
Mitochondrial Lipids: From Membrane Organization to Apoptotic Facilitation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondrial Membrane Physiology and Lipid Composition
2.1. The Mitochondrial Double Membrane: Features, Structure and Function
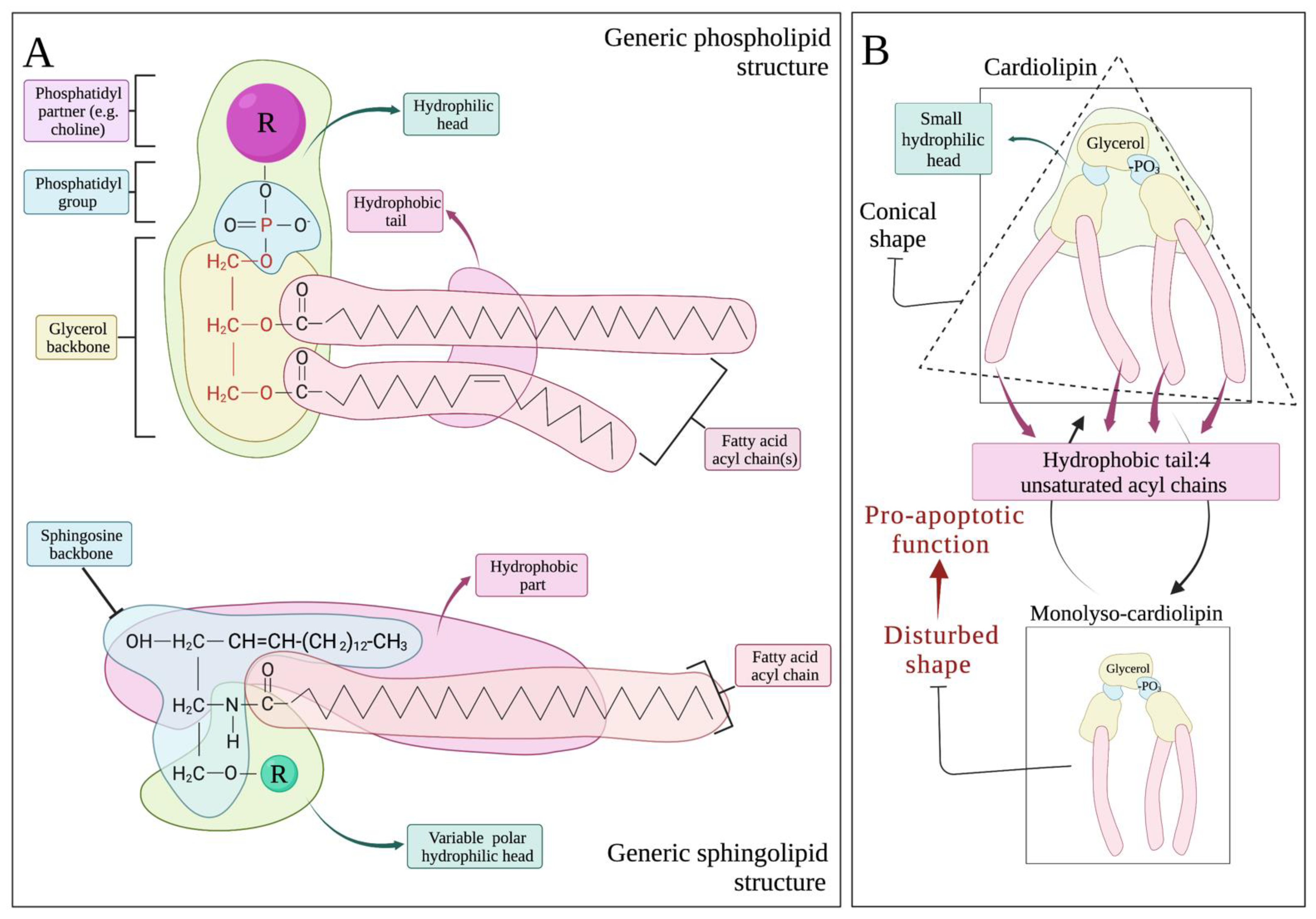
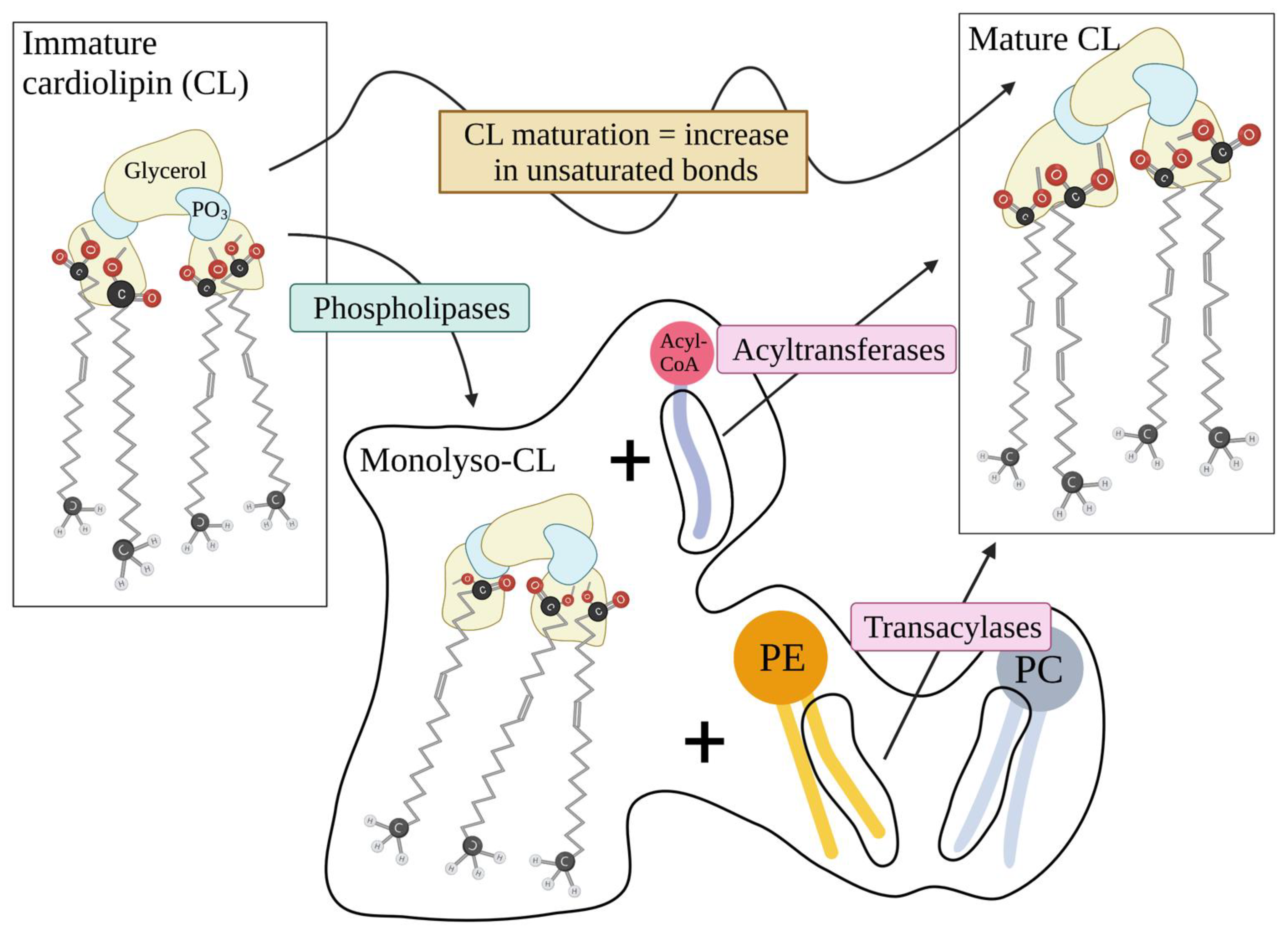
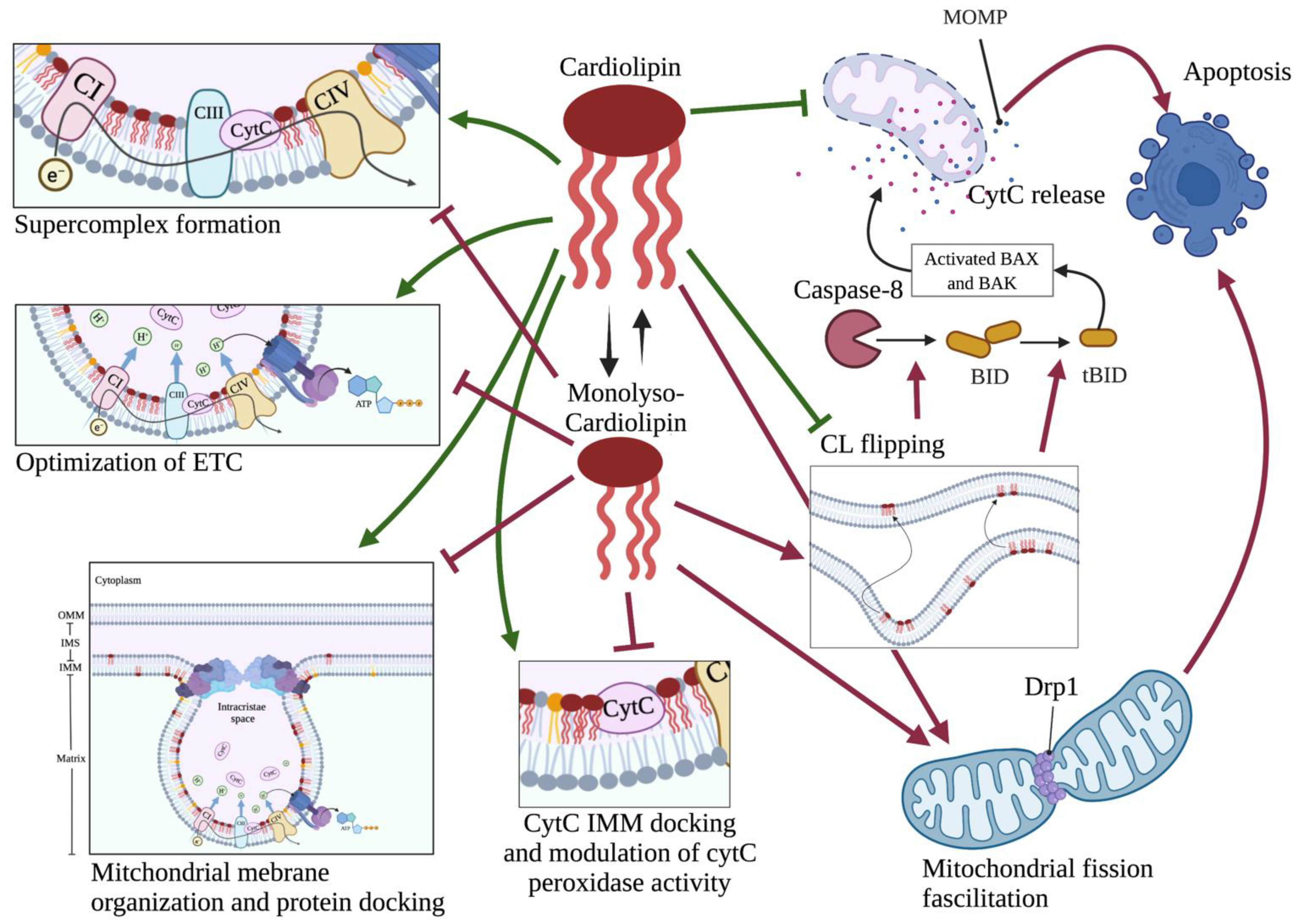
2.2. The Lipid Structure of Mitochondrial Membrane: Cardiolipin Abundance, Sphingolipid Paucity and Respiratory Integrity
3. Mitochondrial Membrane Lipids in Apoptosis
3.1. CL in Apoptosis
3.2. Sphingolipids and Other Non-Mitochondrial Lipids in Apoptosis
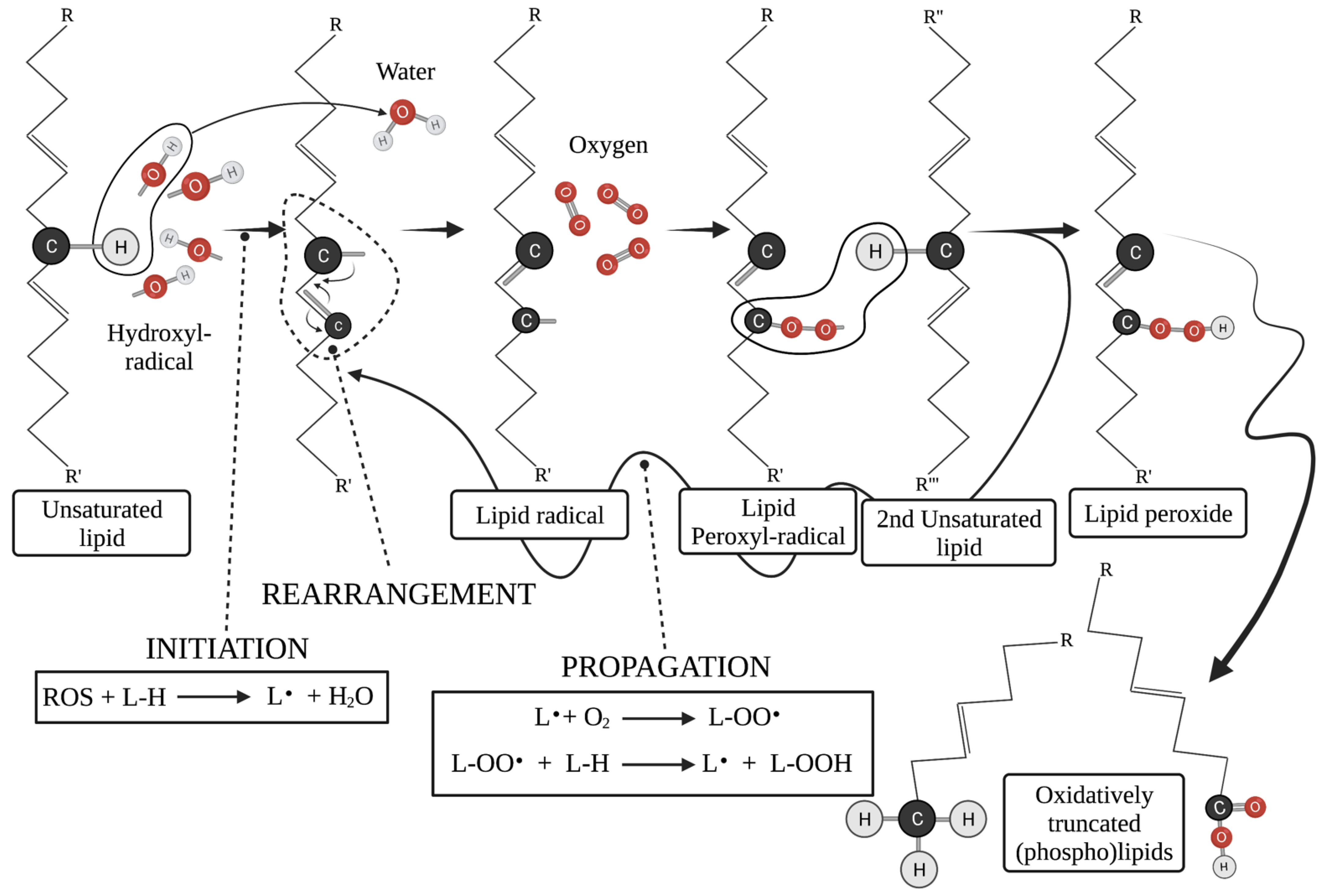
3.2.1. Oxidatively Truncated Phospholipids
3.2.2. Ceramides
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Kell, D.B.; Oliver, S.G. The metabolome 18 years on: A concept comes of age. Metabolomics 2016, 12, 148. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Kornbluth, S. The Tangled Circuitry of Metabolism and Apoptosis. Mol. Cell 2013, 49, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wang, L.; Yang, C.; Pu, S.; Guo, Z.; Wu, Q.; Zhou, Z.; Zhao, H. Mitochondrial Membrane Remodeling. Front. Bioeng. Biotechnol. 2022, 9, 786806. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.E.; Daum, G. Lipids of mitochondria. Prog. Lipid Res. 2013, 52, 590–614. [Google Scholar] [CrossRef] [PubMed]
- Nielson, J.R.; Rutter, J.P. Lipid-mediated signals that regulate mitochondrial biology. J. Biol. Chem. 2018, 293, 7517–7521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.P.; Liesa, M. The Role of Mitochondrial Fat Oxidation in Cancer Cell Proliferation and Survival. Cells 2020, 9, 2600. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Srivastava, P.; Mathur, S.; Abbas, S.; Rai, N.; Tiwari, S.; Tiwari, M.; Sharma, L. Lipid metabolism and mitochondria: Cross talk in cancer. Curr. Drug Targets 2021, 22, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Iovine, J.C.; Claypool, S.M.; Alder, N.N. Mitochondrial compartmentalization: Emerging themes in structure and function. Trends Biochem. Sci. 2021, 46, 902–917. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Herrmann, J.M. Protein Translocation into the Intermembrane Space and Matrix of Mitochondria: Mechanisms and Driving Forces. Front. Mol. Biosci. 2017, 4, 83. [Google Scholar] [CrossRef]
- Edwards, R.; Eaglesfield, R.; Tokatlidis, K. The mitochondrial intermembrane space: The most constricted mitochondrial sub-compartment with the largest variety of protein import pathways. Open Biol. 2021, 11, 210002. [Google Scholar] [CrossRef]
- Porcelli, A.M.; Ghelli, A.; Zanna, C.; Pinton, P.; Rizzuto, R.; Rugolo, M. pH difference across the outer mitochondrial membrane measured with a green fluorescent protein mutant. Biochem. Biophys. Res. Commun. 2005, 326, 799–804. [Google Scholar] [CrossRef]
- Gohil, V.M.; Greenberg, M.L. Mitochondrial membrane biogenesis: Phospholipids and proteins go hand in hand. J. Cell Biol. 2009, 184, 469–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cogliati, S.; Enríquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin-dependent formation of mitochondrial respiratory supercomplexes. Chem. Phys. Lipids 2013, 179, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Mejia, E.M.; Hatch, G.M. Mitochondrial phospholipids: Role in mitochondrial function. J. Bioenerg. Biomembr. 2016, 48, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Hoch, F.L. Cardiolipins and biomembrane function. Biochim. et Biophys. Acta Rev. Biomembr. 1992, 1113, 71–133. [Google Scholar] [CrossRef] [Green Version]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed] [Green Version]
- Breslow, D.K.; Weissman, J.S. Membranes in Balance: Mechanisms of Sphingolipid Homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Rousseau, D.; Boissan, M.; Mannella, C.; Epand, R.; Lacombe, M.L. Mitochondrial cardiolipin/phospholipid trafficking: The role of membrane contact site complexes and lipid transfer proteins. Chem. Phys. Lipids 2014, 179, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Petrungaro, C.; Kornmann, B. Lipid exchange at ER-mitochondria contact sites: A puzzle falling into place with quite a few pieces missing. Curr. Opin. Cell Biol. 2019, 57, 71–76. [Google Scholar] [CrossRef]
- Rainbolt, T.; Saunders, J.M.; Wiseman, R.L. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Ball, W.B.; Neff, J.; Gohil, V.M. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett. 2018, 592, 1273–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohil, V.; Thompson, M.N.; Greenberg, M.L. Synthetic Lethal Interaction of the Mitochondrial Phosphatidylethanolamine and Cardiolipin Biosynthetic Pathways in Saccharomyces cerevisiae. J. Biol. Chem. 2005, 280, 35410–35416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Schlame, M. The turnover of glycerol and acyl moieties of cardiolipin. Chem. Phys. Lipids 2014, 179, 17–24. [Google Scholar] [CrossRef]
- Saini-Chohan, H.K.; Mitchell, R.W.; Vaz, F.; Zelinski, T.; Hatch, G.M. Delineating the role of alterations in lipid metabolism to the pathogenesis of inherited skeletal and cardiac muscle disorders. J. Lipid Res. 2012, 53, 4–27. [Google Scholar] [CrossRef] [Green Version]
- Crimi, M.; Esposti, M.D. Apoptosis-induced changes in mitochondrial lipids. Biochim. Biophys. Acta 2011, 1813, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Francy, C.A.; Clinton, R.W.; Frohlich, C.; Murphy, C.; Mears, J.A. Cryo-EM Studies of Drp1 Reveal Cardiolipin Interactions that Activate the Helical Oligomer. Sci. Rep. 2017, 7, 10744. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, D.B.; Sparagna, G.C.; Amoscato, A.A.; McMillin, J.B.; Dowhan, W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 2001, 276, 38061–38067. [Google Scholar] [CrossRef]
- Xu, Y.; Phoon, C.K.; Berno, B.; D’Souza, K.; Hoedt, E.; Zhang, G.; Neubert, E.H.G.Z.T.A.; Epand, R.; Ren, M.; Schlame, Y.X.M.R.M. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat. Chem. Biol. 2016, 12, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Zhu, M. Free radical oxidation of cardiolipin: Chemical mechanisms, detection and implication in apoptosis, mitochondrial dysfunction and human diseases. Free Radic. Res. 2012, 46, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Bayir, H.; Fadeel, B.; Palladino, M.; Witasp, E.; Kurnikov, I.; Tyurina, Y.; Tyurin, V.; Amoscato, A.; Jiang, J.; Kochanek, P.; et al. Apoptotic interactions of cytochrome c: Redox flirting with anionic phospholipids within and outside of mitochondria. Biochim. Biophys. Acta 2006, 1757, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef]
- Kawai, C.; Ferreira, J.C.; Baptista, M.S.; Nantes, I.L. Not Only Oxidation of Cardiolipin Affects the Affinity of Cytochrome c for Lipid Bilayers. J. Phys. Chem. B 2014, 118, 11863–11872. [Google Scholar] [CrossRef] [PubMed]
- Basova, L.V.; Kurnikov, I.V.; Wang, L.; Ritov, V.B.; Belikova, N.A.; Vlasova, I.I.; Pacheco, A.A.; Winnica, D.E.; Peterson, J.; Bayir, H.; et al. Cardiolipin Switch in Mitochondria: Shutting off the Reduction of Cytochrome c and Turning on the Peroxidase Activity. Biochemistry 2007, 46, 3423–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, M.; Niibayashi, R.; Koubori, S.; Moriyama, I.; Miyoshi, H. Molecular Mechanisms for the Induction of Peroxidase Activity of the Cytochrome c–Cardiolipin Complex. Biochemistry 2011, 50, 8383–8391. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B. Cardiolipin oxidation sets cytochrome c free. Nat. Chem. Biol. 2005, 1, 188–189. [Google Scholar] [CrossRef]
- Li, X.X.; Tsoi, B.; Li, Y.F.; Kurihara, H.; He, R.R. Cardiolipin and Its Different Properties in Mitophagy and Apoptosis. J. Histochem. Cytochem. 2015, 63, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, T.; Giammarioli, A.M.; Misasi, R.; Tinari, A.; Manganelli, V.; Gambardella, L.; Pavan, A.; Malorni, W.; Sorice, M. Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 2005, 12, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009, 583, 2447–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.; Newmeyer, D.D. Bid, Bax, and Lipids Cooperate to Form Supramolecular Openings in the Outer Mitochondrial Membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Martinou, J.-C.; Fornallaz-Mulhauser, M.; Hughes, D.W.; Epand, R.M. The Apoptotic Protein tBid Promotes Leakage by Altering Membrane Curvature. J. Biol. Chem. 2002, 277, 32632–32639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goonesinghe, A.; Mundy, E.S.; Smith, M.; Khosravi-Far, R.; Martinou, J.-C.; Esposti, M.D. Pro-apoptotic Bid induces membrane perturbation by inserting selected lysolipids into the bilayer. Biochem. J. 2005, 387, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.; Tyurina, Y.; Bayir, H.; Chu, C.; Kapralov, A.; Vlasova, I.; Belikova, N.; Tyurin, V.; Amoscato, A.; Epperly, M.; et al. The “pro-apoptotic genies” get out of mitochondria: Oxidative lipidomics and redox activity of cytochrome c/cardiolipin complexes. Chem. Interact. 2006, 163, 15–28. [Google Scholar] [CrossRef]
- Chen, R.; Feldstein, A.E.; McIntyre, T.M. Suppression of Mitochondrial Function by Oxidatively Truncated Phospholipids Is Reversible, Aided by Bid, and Suppressed by Bcl-XL. J. Biol. Chem. 2009, 284, 26297–26308. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Fruhwirth, G.O.; Loidl, A.; Hermetter, A. Oxidized phospholipids: From molecular properties to disease. Biochim. Biophys. Acta 2007, 1772, 718–736. [Google Scholar] [CrossRef] [Green Version]
- Latchoumycandane, C.; Marathe, G.K.; Zhang, R.; McIntyre, T.M. Oxidatively truncated phospholipids are required agents of tumor necrosis factor alpha (TNFalpha)-induced apoptosis. J. Biol. Chem. 2012, 287, 17693–17705. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Stemmer, U.; A Dunai, Z.; Koller, D.; Pürstinger, G.; Zenzmaier, E.; Deigner, H.P.; Aflaki, E.; Kratky, D.; Hermetter, A. Toxicity of oxidized phospholipids in cultured macrophages. Lipids Health Dis. 2012, 11, 110. [Google Scholar] [CrossRef] [Green Version]
- Halasiddappa, L.M.; Koefeler, H.; Futerman, A.H.; Hermetter, A. Oxidized Phospholipids Induce Ceramide Accumulation in RAW 264.7 Macrophages: Role of Ceramide Synthases. PLoS ONE 2013, 8, e70002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Yang, L.; McIntyre, T.M. Cytotoxic phospholipid oxidation products. Cell death from mitochondrial damage and the intrinsic caspase cascade. J. Biol. Chem. 2007, 282, 24842–24850. [Google Scholar] [CrossRef] [Green Version]
- Aufschnaiter, A.; Kohler, V.; Diessl, J.; Peselj, C.; Carmona-Gutierrez, D.; Keller, W.; Büttner, S. Mitochondrial lipids in neurodegeneration. Cell. Tissue Res. 2017, 367, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide forms channels in mitochondrial outer membranes at physiologically relevant concentrations. Mitochondrion 2006, 6, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Colombini, M. Ceramide channels and their role in mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2010, 1797, 1239–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and Mitochondria in Ischemia/Reperfusion. J. Cardiovasc. Pharmacol. 2009, 53, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Wattenberg, B.W. The long and the short of ceramides. J. Biol. Chem. 2018, 293, 9922–9923. [Google Scholar] [CrossRef] [Green Version]
- Knupp, J.; Martínez-Montañés, F.; Bergh, F.V.D.; Cottier, S.; Schneiter, R.; Beard, D.; Chang, A. Sphingolipid accumulation causes mitochondrial dysregulation and cell death. Cell Death Differ. 2017, 24, 2044–2053. [Google Scholar] [CrossRef] [Green Version]
- Siskind, L.J. Mitochondrial Ceramide and the Induction of Apoptosis. J. Bioenerg. Biomembr. 2005, 37, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fugio, L.B.; Coeli-Lacchini, F.; Leopoldino, A.M. Sphingolipids and Mitochondrial Dynamic. Cells 2020, 9, 581. [Google Scholar] [CrossRef] [Green Version]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.M.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Muller, D.; Schneider, M.; et al. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Park, W.; Park, J. The role of sphingolipids in endoplasmic reticulum stress. FEBS Lett. 2020, 594, 3632–3651. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Lisbona, F.; Rivera, D.R.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Kitatani, K.; Kondo, T.; Hashimoto-Nishimura, M.; Asano, S.; Hayashi, A.; Mitsutake, S.; Igarashi, Y.; Umehara, H.; Takeya, H.; et al. Regulation of autophagy and its associated cell death by sphingolipid rheostat: Reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J. Biol. Chem. 2012, 287, 39898–39910. [Google Scholar] [CrossRef] [Green Version]
- Gonzalvez, F.; D’Aurelio, M.; Boutant, M.; Moustapha, A.; Puech, J.P.; Landes, T.; Arnaune-Pelloquin, L.; Vial, G.; Taleux, N.; Slomianny, C.; et al. Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation. Biochim. Biophys. Acta 2013, 1832, 1194–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falabella, M.; Vernon, H.J.; Hanna, M.G.; Claypool, S.M.; Pitceathly, R.D.S. Cardiolipin, Mitochondria, and Neurological Disease. Trends Endocrinol. Metab. 2021, 32, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Kiebish, M.A.; Han, X.; Cheng, H.; Chuang, J.H.; Seyfried, T.N. Cardiolipin and electron transport chain abnormalities in mouse brain tumor mitochondria: Lipidomic evidence supporting the Warburg theory of cancer. J. Lipid Res. 2008, 49, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Poulaki, A.; Katsila, T.; Stergiou, I.E.; Giannouli, S.; Gomicronmez-Tamayo, J.C.; Piperaki, E.T.; Kambas, K.; Dimitrakopoulou, A.; Patrinos, G.P.; Tzioufas, A.G.; et al. Bioenergetic Profiling of the Differentiating Human MDS Myeloid Lineage with Low and High Bone Marrow Blast Counts. Cancers 2020, 12, 3520. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poulaki, A.; Giannouli, S. Mitochondrial Lipids: From Membrane Organization to Apoptotic Facilitation. Int. J. Mol. Sci. 2022, 23, 3738. https://doi.org/10.3390/ijms23073738
Poulaki A, Giannouli S. Mitochondrial Lipids: From Membrane Organization to Apoptotic Facilitation. International Journal of Molecular Sciences. 2022; 23(7):3738. https://doi.org/10.3390/ijms23073738
Chicago/Turabian StylePoulaki, Aikaterini, and Stavroula Giannouli. 2022. "Mitochondrial Lipids: From Membrane Organization to Apoptotic Facilitation" International Journal of Molecular Sciences 23, no. 7: 3738. https://doi.org/10.3390/ijms23073738
APA StylePoulaki, A., & Giannouli, S. (2022). Mitochondrial Lipids: From Membrane Organization to Apoptotic Facilitation. International Journal of Molecular Sciences, 23(7), 3738. https://doi.org/10.3390/ijms23073738