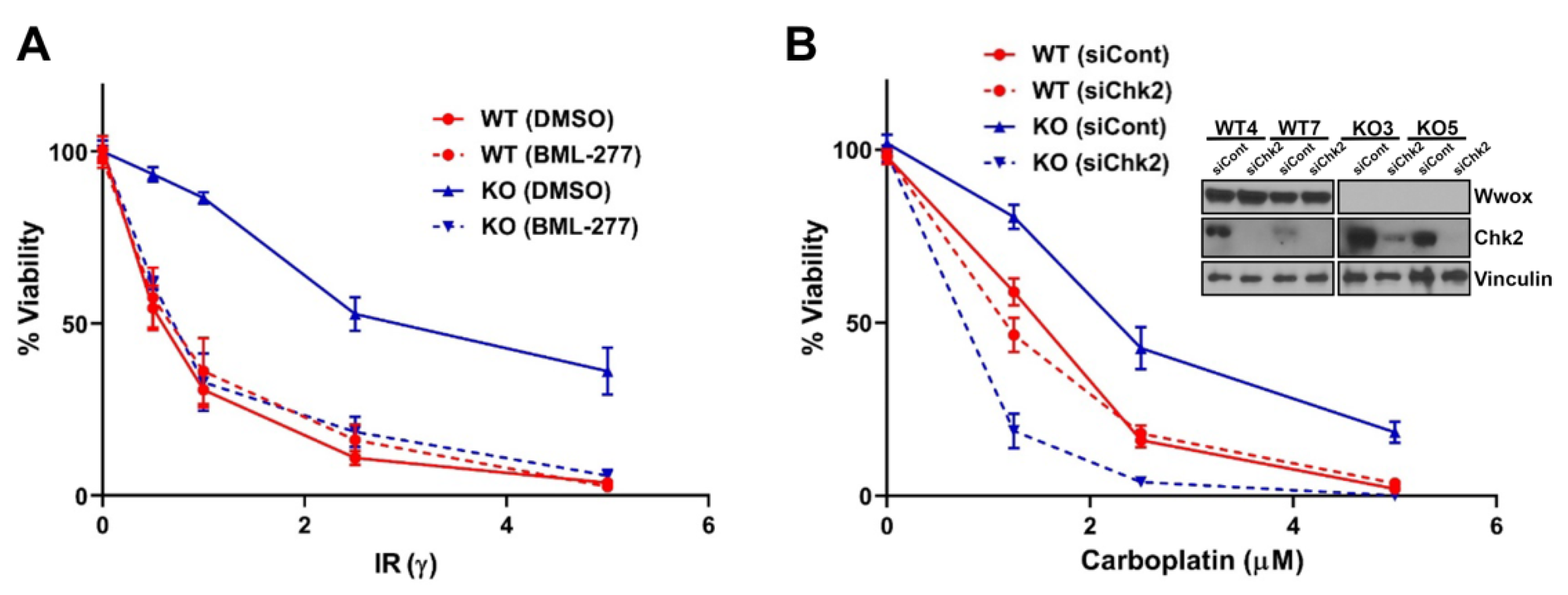
2.5. Loss of Wwox Expression in Mouse Cells Supports Initiation of HR through Rapid Engagement of End-Resection
2.6. Inhibition of End-Resection by Chk2 Silencing Sensitizes Wwox KO Cells to DNA Damaging Agents
3. Discussion
3.1. The Murine Brca1-BRCT Tandem Domain Is Required for Binding of Wwox to Brca1
3.2. Wwox Loss Accelerates Brca1-CtIP and MRN Complex Formation
3.3. Wwox Absence in Mouse Cells, as in Human, Favors the Choice of HR Repair Early after DSB Induction
4. Materials and Methods
4.1. Cell Lines and Transfection
4.2. Immunofluorescence Staining and Duolink In Situ Proximity Ligation Assay (PLA)
4.3. Western Blot and Immunoprecipitation Assays
4.4. Isolation of Soluble Nuclear Fraction
4.5. DR-GFP Assay
4.6. Chemicals and Antisera
4.7. Single Molecule Analysis of Resection Tracks (SMART) Assay
4.8. Colony Formation and Mutation Assays
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richards, R.I.; Choo, A.; Lee, C.S.; Dayan, S.; O’Keefe, L. WWOX, the chromosomal fragile site FRA16D spanning gene: Its role in metabolism and contribution to cancer. Exp. Biol. Med. 2015, 240, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bednarek, A.K.; Laflin, K.J.; Daniel, R.L.; Liao, Q.; Hawkins, K.A.; Aldaz, C.M. WWOX, a novel WW domain-containing protein mapping to human chromosome 16q23.3-24.1, a region frequently affected in breast cancer. Cancer Res. 2000, 60, 2140–2145. [Google Scholar] [PubMed]
- Ramos, D.; Abba, M.; López-Guerrero, J.A.; Rubio, J.; Solsona, E.; Almenar, S.; Llombart-Bosch, A.; Aldaz, C.M. Low levels of WWOX protein immunoexpression correlate with tumour grade and a less favourable outcome in patients with urinary bladder tumours. Histopathology 2008, 52, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Płuciennik, E.; Kusińska, R.; Potemski, P.; Kubiak, R.; Kordek, R.; Bednarek, A.K. WWOX--the FRA16D cancer gene: Expression correlation with breast cancer progression and prognosis. Eur. J. Surg. Oncol. 2006, 32, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Paige, A.J.; Zucknick, M.; Janczar, S.; Paul, J.; Mein, C.A.; Taylor, K.J.; Stewart, M.; Gourley, C.; Richardson, S.; Perren, T.; et al. WWOX tumour suppressor gene polymorphisms and ovarian cancer pathology and prognosis. Eur. J. Cancer 2010, 46, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Liu, B.; Shrock, M.S.; Williams, T.; Aldaz, C.M. WWOX, the FRA16D gene: A target of and a contributor to genomic instability. Genes Chromosomes Cancer 2019, 58, 324–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrock, M.S.; Batar, B.; Lee, J.; Druck, T.; Ferguson, B.; Cho, J.H.; Akakpo, K.; Hagrass, H.; Heerema, N.A.; Xia, F.; et al. Wwox-Brca1 interaction: Role in DNA repair pathway choice. Oncogene 2017, 36, 2215–2227. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.; Himmetoglu, C.; Jimenez, R.E.; Geyer, S.M.; Wang, W.P.; Costinean, S.; Pilarski, R.T.; Morrison, C.; Suren, D.; Liu, J.; et al. Aberrant expression of DNA damage response proteins is associated with breast cancer subtype and clinical features. Breast Cancer Res. Treat. 2011, 129, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Abu-Odeh, M.; Hereema, N.A.; Aqeilan, R.I. WWOX modulates the ATR-mediated DNA damage checkpoint response. Oncotarget 2016, 7, 4344–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.; Gharghabi, M.; Schrock, M.S.; Plow, R.; Druck, T.; Yungvirt, C.; Aldaz, C.M.; Huebner, K. Interaction of Wwox with Brca1 and associated complex proteins prevents premature resection at double-strand breaks and aberrant homologous recombination. DNA Repair 2021, 110, 103264. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collins, K.M.; Brown, A.L.; Lee, C.H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B. BRCA1 tumor suppressor network: Focusing on its tail. Cell Biosci. 2012, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Manke, I.A.; Lowery, D.M.; Nguyen, A.; Yaffe, M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 2003, 302, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Yu, X.; Chen, J.; Songyang, Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J. Biol. Chem. 2003, 278, 52914–52918. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef] [Green Version]
- Clapperton, J.A.; Manke, I.A.; Lowery, D.M.; Ho, T.; Haire, L.F.; Yaffe, M.B.; Smerdon, S.J. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat. Struct. Mol. Biol. 2004, 11, 512–518. [Google Scholar] [CrossRef]
- Wu, Q.; Paul, A.; Su, D.; Mehmood, S.; Foo, T.K.; Ochi, T.; Bunting, E.L.; Xia, B.; Robinson, C.V.; Wang, B.; et al. Structure of BRCA1-BRCT/Abraxas Complex Reveals Phosphorylation-Dependent BRCT Dimerization at DNA Damage Sites. Mol. Cell 2016, 61, 434–448. [Google Scholar] [CrossRef] [Green Version]
- Dever, S.M.; Golding, S.E.; Rosenberg, E.; Adams, B.R.; Idowu, M.O.; Quillin, J.M.; Valerie, N.; Xu, B.; Povirk, L.F.; Valerie, K. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging 2011, 3, 515–532. [Google Scholar] [CrossRef] [Green Version]
- Glover, J.N. Insights into the molecular basis of human hereditary breast cancer from studies of the BRCA1 BRCT domain. Fam. Cancer 2006, 5, 89–93. [Google Scholar] [CrossRef]
- Lobachev, K.S.; Gordenin, D.A.; Resnick, M.A. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell 2002, 108, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019, 38, e101005. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Szabolcs, M.; Stark, J.M.; Ludwig, T.; Baer, R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol. 2013, 201, 693–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reczek, C.R. The Role of CtIP in Brca1-Mediated Tumor Suppression. Ph.D. Thesis, Columbia University, New York, NY, USA, 2012. [Google Scholar] [CrossRef]
- Cruz-García, A.; López-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, T. BRCA1 phosphorylation: Biological consequences. Cancer Biol. Ther. 2006, 5, 470–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.L.; Greenberg, R.A. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem. Sci. 2012, 37, 418–424. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, B.; Chiang, H.C.; Lu, Y.; Coates, J.; Deng, C.X.; Baer, R.; Lin, H.K.; Li, R.; Paull, T.T.; Hu, Y. Damage-induced BRCA1 phosphorylation by Chk2 contributes to the timing of end resection. Cell Cycle 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.S.; de Rooij, D.G.; Hirsch, S.; Ravi, K.; Hicks, J.B.; et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science 2011, 334, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, T.; Lee, J.; Abba, M.C.; Chen, J.; Aldaz, C.M. Delineating WWOX Protein Interactome by Tandem Affinity Purification-Mass Spectrometry: Identification of Top Interactors and Key Metabolic Pathways Involved. Front. Oncol. 2018, 8, 591. [Google Scholar] [CrossRef]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Nagaraju, G. FANCJ helicase promotes DNA end resection by facilitating CtIP recruitment to DNA double-strand breaks. PLoS Genet. 2020, 16, e1008701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joukov, V.; Chen, J.; Fox, E.A.; Green, J.B.; Livingston, D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA 2001, 98, 12078–12083. [Google Scholar] [CrossRef] [Green Version]
- Botuyan, M.V.; Nominé, Y.; Yu, X.; Juranic, N.; Macura, S.; Chen, J.; Mer, G. Structural basis of BACH1 phosphopeptide recognition by BRCA1 tandem BRCT domains. Structure 2004, 12, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Reid, L.J.; Shakya, R.; Modi, A.P.; Lokshin, M.; Cheng, J.T.; Jasin, M.; Baer, R.; Ludwig, T. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 20876–20881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daley, J.M.; Jimenez-Sainz, J.; Wang, W.; Miller, A.S.; Xue, X.; Nguyen, K.A.; Jensen, R.B.; Sung, P. Enhancement of BLM-DNA2-Mediated Long-Range DNA End Resection by CtIP. Cell Rep. 2017, 21, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altan, B.; Yokobori, T.; Ide, M.; Bai, T.; Yanoma, T.; Kimura, A.; Kogure, N.; Suzuki, M.; Bao, P.; Mochiki, E.; et al. High Expression of MRE11-RAD50-NBS1 Is Associated with Poor Prognosis and Chemoresistance in Gastric Cancer. Anticancer Res. 2016, 36, 5237–5247. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Bergin, S.M.; Jones, D.; Ru, P.; Koivisto, C.S.; Jeon, Y.J.; Sizemore, G.M.; Kladney, R.D.; Hadjis, A.; Shakya, R.; et al. Ablation of the Brca1-Palb2 Interaction Phenocopies Fanconi Anemia in Mice. Cancer Res. 2020, 80, 4172–4184. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Shakya, R.; Koivisto, C.; Pitarresi, J.R.; Szabolcs, M.; Kladney, R.; Hadjis, A.; Mace, T.A.; Ludwig, T. Murine models for familial pancreatic cancer: Histopathology, latency and drug sensitivity among cancers of Palb2, Brca1 and Brca2 mutant mouse strains. PLoS ONE 2019, 14, e0226714. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antisera | Source | WB | IP | IF/PLA |
|---|---|---|---|---|
| Brca1 | 57X from Ludwig Lab | 1:2000 | 1:500 | |
| Abraxas | Ludwig lab | 1:5000 | 1:3000 | |
| CtIP (mab) | 11-1 from R. Baer Lab | 1:50 | 1:25 | |
| CtIP | Santa Cruz, SC-271339 | 1:300 | 1:100 | |
| Rad51 | Abcam, ab133534 | 1:500 | ||
| Phospho RPA32 (S4/S8) | Bethyl, A300-245A | 1:1000 | ||
| Mre11 | Cell signaling, #4895S | 1:1000 | ||
| Rad50 | Cell signaling, #3427S | 1:1000 | ||
| Nbs1 | Novus, NB100-143 | 1:2000 | ||
| Wwox | Huebner Lab | 1:5000 | 1:125 | |
| Vinculin | Invitrogen, 700062 | 1:5000 | ||
| HA | Roche, 11867423001 | 1:500 | ||
| BrdU | Abcam, ab6326 | 1:100 | ||
| Chk2 | Cell signaling, #3440S | 1:1000 | ||
| Gamma H2AX | Cell signaling, #9718S | 1:500 | ||
| Gamma H2AX | Sigma, #05-636 | 1:5000 | ||
| HA magnetic beads | Pierce, 88837 | 20 μL | ||
| Myc magnetic beads | Pierce, 88842 | 20 μL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, D.; Gharghabi, M.; Reczek, C.R.; Plow, R.; Yungvirt, C.; Aldaz, C.M.; Huebner, K. Wwox Binding to the Murine Brca1-BRCT Domain Regulates Timing of Brip1 and CtIP Phospho-Protein Interactions with This Domain at DNA Double-Strand Breaks, and Repair Pathway Choice. Int. J. Mol. Sci. 2022, 23, 3729. https://doi.org/10.3390/ijms23073729
Park D, Gharghabi M, Reczek CR, Plow R, Yungvirt C, Aldaz CM, Huebner K. Wwox Binding to the Murine Brca1-BRCT Domain Regulates Timing of Brip1 and CtIP Phospho-Protein Interactions with This Domain at DNA Double-Strand Breaks, and Repair Pathway Choice. International Journal of Molecular Sciences. 2022; 23(7):3729. https://doi.org/10.3390/ijms23073729
Chicago/Turabian StylePark, Dongju, Mehdi Gharghabi, Colleen R. Reczek, Rebecca Plow, Charles Yungvirt, C. Marcelo Aldaz, and Kay Huebner. 2022. "Wwox Binding to the Murine Brca1-BRCT Domain Regulates Timing of Brip1 and CtIP Phospho-Protein Interactions with This Domain at DNA Double-Strand Breaks, and Repair Pathway Choice" International Journal of Molecular Sciences 23, no. 7: 3729. https://doi.org/10.3390/ijms23073729
APA StylePark, D., Gharghabi, M., Reczek, C. R., Plow, R., Yungvirt, C., Aldaz, C. M., & Huebner, K. (2022). Wwox Binding to the Murine Brca1-BRCT Domain Regulates Timing of Brip1 and CtIP Phospho-Protein Interactions with This Domain at DNA Double-Strand Breaks, and Repair Pathway Choice. International Journal of Molecular Sciences, 23(7), 3729. https://doi.org/10.3390/ijms23073729