
Identification and New Indication of Melanin-Concentrating Hormone Receptor 1 (MCHR1) Antagonist Derived from Machine Learning and Transcriptome-Based Drug Repositioning Approaches

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
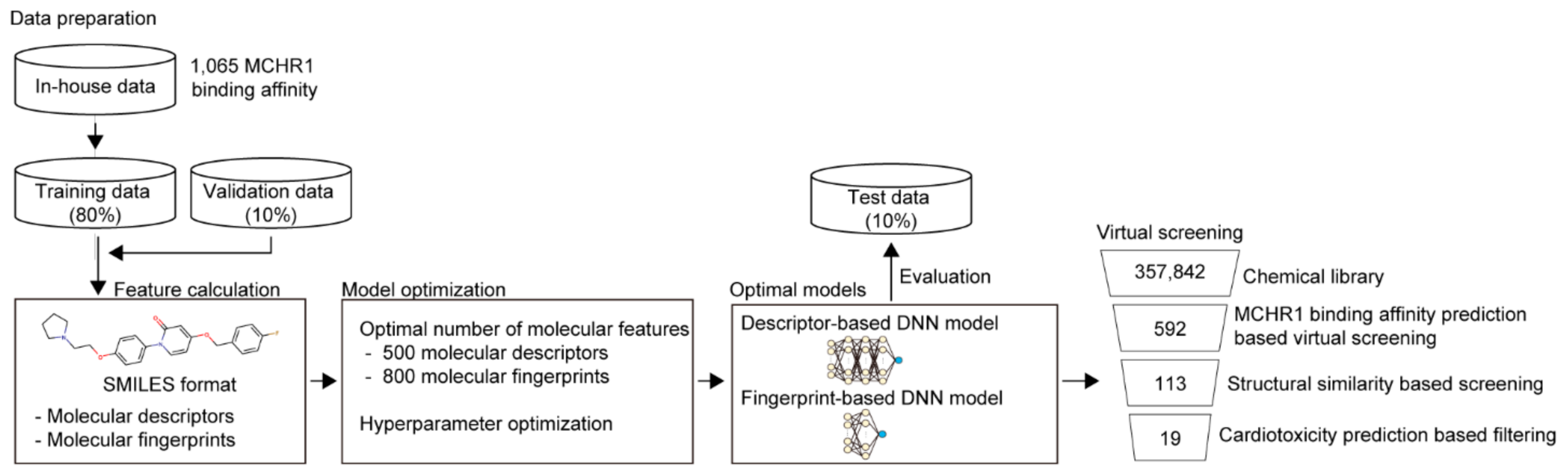
2.1. Development of Prediction Model for MCHR1 Binding Activity
2.2. Virtual Screening of MCHR1 Antagonists Using Ligand-Based Machine Learning Models
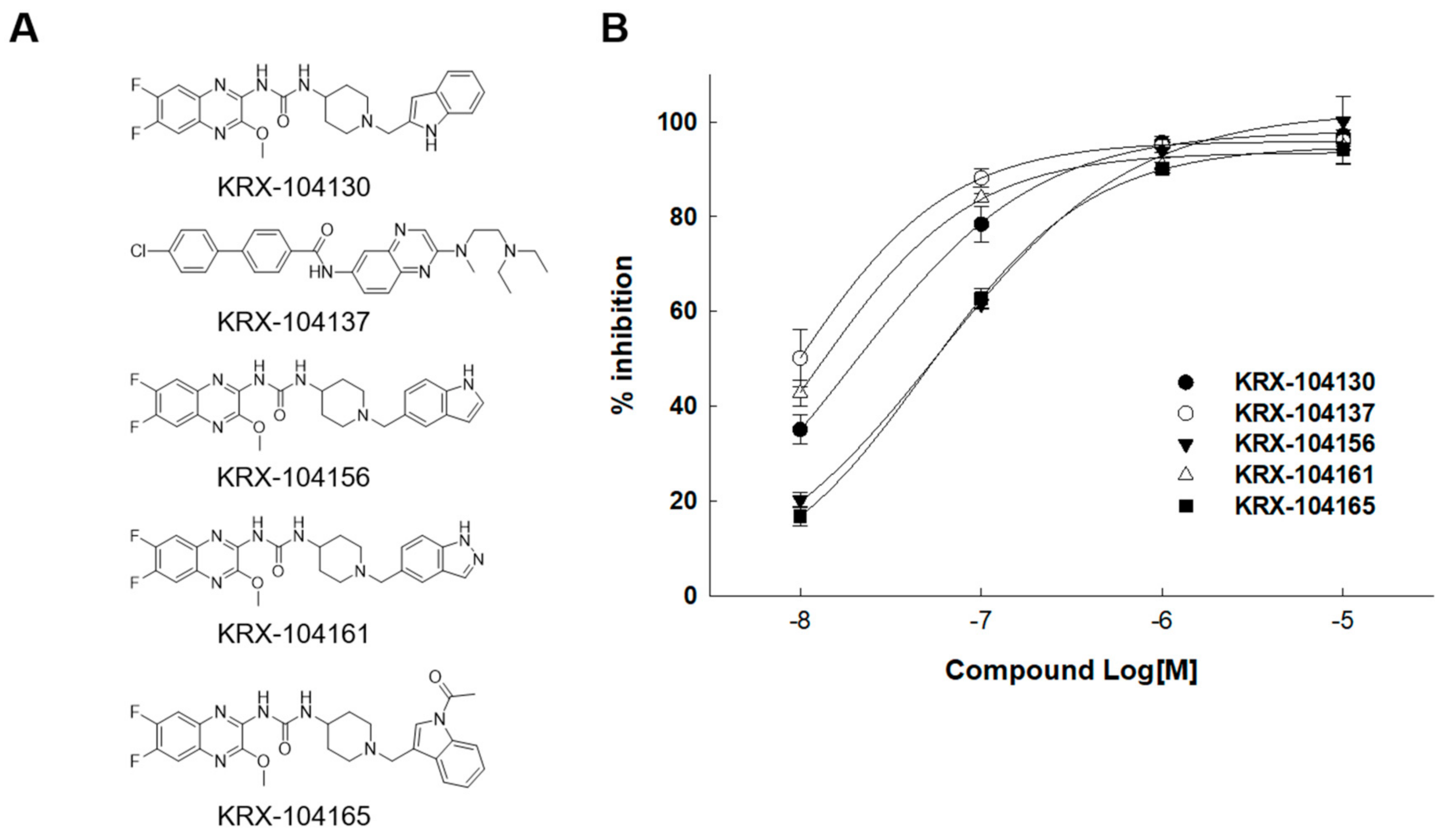
2.3. Identification of Small-Molecule Antagonist of MCHR1
2.4. Plasma Pharmacokinetic (PK) Profile of KRX-104130
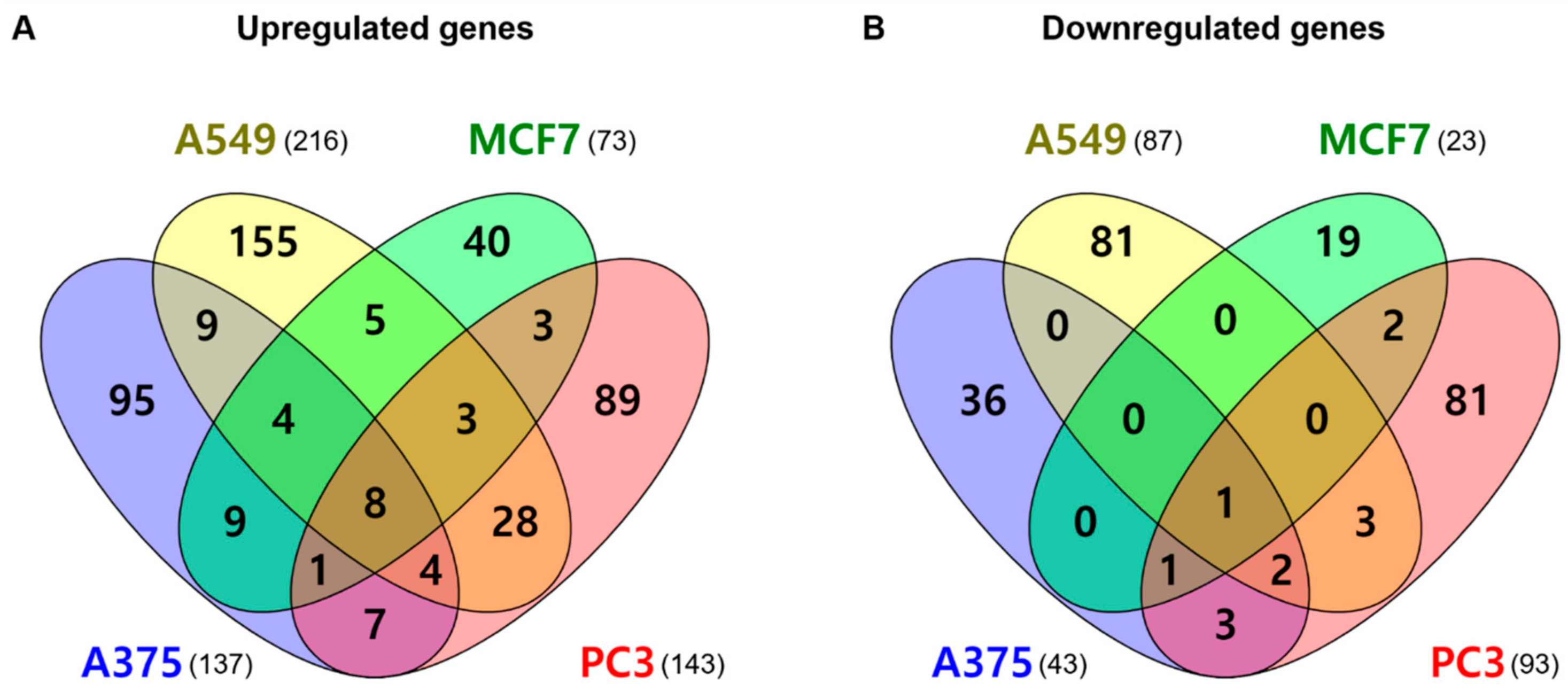
2.5. mRNA Gene Expression Analysis
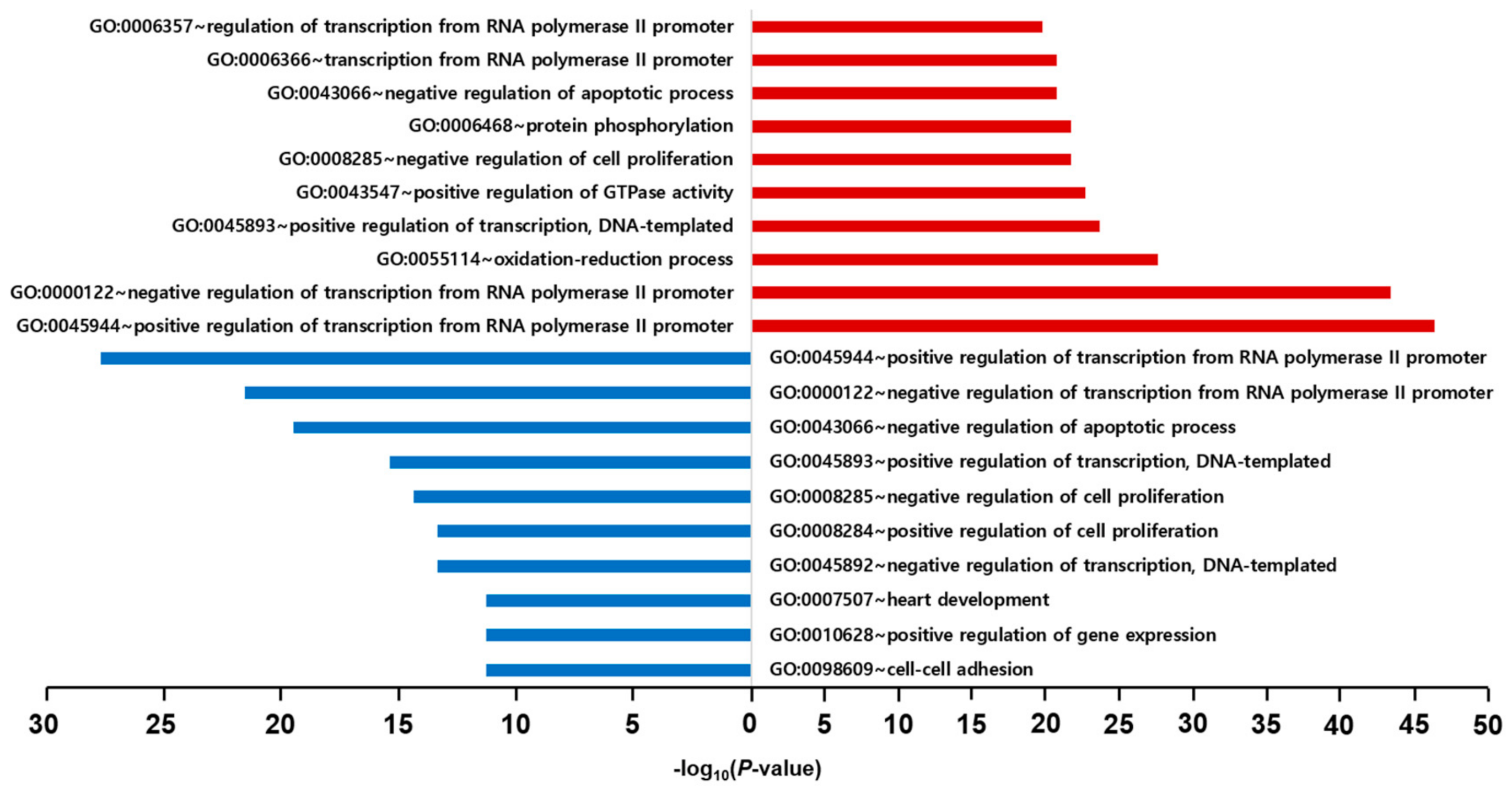
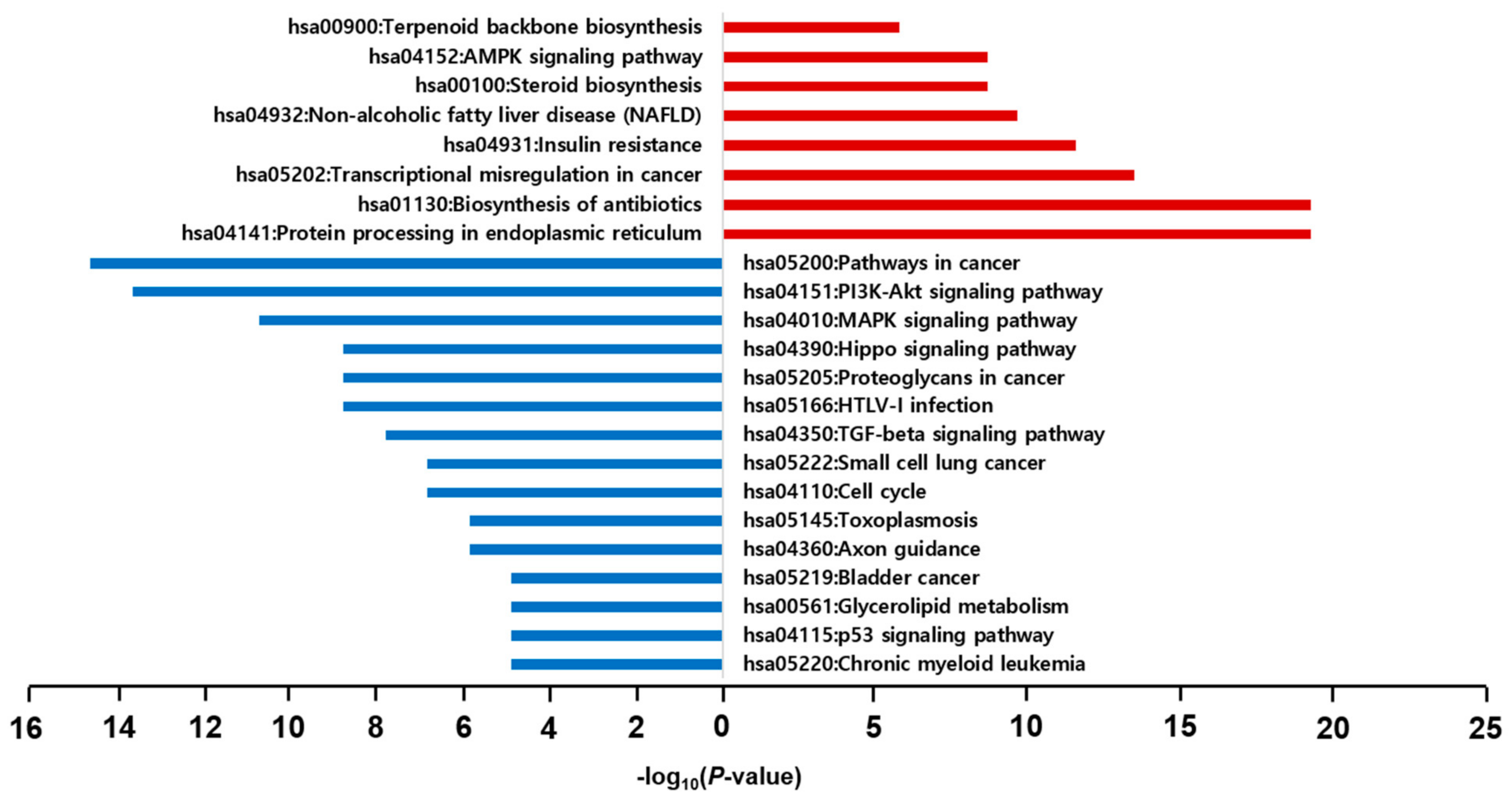
2.6. GO and KEGG Pathway Enrichment Analyses
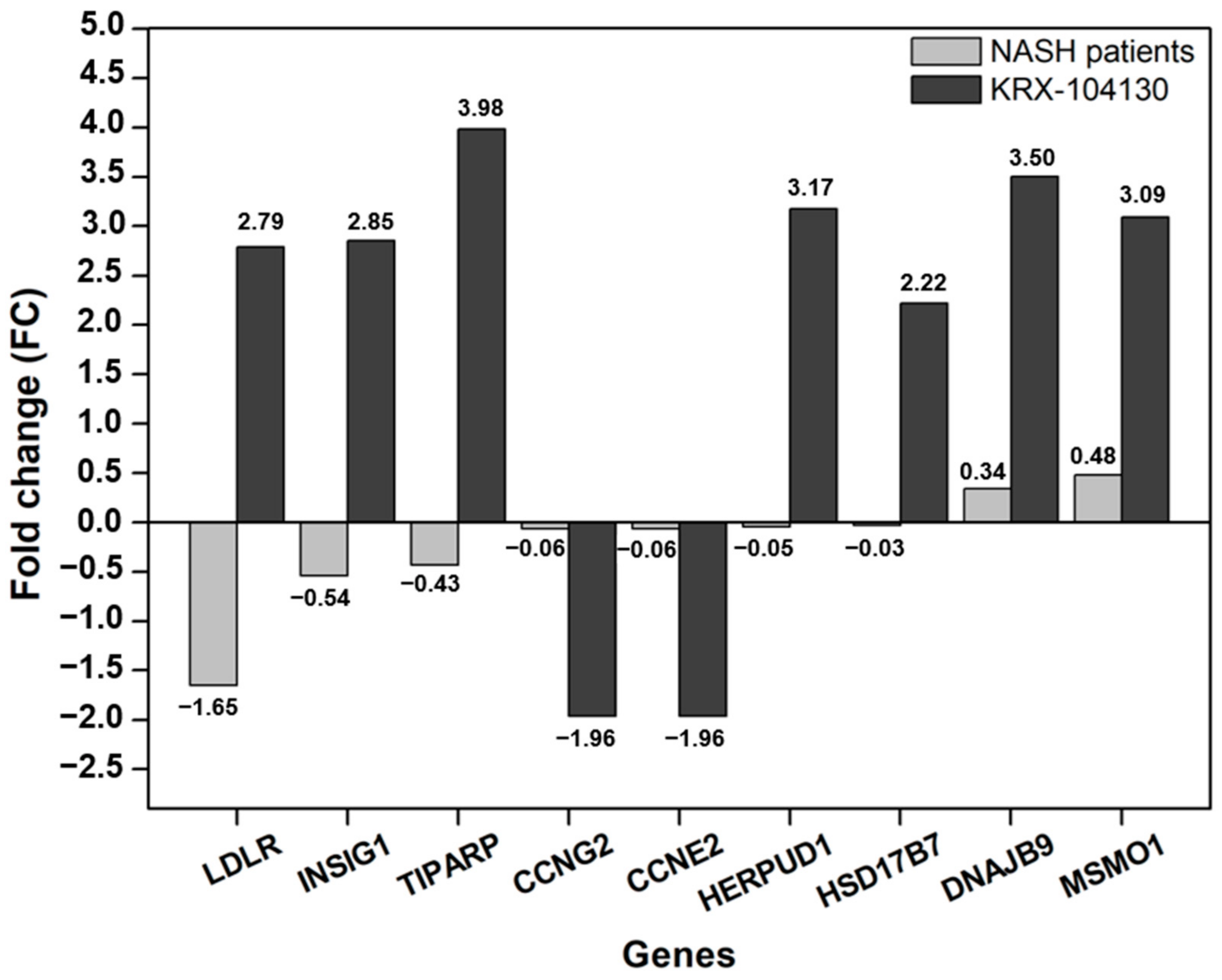
2.7. Comparison of KRX-104130-Induced Gene Expression in NASH Patients
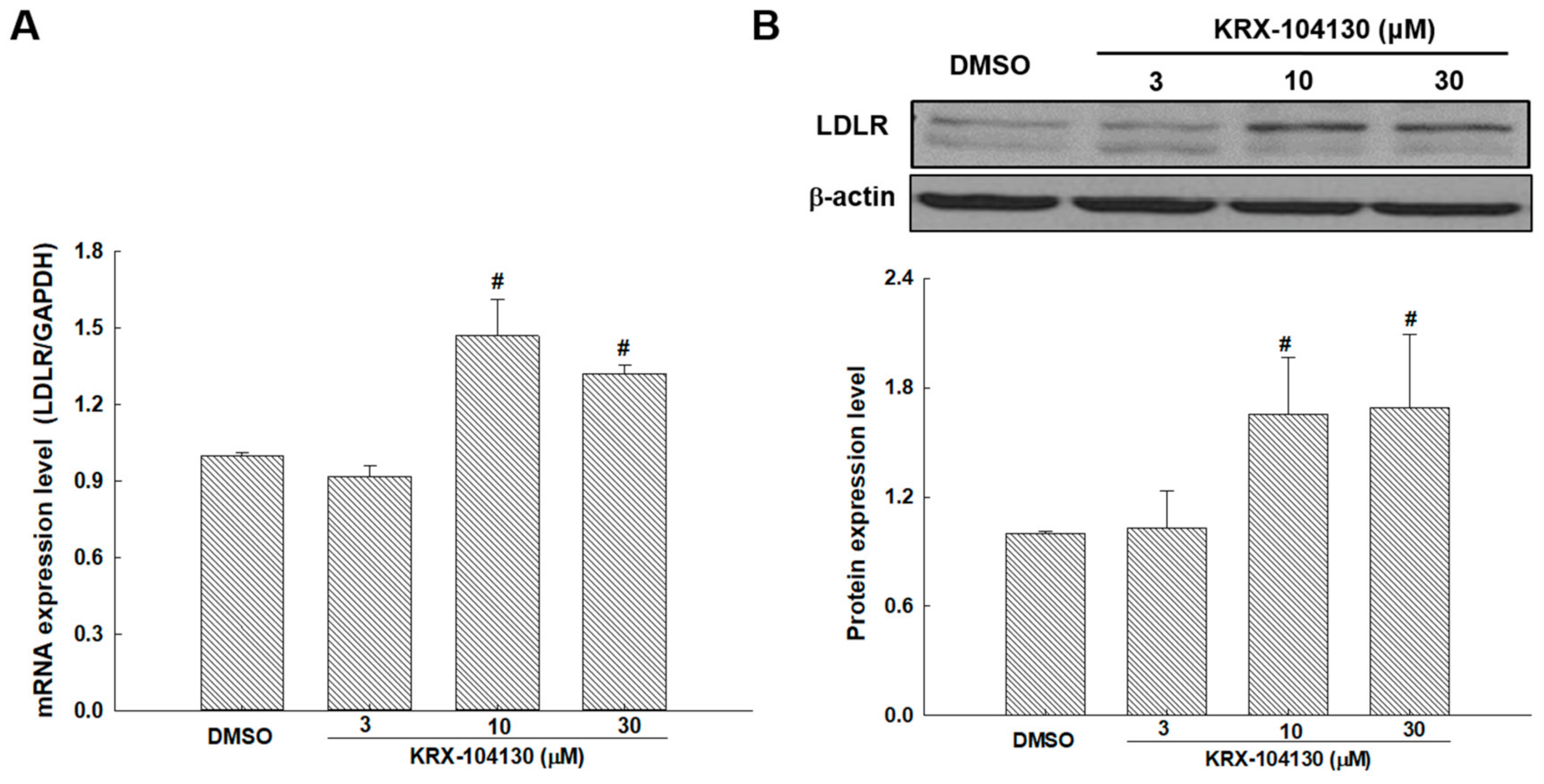
2.8. The Effects of KRX-104130 on the mRNA and Protein Expression Levels of LDLR
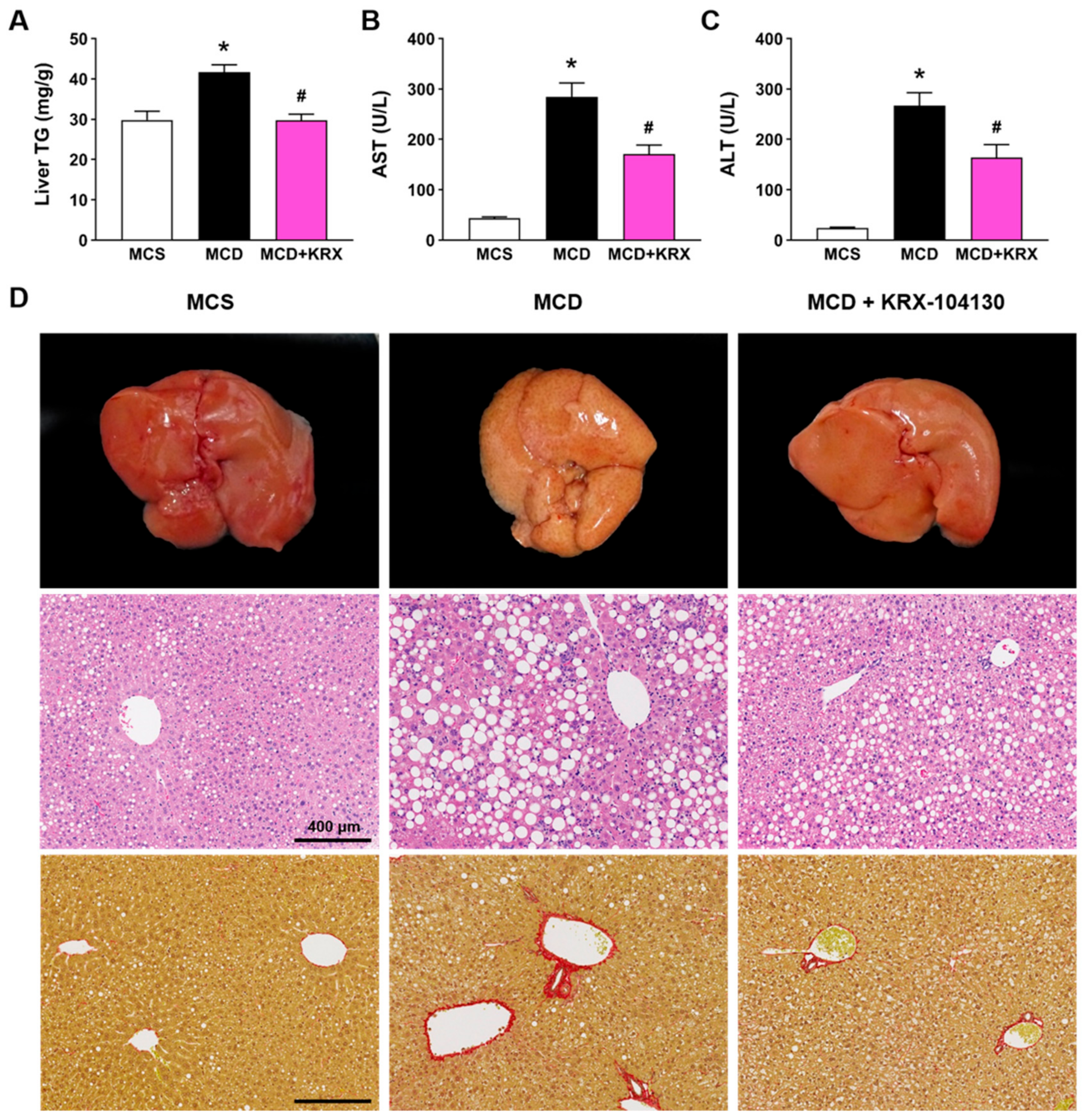
2.9. Effect of KRX-104130 in a NASH Animal Model
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Data Preparation
5.2. Preparation of Molecular Features
5.3. Model Development
5.4. MCHR1 Binding Assay
5.5. Patch Clamp Assay
5.6. Cell Culture and Sample Preparation for Gene Expression Analysis
5.7. mRNA Expression Data Analysis
5.8. Real-Time Quantitative Transcription PCR (RT-qPCR) Analysis for LDLR mRNA Expression Level
5.9. Western Blot Assay for LDLR Protein Expression Level
5.10. Animal Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Skofitsch, G.; Jacobowitz, D.M.; Zamir, N. Immunohistochemical localization of a melanin concentrating hormone-like peptide in the rat brain. Brain Res. Bull. 1985, 15, 635–649. [Google Scholar] [CrossRef]
- Bittencourt, J.C.; Presse, F.; Arias, C.; Peto, C.; Vaughan, J.; Nahon, J.L.; Vale, W.; Sawchenko, P.E. The melanin-concentrating hormone system of the rat brain: An immuno- and hybridization histochemical characterization. J. Comp. Neurol. 1992, 319, 218–245. [Google Scholar] [CrossRef]
- Qu, D.; Ludwig, D.S.; Gammeltoft, S.; Piper, M.; Pelleymounter, M.A.; Cullen, M.J.; Mathes, W.F.; Przypek, R.; Kanarek, R. Maratos-Flier, E. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature 1996, 380, 243–247. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Mountjoy, K.G.; Tatro, J.B.; Gillette, J.A.; Frederich, R.C.; Flier, J.S.; Maratos-Flier, E. Melanin-concentrating hormone: A functional melanocortin antagonist in the hypothalamus. Am. J. Physiol. 1998, 274, E627–E633. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Tritos, N.A.; Mastaitis, J.W.; Kulkarni, R.; Kokkotou, E.; Elmquist, J.; Lowell, B.; Flier, J.S.; Maratos-Flier, E. Melanin-concentrating hormone overexpression in transgenic mice leads to obesity and insulin resistance. J. Clin. Investig. 2001, 107, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.J.; Kim, N.; Lee, E.K.; Lee, B.H.; Oh, K.-S.; Yoo, S.-E.; Yi, K.Y. Synthesis and SAR investigations of novel 2-arylbenzimidazole derivatives as melanin-concentrating hormone receptor 1 (MCH-R1) antagonists. Bioorganic Med. Chem. Lett. 2011, 21, 2309–2312. [Google Scholar] [CrossRef]
- Lim, C.J.; Kim, S.H.; Lee, B.H.; Oh, K.-S.; Yi, K.Y. 4-Arylphthalazin-1(2H)-one derivatives as potent antagonists of the melanin concentrating hormone receptor 1 (MCH-R1). Bioorganic Med. Chem. Lett. 2012, 22, 427–430. [Google Scholar] [CrossRef]
- Lim, C.J.; Kim, J.Y.; Lee, B.H.; Oh, K.-S.; Yi, K.Y. Synthesis and SAR study of pyrrolo[3,4-b]pyridin-7(6H)-one derivatives as melanin concentrating hormone receptor 1 (MCH-R1) antagonists. Bioorganic Med. Chem. Lett. 2013, 23, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Hogberg, T.; Frimurer, T.M.; Sasmal, P.K. Melanin concentrating hormone receptor 1 (MCHR1) antagonists-Still a viable approach for obesity treatment? Bioorg. Med. Chem. Lett. 2012, 22, 6039–6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; Keane, A.; Dumesic, J.A.; Huber, G.W.; Zavala, V.M. A machine learning framework for the analysis and prediction of catalytic activity from experimental data. Appl. Catal. B Environ. 2020, 263, 118257. [Google Scholar] [CrossRef]
- Burbidge, R.; Trotter, M.; Buxton, B.; Holden, S. Drug design by machine learning: Support vector machines for pharmaceutical data analysis. Comput. Chem. 2001, 26, 5–14. [Google Scholar] [CrossRef]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Lee, M.Y.; Lee, J.H.; Lee, B.H.; Oh, K.S. DeepHIT: A deep learning framework for prediction of hERG-induced cardiotoxicity. Bioinformatics 2020, 36, 3049–3055. [Google Scholar] [CrossRef]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. [Google Scholar] [CrossRef]
- Lim, G.; Lim, C.J.; Lee, J.H.; Lee, B.H.; Ryu, J.Y.; Oh, K.-S. Identification of new target proteins of a Urotensin-II receptor antagonist using transcriptome-based drug repositioning approach. Sci. Rep. 2021, 11, 17138. [Google Scholar] [CrossRef]
- Rajendran, K.; Kumar, V.; Raja, I.; Kumariah, M.; Tennyson, J. Identification of small non-coding RNAs from Rhizobium etli by integrated genome-wide and transcriptome-based methods. ExRNA 2020, 2, 14. [Google Scholar] [CrossRef]
- Xie, L.; He, S.; Song, X.; Bo, X.; Zhang, Z. Deep learning-based transcriptome data classification for drug-target interaction prediction. BMC Genom. 2018, 19 (Suppl. 7), 667. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Han, H.W.; Hahn, S.; Jeong, H.Y.; Jee, J.H.; Nam, M.O.; Kim, H.K.; Lee, D.H.; Lee, S.Y.; Choi, D.K.; Yu, J.H.; et al. LINCS L1000 dataset-based repositioning of CGP-60474 as a highly potent anti-endotoxemic agent. Sci. Rep. 2018, 8, 14969. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; He, S.; Zhou, Z.; Bo, X.; Qi, D.; Fu, X.; Wang, Z.; Yang, J.; Wang, S. LINCS dataset-based repositioning of rosiglitazone as a potential anti-human adenovirus drug. Antivir. Res. 2020, 179, 104789. [Google Scholar] [CrossRef]
- Dey-Rao, R.; Smith, J.R.; Chow, S.; Sinha, A.A. Differential gene expression analysis in CCLE lesions provides new insights regarding the genetics basis of skin vs. systemic disease. Genomics 2014, 104, 144–155. [Google Scholar] [CrossRef]
- Chen, Y.; McGee, J.; Chen, X.; Doman, T.N.; Gong, X.; Zhang, Y.; Hamm, N.; Ma, X.; Higgs, R.E.; Bhagwat, S.V.; et al. Identification of druggable cancer driver genes amplified across TCGA datasets. PLoS ONE 2014, 9, e98293. [Google Scholar] [CrossRef]
- Jeon, H.; Blacklow, S.C. Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem. 2005, 74, 535–562. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Kawarabayasi, Y.; Nakai, T.; Sakai, J.; Yamamoto, T. Rabbit very low density lipoprotein receptor: A low density lipoprotein receptor-like protein with distinct ligand specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 9252. [Google Scholar] [CrossRef] [Green Version]
- Naganuma, F.; Bandaru, S.S.; Absi, G.; Mahoney, C.E.; Scammell, T.E.; Vetrivelan, R. Melanin-concentrating hormone neurons contribute to dysregulation of rapid eye movement sleep in narcolepsy. Neurobiol. Dis. 2018, 120, 12–20. [Google Scholar] [CrossRef]
- He, Y.; Rodrigues, R.M.; Wang, X.; Seo, W.; Ma, J.; Hwang, S.; Fu, Y.; Trojnar, E.; Matyas, C.; Zhao, S.; et al. Neutrophil-to-hepatocyte communication via LDLR-dependent miR-223-enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. J. Clin. Investig. 2021, 131, e141513. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Lee, J.H.; Lee, B.H.; Song, J.S.; Ahn, S.; Oh, K.S. PredMS: A random Forest model for predicting metabolic stability of drug candidates in human liver microsomes. Bioinformatics 2021, 38, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Kerr, T.A.; Davidson, N.O. Cholesterol and nonalcoholic fatty liver disease: Renewed focus on an old villain. Hepatology 2012, 56, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-K.; Kapoor, A.; Fuchs, M.; Mirshahi, F.; Zhou, H.; Maher, J.; Kellum, J.; Warnick, R.; Contos, M.J.; Sanyal, A.J. Increased Hepatic Synthesis and Dysregulation of Cholesterol Metabolism Is Associated with the Severity of Nonalcoholic Fatty Liver Disease. Cell Metab. 2012, 15, 665–674. [Google Scholar] [CrossRef] [Green Version]
- De Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C.; et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2021, 41, 754–763. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol 2013, 58, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Yao, Z.J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.P.; Chen, A.F.; Cao, D.S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Pereira, F.; Burges, C.J.; Bottou, L.; Weinberger, K.Q. Practical Bayesian optimization of machine learning algorithms. Neural Inf. Processing Syst. 2012, 25, 2960–2968. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-scale machine learning on heterogeneous distributed systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | KRX-104130 | |
|---|---|---|
| Route | I.V. | P.O. |
| Dose (mg/kg) | 10 | 20 |
| n | 3 | 3 |
| AUC0–t (μg∙h/mL) | 4.53 ± 0.22 | 1.72 ± 0.85 |
| T1/2 (h) | 5.71 ± 1.03 | 10.22 |
| Tmax (h) | 6.67 ± 2.31 | |
| Cmax (μg/mL) | 0.13 ± 0.05 | |
| CL (L/kg/h) | 2.21 ± 0.11 | |
| Vss (L/kg) | 14.16 ± 0.72 | |
| F (%) | 20.3 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, G.; You, K.Y.; Lee, J.H.; Jeon, M.K.; Lee, B.H.; Ryu, J.Y.; Oh, K.-S. Identification and New Indication of Melanin-Concentrating Hormone Receptor 1 (MCHR1) Antagonist Derived from Machine Learning and Transcriptome-Based Drug Repositioning Approaches. Int. J. Mol. Sci. 2022, 23, 3807. https://doi.org/10.3390/ijms23073807
Lim G, You KY, Lee JH, Jeon MK, Lee BH, Ryu JY, Oh K-S. Identification and New Indication of Melanin-Concentrating Hormone Receptor 1 (MCHR1) Antagonist Derived from Machine Learning and Transcriptome-Based Drug Repositioning Approaches. International Journal of Molecular Sciences. 2022; 23(7):3807. https://doi.org/10.3390/ijms23073807
Chicago/Turabian StyleLim, Gyutae, Ka Young You, Jeong Hyun Lee, Moon Kook Jeon, Byung Ho Lee, Jae Yong Ryu, and Kwang-Seok Oh. 2022. "Identification and New Indication of Melanin-Concentrating Hormone Receptor 1 (MCHR1) Antagonist Derived from Machine Learning and Transcriptome-Based Drug Repositioning Approaches" International Journal of Molecular Sciences 23, no. 7: 3807. https://doi.org/10.3390/ijms23073807
APA StyleLim, G., You, K. Y., Lee, J. H., Jeon, M. K., Lee, B. H., Ryu, J. Y., & Oh, K.-S. (2022). Identification and New Indication of Melanin-Concentrating Hormone Receptor 1 (MCHR1) Antagonist Derived from Machine Learning and Transcriptome-Based Drug Repositioning Approaches. International Journal of Molecular Sciences, 23(7), 3807. https://doi.org/10.3390/ijms23073807