
The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models


Abstract
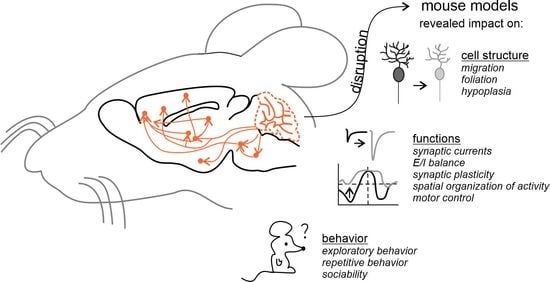
:
1. Introduction
2. Neural Bases for Impaired Social Cognition in ASD
3. Cerebellar Involvement in ASD
3.1. Cerebellar Circuit Microanatomy
3.2. Cerebellar Connectivity to Social Brain Areas
3.3. Cerebellar Structural Abnormalities in ASD
3.4. Cerebellar Functional Abnormalities in ASD
3.5. Cerebellar Neurochemical Abnormalities in ASD
3.6. Cerebellar Inflammation in ASD
4. Cerebellar Monogenic Mouse Models of ASD
4.1. Models Involving Cerebellar Development
4.2. Models of Syndromic ASD
4.3. Models of Non-Syndromic ASD
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- DSM-5. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association, Ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Adolphs, R. The neurobiology of social cognition. Curr. Opin. Neurobiol. 2001, 11, 231–239. [Google Scholar] [CrossRef]
- Baron, R.; Bryne, D. Social Psycology: Understanding Human Interaction, 9th ed.; Bacon, B.A.A., Ed.; Allyn and Bacon: Boston, MA, USA, 1991. [Google Scholar]
- Sodian, B.; Thoermer, C. Precursors to a Theory of Mind in infancy: Perspectives for Research on Autism. Q. J. Exp. Psychol. 2008, 61, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Happé, F.; Cook, J.L.; Bird, G. The Structure of Social Cognition: In(ter)dependence of Sociocognitive Processes. Annu. Rev. Psychol. 2017, 68, 243–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron-Cohen, S.; Bowen, D.C.; Holt, R.J.; Allison, C.; Auyeung, B.; Lombardo, M.; Smith, P.; Lai, M.-C. The “Reading the Mind in the Eyes” Test: Complete Absence of Typical Sex Difference in ~400 Men and Women with Autism. PLoS ONE 2015, 10, e0136521. [Google Scholar] [CrossRef]
- Simonoff, E.; Pickles, A.; Charman, T.; Chandler, S.; Loucas, T.; Baird, G. Psychiatric Disorders in Children with Autism Spectrum Disorders: Prevalence, Comorbidity, and Associated Factors in a Population-Derived Sample. J. Am. Acad. Child Adolesc. Psychiatry 2008, 47, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Geschwind, D.H. Advances in Autism. Annu. Rev. Med. 2009, 60, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.F.; Nadeem, A.; Ansari, M.A.; Bakheet, S.A.; Al-Ayadhi, L.Y.; Attia, S.M. Upregulation of IL-9 and JAK-STAT signaling pathway in children with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 472–480. [Google Scholar] [CrossRef]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Meltzer, A.; Van De Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Tsilioni, I.; Patel, A.B.; Doyle, R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl. Psychiatry 2016, 6, e844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Castelbaum, L.; Sylvester, C.M.; Zhang, Y.; Yu, Q.; Constantino, J.N. On the Nature of Monozygotic Twin Concordance and Discordance for Autistic Trait Severity: A Quantitative Analysis. Behav. Genet. 2020, 50, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef]
- Dias, C.M.; Walsh, C.A. Recent Advances in Understanding the Genetic Architecture of Autism. Annu. Rev. Genom. Hum. Genet. 2020, 21, 289–304. [Google Scholar] [CrossRef]
- Yoo, H. Genetics of Autism Spectrum Disorder: Current Status and Possible Clinical Applications. Exp. Neurobiol. 2015, 24, 257–272. [Google Scholar] [CrossRef] [Green Version]
- De Rubeis, S.; Buxbaum, J.D. Genetics and genomics of autism spectrum disorder: Embracing complexity. Hum. Mol. Genet. 2015, 24, R24–R31. [Google Scholar] [CrossRef]
- Berg, J.M.; Geschwind, D.H. Autism genetics: Searching for specificity and convergence. Genome Biol. 2012, 13, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gaugler, T.; Klei, L.; Sanders, S.; Bodea, C.A.; Goldberg, A.P.; Lee, A.B.; Mahajan, M.C.; Manaa, D.; Pawitan, Y.; Reichert, J.G.; et al. Most genetic risk for autism resides with common variation. Nat. Genet. 2014, 46, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Bölte, S.; Girdler, S.; Marschik, P.B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell. Mol. Life Sci. 2019, 76, 1275–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loke, Y.J.; Hannan, A.J.; Craig, J.M. The Role of Epigenetic Change in Autism Spectrum Disorders. Front. Neurol. 2015, 6, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devlin, B.; Scherer, S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012, 22, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Woodbury-Smith, M.; Scherer, S.W. Progress in the genetics of autism spectrum disorder. Dev. Med. Child Neurol. 2018, 60, 445–451. [Google Scholar] [CrossRef]
- Misra, V. The Social Brain Network and Autism. Ann. Neurosci. 2014, 21, 69–73. [Google Scholar] [CrossRef]
- Pelphrey, K.; Adolphs, R.; Morris, J.P. Neuroanatomical substrates of social cognition dysfunction in autism. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 259–271. [Google Scholar] [CrossRef]
- Dunbar, R. The social brain hypothesis. Evol. Anthropol. 1998, 6, 178–190. [Google Scholar] [CrossRef]
- D’Angelo, E. The cerebellum gets social. Science 2019, 363, 229. [Google Scholar] [CrossRef]
- Hoche, F.; Guell, X.; Sherman, J.C.; Vangel, M.G.; Schmahmann, J.D. Cerebellar Contribution to Social Cognition. Cerebellum 2016, 15, 732–743. [Google Scholar] [CrossRef]
- D’Angelo, E.; Casali, S. Seeking a unified framework for cerebellar function and dysfunction: From circuit operations to cognition. Front. Neural Circuits 2013, 6, 116. [Google Scholar] [CrossRef] [Green Version]
- Redcay, E.; Moraczewski, D. Social cognition in context: A naturalistic imaging approach. NeuroImage 2020, 216, 116392. [Google Scholar] [CrossRef] [PubMed]
- Doruyter, A.; Groenewold, N.A.; Dupont, P.; Stein, D.J.; Warwick, J. Resting-state fMRI and social cognition: An opportunity to connect. Hum. Psychopharmacol. Clin. Exp. 2017, 32, e2627. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Brown, W.H.; Egleston, P.N.; Green, R.D.; Farrow, T.F.; Hunter, M.D.; Parks, R.W.; Wilkinson, I.D.; Spence, S.A.; Woodruff, P.W. A Functional Magnetic Resonance Imaging Study of Social Cognition in Schizophrenia During an Acute Episode and After Recovery. Am. J. Psychiatry 2006, 163, 1926–1933. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.G.; Maresca, G.; Stagnitti, M.C.; Anchesi, S.; Casella, C.; Pajno, V.; De Luca, R.; Manuli, A.; Calabrò, R.S. Social cognition in patients with acquired brain lesions: An overview on an under-reported problem. Appl. Neuropsychol. Adult 2020. ahead of print. [Google Scholar] [CrossRef]
- Sokolov, A.A. The Cerebellum in Social Cognition. Front. Cell. Neurosci. 2018, 12, 145. [Google Scholar] [CrossRef] [Green Version]
- Amodio, D.M.; Frith, C.D. Meeting of minds: The medial frontal cortex and social cognition. Nat. Rev. Neurosci. 2006, 7, 268–277. [Google Scholar] [CrossRef]
- Beer, J.S.; Ochsner, K.N. Social cognition: A multi level analysis. Brain Res. 2006, 1079, 98–105. [Google Scholar] [CrossRef]
- Mar, R.A. The Neural Bases of Social Cognition and Story Comprehension. Annu. Rev. Psychol. 2011, 62, 103–134. [Google Scholar] [CrossRef] [Green Version]
- Kipps, C.M.; Duggins, A.J.; McCusker, E.A.; Calder, A.J. Disgust and Happiness Recognition Correlate with Anteroventral Insula and Amygdala Volume Respectively in Preclinical Huntington’s Disease. J. Cogn. Neurosci. 2007, 19, 1206–1217. [Google Scholar] [CrossRef]
- Kelly, R.M.; Strick, P.L. Cerebellar Loops with Motor Cortex and Prefrontal Cortex of a Nonhuman Primate. J. Neurosci. 2003, 23, 8432–8444. [Google Scholar] [CrossRef] [Green Version]
- Middleton, F.; Strick, P.L. Cerebellar Projections to the Prefrontal Cortex of the Primate. J. Neurosci. 2001, 21, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Passingham, R.E.; Stephan, K.E.; Kötter, R. The anatomical basis of functional localization in the cortex. Nat. Rev. Neurosci. 2002, 3, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D. An Emerging Concept. The cerebellar contribution to higher function. Arch. Neurol. 1991, 48, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Laurita, A.; Spreng, N. The Hippocampus and Social Cognition. In The Hippocampus from Cells to Systems; Springer: Berlin/Heidelberg, Germany, 2017; pp. 537–558. [Google Scholar]
- Settell, M.; Testini, P.; Cho, S.; Lee, J.H.; Blaha, C.D.; Jo, H.J.; Lee, K.H.; Min, H.-K. Functional Circuitry Effect of Ventral Tegmental Area Deep Brain Stimulation: Imaging and Neurochemical Evidence of Mesocortical and Mesolimbic Pathway Modulation. Front. Neurosci. 2017, 11, 104. [Google Scholar] [CrossRef]
- Gunaydin, L.A.; Grosenick, L.; Finkelstein, J.C.; Kauvar, I.V.; Fenno, L.E.; Adhikari, A.; Lammel, S.; Mirzabekov, J.J.; Airan, R.D.; Zalocusky, K.A.; et al. Natural Neural Projection Dynamics Underlying Social Behavior. Cell 2014, 157, 1535–1551. [Google Scholar] [CrossRef] [Green Version]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef]
- Courchesne, E.; Mouton, P.R.; Calhoun, M.E.; Semendeferi, K.; Ahrens-Barbeau, C.; Hallet, M.J.; Barnes, C.C.; Pierce, K. Neuron Number and Size in Prefrontal Cortex of Children with Autism. JAMA 2011, 306, 2001–2010. [Google Scholar] [CrossRef]
- Casanova, M.F. White matter volume increase and minicolumns in autism. Ann. Neurol. 2004, 56, 453. [Google Scholar] [CrossRef]
- Richter, J.; Henze, R.; Vomstein, K.; Stieltjes, B.; Parzer, P.; Haffner, J.; Brandeis, D.; Poustka, L. Reduced cortical thickness and its association with social reactivity in children with autism spectrum disorder. Psychiatry Res. Neuroimaging 2015, 234, 15–24. [Google Scholar] [CrossRef]
- Casanova, M.F.; Buxhoeveden, D.P.; Switala, A.E.; Roy, E. Minicolumnar pathology in autism. Neurology 2002, 58, 428–432. [Google Scholar] [CrossRef]
- Biswal, B.B. Resting state fMRI: A personal history. NeuroImage 2012, 62, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Buckner, R.L.; Krienen, F.M.; Yeo, B.T. Opportunities and limitations of intrinsic functional connectivity MRI. Nat. Neurosci. 2013, 16, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Just, M.A.; Cherkassky, V.L.; Keller, T.A.; Minshew, N.J. Cortical activation and synchronization during sentence comprehension in high-functioning autism: Evidence of underconnectivity. Brain 2004, 127, 1811–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just, M.A.; Keller, T.A.; Malave, V.L.; Kana, R.K.; Varma, S. Autism as a neural systems disorder: A theory of frontal-posterior underconnectivity. Neurosci. Biobehav. Rev. 2012, 36, 1292–1313. [Google Scholar] [CrossRef] [Green Version]
- Courchesne, E.; Pierce, K. Why the frontal cortex in autism might be talking only to itself: Local over-connectivity but long-distance disconnection. Curr. Opin. Neurobiol. 2005, 15, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Kana, R.K.; Keller, T.A.; Minshew, N.J.; Just, M.A. Inhibitory Control in High-Functioning Autism: Decreased Activation and Underconnectivity in Inhibition Networks. Biol. Psychiatry 2007, 62, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just, M.A.; Cherkassky, V.L.; Keller, T.A.; Kana, R.K.; Minshew, N.J. Functional and Anatomical Cortical Underconnectivity in Autism: Evidence from an fMRI Study of an Executive Function Task and Corpus Callosum Morphometry. Cereb. Cortex 2007, 17, 951–961. [Google Scholar] [CrossRef]
- Assaf, M.; Jagannathan, K.; Calhoun, V.D.; Miller, L.; Stevens, M.; Sahl, R.; O’Boyle, J.G.; Schultz, R.T.; Pearlson, G.D. Abnormal functional connectivity of default mode sub-networks in autism spectrum disorder patients. NeuroImage 2010, 53, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Wass, S. Distortions and disconnections: Disrupted brain connectivity in autism. Brain Cogn. 2011, 75, 18–28. [Google Scholar] [CrossRef]
- Rane, P.; Cochran, D.; Hodge, S.M.; Haselgrove, C.; Kennedy, D.N.; Frazier, J.A. Connectivity in Autism: A Review of MRI Con-nectivity Studies. Harv. Rev. Psychiatry 2015, 23, 223–244. [Google Scholar] [CrossRef] [Green Version]
- Zikopoulos, B.; Barbas, H. Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci. 2013, 7, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Z.; Duan, X.; Mantini, D.; Chen, H. Alteration of functional connectivity in autism spectrum disorder: Effect of age and anatomical distance. Sci. Rep. 2016, 6, 26527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, C.S.; Weng, S.-J.; Wiggins, J.L.; Kurapati, N.; Louro, H.M.; Carrasco, M.; Maslowsky, J.; Risi, S.; Lord, C. Neural circuitry of emotional face processing in autism spectrum disorders. J. Psychiatry Neurosci. 2010, 35, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostofsky, S.H.; Powell, S.K.; Simmonds, D.J.; Goldberg, M.C.; Caffo, B.; Pekar, J. Decreased connectivity and cerebellar activity in autism during motor task performance. Brain 2009, 132, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, M.V.; Eyler, L.; Moore, A.; Datko, M.; Barnes, C.C.; Cha, D.; Courchesne, E.; Pierce, K. Default mode-visual network hypoconnectivity in an autism subtype with pronounced social visual engagement difficulties. eLife 2019, 8, e47427. [Google Scholar] [CrossRef]
- Maximo, J.; Cadena, E.J.; Kana, R.K. The Implications of Brain Connectivity in the Neuropsychology of Autism. Neuropsychol. Rev. 2014, 24, 16–31. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, A.; Villalobos, M.E.; Davies, M.M.; Dahl, B.C.; Müller, R.-A. Partially enhanced thalamocortical functional connectivity in autism. Brain Res. 2006, 1104, 160–174. [Google Scholar] [CrossRef]
- Di Martino, A.; Kelly, C.; Grzadzinski, R.; Zuo, X.-N.; Mennes, M.; Mairena, M.A.; Lord, C.; Castellanos, F.; Milham, M.P. Aberrant Striatal Functional Connectivity in Children with Autism. Biol. Psychiatry 2011, 69, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Shih, P.; Shen, M.D.; Öttl, B.; Keehn, B.; Gaffrey, M.S.; Müller, R.-A. Atypical network connectivity for imitation in autism spectrum disorder. Neuropsychologia 2010, 48, 2931–2939. [Google Scholar] [CrossRef] [Green Version]
- Maximo, J.O.; Keown, C.L.; Nair, A.; Müller, R.-A. Approaches to local connectivity in autism using resting state functional connectivity MRI. Front. Hum. Neurosci. 2013, 7, 605. [Google Scholar] [CrossRef] [Green Version]
- Keown, C.L.; Shih, P.; Nair, A.; Peterson, N.; Mulvey, M.E.; Müller, R.-A. Local Functional Overconnectivity in Posterior Brain Regions Is Associated with Symptom Severity in Autism Spectrum Disorders. Cell Rep. 2013, 5, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barttfeld, P.; Wicker, B.; Cukier, S.; Navarta, S.; Lew, S.; Sigman, M. A big-world network in ASD: Dynamical connectivity analysis reflects a deficit in long-range connections and an excess of short-range connections. Neuropsychologia 2011, 49, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E.R.; Foss-Feig, J.; Kenworthy, L.; Gaillard, W.D.; Vaidya, C.J. Atypical Functional Connectivity of the Amygdala in Childhood Autism Spectrum Disorders during Spontaneous Attention to Eye-Gaze. Autism Res. Treat. 2012, 2012, 652408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, P.; Keehn, B.; Oram, J.K.; Leyden, K.M.; Keown, C.L.; Müller, R.-A. Functional Differentiation of Posterior Superior Temporal Sulcus in Autism: A Functional Connectivity Magnetic Resonance Imaging Study. Biol. Psychiatry 2011, 70, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Gramfort, A.; Shetty, N.R.; Kitzbichler, M.G.; Ganesan, S.; Moran, J.M.; Lee, S.M.; Gabrieli, J.D.E.; Tager-Flusberg, H.B.; Joseph, R.M.; et al. Local and long-range functional connectivity is reduced in concert in autism spectrum disorders. Proc. Natl. Acad. Sci. USA 2013, 110, 3107–3112. [Google Scholar] [CrossRef] [Green Version]
- Coskun, M.A.; Loveland, K.A.; Pearson, D.A.; Papanicolaou, A.C.; Sheth, B.R. Functional Assays of Local Connectivity in the Somatosensory Cortex of Individuals with Autism. Autism Res. 2013, 6, 190–200. [Google Scholar] [CrossRef] [Green Version]
- Murias, M.; Webb, S.J.; Greenson, J.; Dawson, G. Resting State Cortical Connectivity Reflected in EEG Coherence in Individuals with Autism. Biol. Psychiatry 2007, 62, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, J.L.R.; Merzenich, M.M. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003, 2, 255–267. [Google Scholar] [CrossRef]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [Green Version]
- Trakoshis, S.; Martínez-Cañada, P.; Rocchi, F.; Canella, C.; You, W.; Chakrabarti, B.; Ruigrok, A.N.; Bullmore, E.T.; Suckling, J.; Markicevic, M.; et al. Intrinsic excitation-inhibition imbalance affects medial prefrontal cortex differently in autistic men versus women. eLife 2020, 9, e55684. [Google Scholar] [CrossRef]
- Ecellot, G.; Echerubini, E. GABAergic Signaling as Therapeutic Target for Autism Spectrum Disorders. Front. Pediatr. 2014, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Hussman, J. Suppressed GABAergic Inhibition as a Common Factor in Suspected Etiologies of Autism. J. Autism Dev. Disord. 2001, 31, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.S.; Angelucci, A.; Bressloff, P.C. Anatomical Substrates for Functional Columns in Macaque Monkey Primary Visual Cortex. Cereb. Cortex 2003, 13, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, V.; Zhang, F.; Yizhar, O.; Deisseroth, K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009, 459, 698–702. [Google Scholar] [CrossRef] [Green Version]
- Keil, A.; Muller, M.M.; Ray, W.J.; Gruber, T.; Elbert, T. Human Gamma Band Activity and Perception of a Gestalt. J. Neurosci. 1999, 19, 7152–7161. [Google Scholar] [CrossRef] [Green Version]
- Tiitinen, H.T.; Sinkkonen, J.; Reinikainen, K.; Alho, K.; Lavikainen, J.; Naatanen, R. Selective attention enhances the auditory 40-Hz transient response in humans. Nature 1993, 364, 59–60. [Google Scholar] [CrossRef]
- Gobbelé, R.; Waberski, T.D.; Schmitz, S.; Sturm, W.; Buchner, H. Spatial direction of attention enhances right hemispheric event-related gamma-band synchronization in humans. Neurosci. Lett. 2002, 327, 57–60. [Google Scholar] [CrossRef]
- Fries, P. Neuronal Gamma-Band Synchronization as a Fundamental Process in Cortical Computation. Annu. Rev. Neurosci. 2009, 32, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Roux, F.; Uhlhaas, P.J. Working memory and neural oscillations: Alpha–gamma versus theta–gamma codes for distinct WM information? Trends Cogn. Sci. 2014, 18, 16–25. [Google Scholar] [CrossRef]
- Kucewicz, M.; Berry, B.M.; Kremen, V.; Brinkmann, B.; Sperling, M.R.; Jobst, B.C.; Gross, R.E.; Lega, B.; Sheth, S.A.; Stein, J.M.; et al. Dissecting gamma frequency activity during human memory processing. Brain 2017, 140, 1337–1350. [Google Scholar] [CrossRef] [Green Version]
- Rojas, D.C.; Wilson, L.B. γ-band abnormalities as markers of autism spectrum disorders. Biomark. Med. 2014, 8, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas, D.C.; Teale, P.D.; Maharajh, K.; Kronberg, E.; Youngpeter, K.; Wilson, L.B.; Wallace, A.; Hepburn, S. Transient and steady-state auditory gamma-band responses in first-degree relatives of people with autism spectrum disorder. Mol. Autism 2011, 2, 11. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, C.R.; Villalobos, M.E.; Schultz, R.T.; Herpertz-Dahlmann, B.; Konrad, K.; Kohls, G. Atypical Laterality of Resting Gamma Oscillations in Autism Spectrum Disorders. J. Autism Dev. Disord. 2013, 45, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E. Abnormal early brain development in autism. Mol. Psychiatry 2002, 7 (Suppl. 2), S21–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, G.; Gudsnuk, K.; Kuo, S.-H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-Dependent Macroautophagy Causes Autistic-like Synaptic Pruning Deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, S.J.; Wehner, E.A.; Hagerman, R. The Behavioral Phenotype in Fragile X: Symptoms of Autism in Very Young Children with Fragile X Syndrome, Idiopathic Autism, and Other Developmental Disorders. J. Dev. Behav. Pediatr. 2001, 22, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Folsom, T.D.; Kneeland, R.E.; Liesch, S.B. Metabotropic Glutamate Receptor 5 Upregulation in Children with Autism is Associated with Underexpression of Both Fragile X Mental Retardation Protein and GABAA Receptor Beta 3 in Adults with Autism. Anat. Rec. 2011, 294, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhou, L.; Yuan, H.; Vieira, M.; Sanz-Clemente, A.; Badger, J.D.; Lu, W.; Traynelis, S.F.; Roche, K.W. A Rare Variant Identified Within the GluN2B C-Terminus in a Patient with Autism Affects NMDA Receptor Surface Expression and Spine Density. J. Neurosci. 2017, 37, 4093–4102. [Google Scholar] [CrossRef]
- Gandal, M.J.; Anderson, R.L.; Billingslea, E.N.; Carlson, G.C.; Roberts, T.P.L.; Siegel, S.J. Mice with reduced NMDA receptor expression: More consistent with autism than schizophrenia? Genes Brain Behav. 2012, 11, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Schmahmann, J.D.; Caplan, D. Cognition, emotion and the cerebellum. Brain 2006, 129, 290–292. [Google Scholar] [CrossRef]
- Wolf, U.; Rapoport, M.J.; Schweizer, T.A. Evaluating the Affective Component of the Cerebellar Cognitive Affective Syndrome. J. Neuropsychiatry Clin. Neurosci. 2009, 21, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, D.C.; Donahue, C.J.; Glasser, M.F. Development and Evolution of Cerebral and Cerebellar Cortex. Brain Behav. Evol. 2018, 91, 158–169. [Google Scholar] [CrossRef]
- Pijpers, W.; Apps, R.; Pardoe, J.; Voogd, J.; Ruigrok, T. Precise Spatial Relationships between Mossy Fibers and Climbing Fibers in Rat Cerebellar Cortical Zones. J. Neurosci. 2006, 26, 12067–12080. [Google Scholar] [CrossRef] [PubMed]
- Oberdick, J.; Sillitoe, R.V. Cerebellar Zones: History, Development, and Function. Cerebellum 2011, 10, 301–306. [Google Scholar] [CrossRef]
- Prestori, F.; Mapelli, L.; D’Angelo, E. Diverse Neuron Properties and Complex Network Dynamics in the Cerebellar Cortical Inhibitory Circuit. Front. Mol. Neurosci. 2019, 12, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eito, M. Error detection and representation in the olivo-cerebellar system. Front. Neural Circuits 2013, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, F.; Jorntell, H. Specific Relationship between Excitatory Inputs and Climbing Fiber Receptive Fields in Deep Cerebellar Nuclear Neurons. PLoS ONE 2014, 9, e84616. [Google Scholar] [CrossRef]
- Steuber, V.; Jaeger, D. Modeling the generation of output by the cerebellar nuclei. Neural Netw. 2013, 47, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Eccles, J.; Llinás, R.; Sasaki, K. The inhibitory interneurones within the cerebellar cortex. Exp. Brain Res. 1966, 1, 1–16. [Google Scholar] [CrossRef]
- Eccles, J.; Llinás, R.; Sasak, K. Inhibitory systems in the cerebellar cortex. Proc. Aust. Assoc. Neurol. 1965, 3, 7–14. [Google Scholar]
- Cesana, E.; Pietrajtis, K.; Bidoret, C.; Isope, P.; D’Angelo, E.U.; Dieudonné, S.; Forti, L. Granule Cell Ascending Axon Excitatory Synapses onto Golgi Cells Implement a Potent Feedback Circuit in the Cerebellar Granular Layer. J. Neurosci. 2013, 33, 12430–12446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locatelli, F.; Soda, T.; Montagna, I.; Tritto, S.; Botta, L.; Prestori, F.; D’Angelo, E. Calcium Channel-Dependent Induction of Long-Term Synaptic Plasticity at Excitatory Golgi Cell Synapses of Cerebellum. J. Neurosci. 2021, 41, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Dieudonné, S. Submillisecond kinetics and low efficacy of parallel fibre-Golgi cell synaptic currents in the rat cerebellum. J. Physiol. 1998, 510 Pt 3, 845–866. [Google Scholar] [CrossRef] [PubMed]
- Vos, B.P.; Volny-Luraghi, A.; De Schutter, E. Cerebellar Golgi cells in the rat: Receptive fields and timing of responses to facial stimulation. Eur. J. Neurosci. 1999, 11, 2621–2634. [Google Scholar] [CrossRef] [PubMed]
- Eccles, J.; Llinás, R.; Sasaki, K. The mossy fibre-granule cell relay of the cerebellum and its inhibitory control by Golgi cells. Exp. Brain Res. 1966, 1, 82–101. [Google Scholar] [CrossRef]
- Evarts, E.V.; Thach, W.T. Motor Mechanisms of the CNS: Cerebrocerebellar Interrelations. Annu. Rev. Physiol. 1969, 31, 451–498. [Google Scholar] [CrossRef]
- Schmahmann, J.D.; Pandya, D.N. The Cerebrocerebellar System. Int. Rev. Neurobiol. 1997, 41, 31–60. [Google Scholar] [CrossRef]
- O’Reilly, J.X.; Beckmann, C.F.; Tomassini, V.; Ramnani, N.; Johansen-Berg, H. Distinct and Overlapping Functional Zones in the Cerebellum Defined by Resting State Functional Connectivity. Cereb. Cortex 2009, 20, 953–965. [Google Scholar] [CrossRef] [Green Version]
- Stoodley, C.J.; Schmahmann, J.D. Functional topography in the human cerebellum: A meta-analysis of neuroimaging studies. NeuroImage 2009, 44, 489–501. [Google Scholar] [CrossRef]
- Strata, P. The Emotional Cerebellum. Cerebellum 2015, 14, 570–577. [Google Scholar] [CrossRef]
- Leggio, M.; Olivito, G. Topography of the cerebellum in relation to social brain regions and emotions. Handb. Clin. Neurol. 2018, 154, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Jones, E. The Thalamus; Springer Science & Business Media: New York, NY, USA, 2007. [Google Scholar]
- Chan-Palay, V.; Palay, S.L.; Brown, J.T.; Van Itallie, C. Sagittal organization of olivocerebellar and reticulocerebellar projections: Autoradiographic studies with 35S-methionine. Exp. Brain Res. 1977, 30, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Aumann, T.D.; Rawson, J.A.; Finkelstein, D.; Horne, M.K. Projections from the lateral and interposed cerebellar nuclei to the thalamus of the rat: A light and electron microscopic study using single and double anterograde labelling. J. Comp. Neurol. 1994, 349, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, I.; Popa, D.; Zagrean, L. The Anatomical and Functional Heterogeneity of the Mediodorsal Thalamus. Brain Sci. 2020, 10, 624. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yoshida, K.; Yoshikawa, H.; Kishimoto, Y.; Oka, H. The medial dorsal nucleus is one of the thalamic relays of the cerebellocerebral responses to the frontal association cortex in the monkey: Horseradish peroxidase and fluorescent dye double staining study. Brain Res. 1992, 579, 315–320. [Google Scholar] [CrossRef]
- Schmahmann, J. From movement to thought: Anatomic substrates of the cerebellar contribution to cognitive processing. Hum. Brain Mapp. 1996, 4, 74–198. [Google Scholar] [CrossRef]
- Palesi, F.; Ferrante, M.; Gaviraghi, M.; Misiti, A.; Savini, G.; Lascialfari, A.; D’Angelo, E.; Wheeler-Kingshott, C.A.M.G. Motor and higher-order functions topography of the human dentate nuclei identified with tractography and clustering methods. Hum. Brain Mapp. 2021, 42, 4348–4361. [Google Scholar] [CrossRef]
- Palesi, F.; De Rinaldis, A.; Castellazzi, G.; Calamante, F.; Muhlert, N.; Chard, D.; Tournier, J.D.; Magenes, G.; D’Angelo, E.; Wheeler-Kingshott, C.A.G. Contralateral cortico-ponto-cerebellar pathways reconstruction in humans in vivo: Implications for reciprocal cerebro-cerebellar structural connectivity in motor and non-motor areas. Sci. Rep. 2017, 7, 12841. [Google Scholar] [CrossRef]
- Snider, R.S.; Maiti, A. Cerebellar contributions to the papez circuit. J. Neurosci. Res. 1976, 2, 133–146. [Google Scholar] [CrossRef]
- Anand, B.K.; Malhotra, C.L.; Singh, B.; Dua, S. Cerebellar Projections to Limbic System. J. Neurophysiol. 1959, 22, 451–457. [Google Scholar] [CrossRef]
- Bohne, P.; Schwarz, M.K.; Herlitze, S.; Mark, M.D. A New Projection from the Deep Cerebellar Nuclei to the Hippocampus via the Ventrolateral and Laterodorsal Thalamus in Mice. Front. Neural Circuits 2019, 13, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombel, C.; Lalonde, R.; Caston, J. The effects of unilateral removal of the cerebellar hemispheres on spatial learning and memory in rats. Brain Res. 2004, 1004, 108–115. [Google Scholar] [CrossRef]
- Burguière, E.; Arabo, A.; Jarlier, F.; De Zeeuw, C.I.; Rondi-Reig, L. Role of the Cerebellar Cortex in Conditioned Goal-Directed Behavior. J. Neurosci. 2010, 30, 13265–13271. [Google Scholar] [CrossRef] [Green Version]
- Barbas, H.; Blatt, G.J. Topographically specific hippocampal projections target functionally distinct prefrontal areas in the rhesus monkey. Hippocampus 1995, 5, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Hoover, W.B.; Vertes, R.P. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 2007, 212, 149–179. [Google Scholar] [CrossRef]
- Habas, C. Research note: A resting-state, cerebello-amygdaloid intrinsically connected network. Cerebellum Ataxias 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, L.; Qin, W.; Liu, Y.; Han, W.; Zhang, Y.; Jiang, T.; Yu, C. Resting-state functional connectivity of the vermal and hemispheric subregions of the cerebellum with both the cerebral cortical networks and subcortical structures. NeuroImage 2012, 61, 1213–1225. [Google Scholar] [CrossRef]
- Heath, R.G.; Harper, J.W. Ascending projections of the cerebellar fastigial nucleus to the hippocampus, amygdala, and other temporal lobe sites: Evoked potential and histological studies in monkeys and cats. Exp. Neurol. 1974, 45, 268–287. [Google Scholar] [CrossRef]
- Morris, J.S.; Frith, C.; Perrett, D.I.; Rowland, D.; Young, A.W.; Calder, A.J.; Dolan, R. A differential neural response in the human amygdala to fearful and happy facial expressions. Nature 1996, 383, 812–815. [Google Scholar] [CrossRef] [Green Version]
- Ernst, T.M.; Brol, A.E.; Gratz, M.; Ritter, C.; Bingel, U.; Schlamann, M.; Maderwald, S.; Quick, H.H.; Merz, C.J.; Timmann, D. The cerebellum is involved in processing of predictions and prediction errors in a fear conditioning paradigm. eLife 2019, 8, e46831. [Google Scholar] [CrossRef]
- Vilensky, J.A.; Van Hoesen, G.W. Corticopontine projections from the cingulate cortex in the rhesus monkey. Brain Res. 1981, 205, 391–395. [Google Scholar] [CrossRef]
- Krienen, F.M.; Buckner, R.L. Segregated Fronto-Cerebellar Circuits Revealed by Intrinsic Functional Connectivity. Cereb. Cortex 2009, 19, 2485–2497. [Google Scholar] [CrossRef] [Green Version]
- Rogers, T.D.; Dickson, P.E.; Heck, D.H.; Goldowitz, D.; Mittleman, G.; Blaha, C.D. Connecting the dots of the cerebro-cerebellar role in cognitive function: Neuronal pathways for cerebellar modulation of dopamine release in the prefrontal cortex. Synapse 2011, 65, 1204–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittleman, G.; Goldowitz, D.; Heck, D.H.; Blaha, C.D. Cerebellar modulation of frontal cortex dopamine efflux in mice: Relevance to autism and schizophrenia. Synapse 2008, 62, 544–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, T.; Becker, N.; Apps, R.; Jones, M. Back to front: Cerebellar connections and interactions with the prefrontal cortex. Front. Syst. Neurosci. 2014, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Forster, G.L.; Blaha, C.D. Pedunculopontine tegmental stimulation evokes striatal dopamine efflux by activation of ace-tylcholine and glutamate receptors in the midbrain and pons of the rat. Eur. J. Neurosci. 2003, 17, 751–762. [Google Scholar] [CrossRef]
- Garcia-Rill, E.; Skinner, R.; Miyazato, H.; Homma, Y. Pedunculopontine stimulation induces prolonged activation of pontine reticular neurons. Neuroscience 2001, 104, 455–465. [Google Scholar] [CrossRef]
- Perciavalle, V.; Berretta, S.; Raffaele, R. Projections from the intracerebellar nuclei to the ventral midbrain tegmentum in the rat. Neuroscience 1989, 29, 109–119. [Google Scholar] [CrossRef]
- Schwarz, C.; Schmitz, Y. Projection from the cerebellar lateral nucleus to precerebellar nuclei in the mossy fiber pathway is glutamatergic: A study combining anterograde tracing with immunogold labeling in the rat. J. Comp. Neurol. 1997, 381, 320–334. [Google Scholar] [CrossRef]
- Pinto, A.; Jankowski, M.; Sesack, S.R. Projections from the paraventricular nucleus of the thalamus to the rat prefrontal cortex and nucleus accumbens shell: Ultrastructural characteristics and spatial relationships with dopamine afferents. J. Comp. Neurol. 2003, 459, 142–155. [Google Scholar] [CrossRef]
- Del Arco, A.; Mora, F. Glutamate-dopamine in vivo interaction in the prefrontal cortex modulates the release of dopamine and acetylcholine in the nucleus accumbens of the awake rat. J. Neural Transm. 2005, 112, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Carta, I.; Chen, C.H.; Schott, A.L.; Dorizan, S.; Khodakhah, K. Cerebellar modulation of the reward circuitry and social behavior. Science 2019, 363, eaav0581. [Google Scholar] [CrossRef]
- D’Mello, A.; Stoodley, C.J. Cerebro-cerebellar circuits in autism spectrum disorder. Front. Neurosci. 2015, 9, 408. [Google Scholar] [CrossRef] [Green Version]
- Rogers, T.D.; Dickson, P.E.; McKimm, E.; Heck, D.H.; Goldowitz, D.; Blaha, C.D.; Mittleman, G. Reorganization of Circuits Underlying Cerebellar Modulation of Prefrontal Cortical Dopamine in Mouse Models of Autism Spectrum Disorder. Cerebellum 2013, 12, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.-H.; Kloth, A.D.; Badura, A. The Cerebellum, Sensitive Periods, and Autism. Neuron 2014, 83, 518–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus Paper: Pathological Role of the Cerebellum in Autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef] [Green Version]
- Reeber, S.L.; Otis, T.S.; Sillitoe, R.V. New roles for the cerebellum in health and disease. Front. Syst. Neurosci. 2013, 7, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauman, M.; Kemper, T.L. Histoanatomic observations of the brain in early infantile autism. Neurology 1985, 35, 866. [Google Scholar] [CrossRef]
- Kemper, T.L.; Bauman, M.L. The Contribution of Neuropathologic Studies to the Understanding of Autism. Neurol. Clin. 1993, 11, 175–187. [Google Scholar] [CrossRef]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.L.; Lantos, P. A clinicopathological study of autism. Brain 1998, 121 Pt 5, 889–905. [Google Scholar] [CrossRef] [Green Version]
- Whitney, E.R.; Kemper, T.L.; Bauman, M.L.; Rosene, D.; Blatt, G.J. Cerebellar Purkinje Cells are Reduced in a Subpopulation of Autistic Brains: A Stereological Experiment Using Calbindin-D28k. Cerebellum 2008, 7, 406–416. [Google Scholar] [CrossRef]
- Skefos, J.; Cummings, C.; Enzer, K.; Holiday, J.; Weed, K.; Levy, E.; Yuce, T.; Kemper, T.; Bauman, M. Regional Alterations in Purkinje Cell Density in Patients with Autism. PLoS ONE 2014, 9, e81255. [Google Scholar] [CrossRef]
- Palmen, S.J.M.C.; van Engeland, H.; Hof, P.R.; Schmitz, C. Neuropathological findings in autism. Brain 2004, 127, 2572–2583. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Halt, A.R.; Realmuto, G.; Earle, J.; Kist, D.A.; Thuras, P.; Merz, A. Purkinje Cell Size Is Reduced in Cerebellum of Patients with Autism. Cell. Mol. Neurobiol. 2002, 22, 171–175. [Google Scholar] [CrossRef]
- Bauman, M.; Kemper, T. (Eds.) Structural brain anatomy in autism: What is evidence? In The Neurobiology of Autism; JHU Press: Baltimore, MD, USA, 2005; pp. 119–145. [Google Scholar]
- Whitney, E.R.; Kemper, T.L.; Rosene, D.L.; Bauman, M.L.; Blatt, G.J. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. J. Neurosci. Res. 2009, 87, 2245–2254. [Google Scholar] [CrossRef] [Green Version]
- Crepel, F.; Mariani, J. Multiple innervation of purkinje cells by climbing fibers in the cerebellum of the weaver mutant mouse. J. Neurobiol. 1976, 7, 579–582. [Google Scholar] [CrossRef]
- Puro, D.G.; Woodward, D.J. The climbing fiber system in the Weaver mutant. Brain Res. 1977, 129, 141–146. [Google Scholar] [CrossRef]
- Mariani, J. Extent of multiple innervation of purkinje cells by climbing fibers in the olivocerebellar system of weaver, reeler, and staggerer mutant mice. J. Neurobiol. 1982, 13, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Cajal, S. Histology of the Nervous System of Man and Vertebrates; Oxford University Press: New York, NY, USA, 1995; pp. 1909–1910. [Google Scholar]
- Blatt, G.J. The Neuropathology of Autism. Scientifica 2012, 2012, 703675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, T.L.; Bauman, M.L. Neuropathology of infantile autism. Mol. Psychiatry 2002, 7 (Suppl. 2), S12–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampson, D.R.; Blatt, G.J. Autism spectrum disorders and neuropathology of the cerebellum. Front. Neurosci. 2015, 9, 420. [Google Scholar] [CrossRef] [Green Version]
- Courchesne, E.; Townsend, J.; Akshoomoff, N.A.; Saitoh, O.; Yeung-Courchesne, R.; Lincoln, A.J.; James, H.E.; Haas, R.H.; Schreibman, L.; Lau, L. Impairment in shifting attention in autistic and cerebellar patients. Behav. Neurosci. 1994, 108, 848–865. [Google Scholar] [CrossRef] [PubMed]
- Courchesne, E.; Yeung-Courchesne, R.; Hesselink, J.; Jernigan, T. Hypoplasia of Cerebellar Vermal Lobules VI and VII in Autism. N. Engl. J. Med. 1988, 318, 1349–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, J.W.; Courchesne, E.; Press, G.A.; Yeung-Courchesne, R.; Hesselink, J.R. Reduced Cerebellar Hemisphere Size and Its Relationship to Vermal Hypoplasia in Autism. Arch. Neurol. 1989, 46, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Cooper, K.L.; Mostofsky, S.H.; Capone, G.T.; Kates, W.R.; Newschaffer, C.J.; Bukelis, I.; Stump, M.H.; Jann, A.E.; Lanham, D.C. Specificity of Cerebellar Vermian Abnormalities in Autism: A Quantitative Magnetic Resonance Imaging Study. J. Child Neurol. 2003, 18, 463–470. [Google Scholar] [CrossRef]
- Scott, J.A.; Schumann, C.M.; Goodlin-Jones, B.L.; Amaral, D.G. A comprehensive volumetric analysis of the cerebellum in children and adolescents with autism spectrum disorder. Autism Res. 2009, 2, 246–257. [Google Scholar] [CrossRef]
- Piven, J.; Saliba, K.; Bailey, J.; Arndt, S. An MRI study of autism: The cerebellum revisited. Neurology 1997, 49, 546–551. [Google Scholar] [CrossRef]
- Sparks, B.F.; Friedman, S.D.; Shaw, D.W.; Aylward, E.H.; Echelard, D.; Artru, A.A.; Maravilla, K.R.; Giedd, J.N.; Munson, J.; Dawson, G.; et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology 2002, 59, 184–192. [Google Scholar] [CrossRef]
- Courchesne, E.; Pierce, K. Brain overgrowth in autism during a critical time in development: Implications for frontal pyramidal neuron and interneuron development and connectivity. Int. J. Dev. Neurosci. 2005, 23, 153–170. [Google Scholar] [CrossRef]
- Courchesne, E.; Karns, C.M.; Davis, H.R.; Ziccardi, R.; Carper, R.A.; Tigue, Z.D.; Chisum, H.J.; Moses, P.; Pierce, K.; Lord, C.; et al. Unusual brain growth patterns in early life in patients with autistic disorder: An MRI study. Neurology 2001, 57, 245–254. [Google Scholar] [CrossRef]
- Khan, A.J.; Nair, A.; Keown, C.L.; Datko, M.C.; Lincoln, A.J.; Müller, R.-A. Cerebro-cerebellar Resting-State Functional Connectivity in Children and Adolescents with Autism Spectrum Disorder. Biol. Psychiatry 2015, 78, 625–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verly, M.; Verhoeven, J.; Zink, I.; Mantini, D.; Peeters, R.; Deprez, S.; Emsell, L.; Boets, B.; Noens, I.; Steyaert, J.; et al. Altered functional connectivity of the language network in ASD: Role of classical language areas and cerebellum. NeuroImage Clin. 2014, 4, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Kyeong, S.; Kim, E.; Cheon, K.-A. Abnormalities of Inter- and Intra-Hemispheric Functional Connectivity in Autism Spectrum Disorders: A Study Using the Autism Brain Imaging Data Exchange Database. Front. Neurosci. 2016, 10, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, C.S.; Peltier, S.J.; Wiggins, J.L.; Weng, S.-J.; Carrasco, M.; Risi, S.; Lord, C. Abnormalities of intrinsic functional connectivity in autism spectrum disorders. NeuroImage 2009, 47, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Uddin, L.Q.; Supekar, K.; Menon, V. Reconceptualizing functional brain connectivity in autism from a developmental perspective. Front. Hum. Neurosci. 2013, 7, 458. [Google Scholar] [CrossRef] [Green Version]
- Hahamy, A.; Behrmann, M.; Malach, R. The idiosyncratic brain: Distortion of spontaneous connectivity patterns in autism spectrum disorder. Nat. Neurosci. 2015, 18, 302–309. [Google Scholar] [CrossRef]
- Nunes, A.S.; Peatfield, N.; Vakorin, V.; Doesburg, S.M. Idiosyncratic organization of cortical networks in autism spectrum disorder. NeuroImage 2019, 190, 182–190. [Google Scholar] [CrossRef]
- Jack, A.; Morris, J.P. Neocerebellar contributions to social perception in adolescents with autism spectrum disorder. Dev. Cogn. Neurosci. 2014, 10, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Fatemi, S.H.; Stary, J.M.; Halt, A.R.; Realmuto, G.R. Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. J. Autism Dev. Disord. 2001, 31, 529–535. [Google Scholar] [CrossRef]
- Chugani, D.C.; Muzik, O.; Rothermel, R.; Behen, M.E.; Chakraborty, P.K.; Mangner, T.J.; Chugani, H.T. Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann. Neurol. 1997, 42, 666–669. [Google Scholar] [CrossRef]
- Chugani, D.C. Role of altered brain serotonin mechanisms in autism. Mol. Psychiatry 2002, 7 (Suppl. 2), S16–S17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vichier-Guerre, C.; Parker, M.; Pomerantz, Y.; Finnell, R.; Cabrera, R.M. Impact of selective serotonin reuptake inhibitors on neural crest stem cell formation. Toxicol. Lett. 2017, 281, 20–25. [Google Scholar] [CrossRef]
- Fricker, A.D.; Rios, C.; Devi, L.A.; Gomes, I. Serotonin receptor activation leads to neurite outgrowth and neuronal survival. Mol. Brain Res. 2005, 138, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Khozhai, L.I.; Otellin, V.A. Synaptogenesis in the dorsal raphe nucleus of rat medulla oblongata in serotonin deficiency. Morfologiia 2012, 142, 20–24. [Google Scholar] [CrossRef]
- Fatemi, S.; Halt, A.R.; Stary, J.M.; Kanodia, R.; Schulz, S.; Realmuto, G.R. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 2002, 52, 805–810. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J.-J.; Blatt, G.J. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: Pathophysiological implications. Acta Neuropathol. 2007, 113, 559–568. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J.-J.; Blatt, G.J. IncreasedGAD67 mRNA expression in cerebellar interneurons in autism: Implications for Purkinje cell dysfunction. J. Neurosci. Res. 2007, 86, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Llinas, R.; Leznik, E.; Makarenko, V. The Olivo-Cerebellar Circuit as a Universal Motor Control System. IEEE J. Ocean. Eng. 2004, 29, 631–639. [Google Scholar] [CrossRef]
- Yip, J.; Soghomonian, J.J.; Blatt, G.J. Decreased GAD65 mRNA levels in select subpopulations of neurons in the cerebellar dentate nuclei in autism: An in situ hybridization study. Autism Res. 2009, 2, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, J.P.; Weber, D.J.; Cirstea, C.M.; Beversdorf, D.Q. Cerebro-Cerebellar Functional Connectivity is Associated with Cerebellar Excitation–Inhibition Balance in Autism Spectrum Disorder. J. Autism Dev. Disord. 2018, 48, 3460–3473. [Google Scholar] [CrossRef]
- Faissner, A.; Reinhard, J. The extracellular matrix compartment of neural stem and glial progenitor cells. Glia 2015, 63, 1330–1349. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010, 11, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zou, H.; Sheikh, A.M.; Malik, M.; Dobkin, C.; Brown, W.T.; Li, X. IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J. Neuroinflamm. 2011, 8, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.-M.; Ji, L.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Lucchina, L.; Depino, A.M. Altered Peripheral and Central Inflammatory Responses in a Mouse Model of Autism. Autism Res. 2014, 7, 273–289. [Google Scholar] [CrossRef]
- Goines, P.; Haapanen, L.; Boyce, R.; Duncanson, P.; Braunschweig, D.; Delwiche, L.; Hansen, R.; Hertz-Picciotto, I.; Ashwood, P.; Van de Water, J. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav. Immun. 2011, 25, 514–523. [Google Scholar] [CrossRef] [Green Version]
- Wills, S.; Cabanlit, M.; Bennett, J.; Ashwood, P.; Amaral, D.G.; Van de Water, J. Detection of autoantibodies to neural cells of the cerebellum in the plasma of subjects with autism spectrum disorders. Brain Behav. Immun. 2009, 23, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.N.; Van de Water, J. Associations of impaired behaviors with elevated plasma chemokines in autism spectrum disorders. J. Neuroimmunol. 2011, 232, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Heuer, L.; Ashwood, P.; Schauer, J.; Goines, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Croen, L.A.; Pessah, I.N.; Van De Water, J. Reduced levels of immunoglobulin in children with autism correlates with behavioral symptoms. Autism Res. 2008, 1, 275–283. [Google Scholar] [CrossRef]
- Wills, S.; Rossi, C.C.; Bennett, J.; Martinez-Cerdeño, V.; Ashwood, P.; Amaral, D.G.; Van de Water, J. Further characterization of autoantibodies to GABAergic neurons in the central nervous system produced by a subset of children with autism. Mol. Autism 2011, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Bolivar, V.J.; Walters, S.R.; Phoenix, J.L. Assessing autism-like behavior in mice: Variations in social interactions among inbred strains. Behav. Brain Res. 2007, 176, 21–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarlane, H.G.; Kusek, G.K.; Yang, M.; Phoenix, J.L.; Bolivar, V.J.; Crawley, J.N. Autism-like behavioral phenotypes in BTBR T + tf/J mice. Genes Brain Behav. 2008, 7, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.; Zhang, Y.; Gao, D.; Miller, V.M.; Lawrence, D.A. Aberrant Immune Responses in a Mouse with Behavioral Disorders. PLoS ONE 2011, 6, e20912. [Google Scholar] [CrossRef]
- Bakheet, S.A.; Alzahrani, M.Z.; Nadeem, A.; Ansari, M.A.; Zoheir, K.; Attia, S.M.; Al-Ayadhi, L.Y.; Ahmad, S.F. Resveratrol treatment attenuates chemokine receptor expression in the BTBR T + tf/J mouse model of autism. Mol. Cell. Neurosci. 2016, 77, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Alzahrani, M.Z.; Alshammari, M.A.; Alanazi, W.A.; Alasmari, A.F.; Attia, S.M. Resveratrol attenuates pro-inflammatory cytokines and activation of JAK1-STAT3 in BTBR T + Itpr3 tf /J autistic mice. Eur. J. Pharmacol. 2018, 829, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Ansari, M.A.; Nadeem, A.; Bakheet, S.A.; Al-Ayadhi, L.Y.; Attia, S.M. Toll-like receptors, NF-κB, and IL-27 mediate adenosine A2A receptor signaling in BTBR T + Itpr3 tf /J mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 79, 184–191. [Google Scholar] [CrossRef]
- Xiao, R.; Zhong, H.; Li, X.; Ma, Y.; Zhang, R.; Wang, L.; Zang, Z.; Fan, X. Abnormal Cerebellar Development Is Involved in Dystonia-Like Behaviors and Motor Dysfunction of Autistic BTBR Mice. Front. Cell Dev. Biol. 2020, 8, 231. [Google Scholar] [CrossRef]
- Batzoglou, S.; Pachter, L.; Mesirov, J.P.; Berger, B.; Lander, E.S. Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction. Genome Res. 2000, 10, 950–958. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Hansel, C. Deregulation of synaptic plasticity in autism. Neurosci. Lett. 2019, 688, 58–61. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Zoghbi, H. Lessons learned from studying syndromic autism spectrum disorders. Nat. Neurosci. 2016, 19, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O. Genetic causes of syndromic and non-syndromic autism. Dev. Med. Child Neurol. 2010, 52, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.W.; Ji, Z.; Millen, K.; Joyner, A.L. The Engrailed-2 homeobox gene and patterning of spinocerebellar mossy fiber afferents. Dev. Brain Res. 1996, 96, 210–218. [Google Scholar] [CrossRef]
- Millen, K.; Wurst, W.; Herrup, K.; Joyner, A. Abnormal embryonic cerebellar development and patterning of postnatal foliation in two mouse Engrailed-2 mutants. Development 1994, 120, 695–706. [Google Scholar] [CrossRef]
- Boukhtouche, F.; Doulazmi, M.; Frédéric, F.; Dusart, I.; Brugg, B.; Mariani, J. RORα, a pivotal nuclear receptor for Purkinje neuron survival and differentiation: From development to ageing. Cerebellum 2006, 5, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Gold, D.A.; Gent, P.M.; Hamilton, B.A. RORα in genetic control of cerebellum development: 50 staggering years. Brain Res. 2007, 1140, 19–25. [Google Scholar] [CrossRef]
- Ferland, R.; Cherry, T.J.; Preware, P.O.; Morrisey, E.E.; Walsh, C.A. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J. Comp. Neurol. 2003, 460, 266–279. [Google Scholar] [CrossRef]
- Rice, D.S.; Curran, T. Role of the Reelin Signaling Pathway in Central Nervous System Development. Annu. Rev. Neurosci. 2001, 24, 1005–1039. [Google Scholar] [CrossRef] [Green Version]
- Ieraci, A.; Forni, P.E.; Ponzetto, C. Viable hypomorphic signaling mutant of the Met receptor reveals a role for hepatocyte growth factor in postnatal cerebellar development. Proc. Natl. Acad. Sci. USA 2002, 99, 15200–15205. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Huentelman, M.; Smith, C.; Qiu, S. MET Receptor Tyrosine Kinase as an Autism Genetic Risk Factor. Int. Rev. Neurobiol. 2013, 113, 135–165. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.; Krimpenfort, P.; Leung, C.; van der Korput, H.A.G.M.; Trapman, J.; Camenisch, I.; Berns, A.; Brandner, S. PTEN is essential for cell migration but not for fate determination and tumourigenesis in the cerebellum. Development 2002, 129, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Sekine, Y.; Saruta, C.; Nishibe, H.; Morita, N.; Sato, Y.; Sadakata, T.; Shinoda, Y.; Kojima, T.; Furuichi, T. Cerebellar development transcriptome database (CDT-DB): Profiling of spatio-temporal gene expression during the postnatal development of mouse cerebellum. Neural Netw. 2008, 21, 1056–1069. [Google Scholar] [CrossRef] [PubMed]
- Sadakata, T.; Furuichi, T. Developmentally Regulated Ca2+-Dependent Activator Protein for Secretion 2 (CAPS2) is Involved in BDNF Secretion and is Associated with Autism Susceptibility. Cerebellum 2009, 8, 312–322. [Google Scholar] [CrossRef] [PubMed]
- DeLorey, T.M.; Sahbaie, P.; Hashemi, E.; Homanics, G.E.; Clark, J.D. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: A potential model of autism spectrum disorder. Behav. Brain Res. 2008, 187, 207–220. [Google Scholar] [CrossRef] [Green Version]
- Owens, D.F.; Kriegstein, A.R. Is there more to gaba than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef]
- Gharani, N.; Benayed, R.; Mancuso, V.; Brzustowicz, L.M.; Millonig, J.H. Association of the homeobox transcription factor, ENGRAILED 2, 3, with autism spectrum disorder. Mol. Psychiatry 2004, 9, 474–484. [Google Scholar] [CrossRef] [Green Version]
- Kuemerle, B.; Zanjani, H.; Joyner, A.; Herrup, K. Pattern Deformities and Cell Loss inEngrailed-2Mutant Mice Suggest Two Separate Patterning Events during Cerebellar Development. J. Neurosci. 1997, 17, 7881–7889. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, G.; Zunino, G.; Genovesi, S.; Sgadó, P.; Bozzi, Y. Mutant mouse models of autism spectrum disorders. Dis. Markers 2012, 33, 225–239. [Google Scholar] [CrossRef]
- Cheh, M.A.; Millonig, J.H.; Roselli, L.M.; Ming, X.; Jacobsen, E.; Kamdar, S.; Wagner, G.C. En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res. 2006, 1116, 166–176. [Google Scholar] [CrossRef]
- Sarachana, T.; Hu, V. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol. Autism 2013, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Sayad, A.; Noroozi, R.; Omrani, M.D.; Taheri, M.; Ghafouri-Fard, S. Retinoic acid-related orphan receptor alpha (RORA) variants are associated with autism spectrum disorder. Metab. Brain Dis. 2017, 32, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Rauch, T.A.; Pfeifer, G.P.; Hu, V.W. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010, 24, 3036–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidman, R.L.; Lane, P.W.; Dickie, M.M. Staggerer, a New Mutation in the Mouse Affecting the Cerebellum. Science 1962, 137, 610–612. [Google Scholar] [CrossRef] [PubMed]
- Dussault, I.; Fawcett, D.; Matthyssen, A.; Bader, J.-A.; Giguère, V. Orphan nuclear receptor RORα-deficient mice display the cerebellar defects of staggerer. Mech. Dev. 1998, 70, 147–153. [Google Scholar] [CrossRef]
- Steinmayr, M.; André, E.; Conquet, F.; Rondi-Reig, L.; Delhaye-Bouchaud, N.; Auclair, N.; Daniel, H.; Crépel, F.; Mariani, J.; Sotelo, C.; et al. staggerer phenotype in retinoid-related orphan receptor α-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3960–3965. [Google Scholar] [CrossRef] [Green Version]
- Toma, C.; Hervás, A.; Torrico, B.; Balmaña, N.; Salgado, M.; Maristany, M.; Vilella, E.; Martínez-Leal, R.; Planelles, M.I.; Cuscó, I.; et al. Analysis of two language-related genes in autism: A case-control association study of FOXP2 and CNTNAP2. Psychiatr. Genet. 2013, 23, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Bowers, J.M.; Konopka, G. The role of the FOXP family of transcription factors in ASD. Dis. Markers 2012, 33, 251–260. [Google Scholar] [CrossRef]
- Fisher, S.E.; Scharff, C. FOXP2 as a molecular window into speech and language. Trends Genet. 2009, 25, 166–177. [Google Scholar] [CrossRef]
- Shu, W.; Cho, J.Y.; Jiang, Y.; Zhang, M.; Weisz, D.; Elder, G.A.; Schmeidler, J.; De Gasperi, R.; Sosa, M.A.G.; Rabidou, D.; et al. Altered ultrasonic vocalization in mice with a disruption in the Foxp2 gene. Proc. Natl. Acad. Sci. USA 2005, 102, 9643–9648. [Google Scholar] [CrossRef] [Green Version]
- Fujita, E.; Tanabe, Y.; Shiota, A.; Ueda, M.; Suwa, K.; Momoi, M.Y.; Momoi, T. Ultrasonic vocalization impairment of Foxp2 (R552H) knockin mice related to speech-language disorder and abnormality of Purkinje cells. Proc. Natl. Acad. Sci. USA 2008, 105, 3117–3122. [Google Scholar] [CrossRef] [Green Version]
- Groszer, M.; Keays, D.A.; Deacon, R.M.; de Bono, J.P.; Prasad-Mulcare, S.; Gaub, S.; Baum, M.G.; French, C.; Nicod, J.; Coventry, J.A.; et al. Impaired Synaptic Plasticity and Motor Learning in Mice with a Point Mutation Implicated in Human Speech Deficits. Curr. Biol. 2008, 18, 354–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatemi, S.H.; Snow, A.V.; Stary, J.M.; Araghi-Niknam, M.; Reutiman, T.J.; Lee, S.; Brooks, A.I.; Pearce, D.A. Reelin signaling is impaired in autism. Biol. Psychiatry 2005, 57, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Falconer, D.S. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J. Genet. 1951, 50, 192–205. [Google Scholar] [CrossRef]
- Mariani, J.; Crepel, F.; Mikoshiba, K.; Changeux, J.P.; Sotelo, C. Anatomical, Physiological and biochemical studies of the cerebellum fromreelermutant mouse. Philos. Trans. R. Soc. B Biol. Sci. 1977, 281, 1–28. [Google Scholar] [CrossRef]
- Imamura, R.; Matsumoto, K. Hepatocyte growth factor in physiology and infectious diseases. Cytokine 2017, 98, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.B.; D’Oronzio, R.; Garbett, K.; Ebert, P.J.; Mirnics, K.; Levitt, P.; Persico, A.M. Disruption of cerebral cortex MET signaling in autism spectrum disorder. Ann. Neurol. 2007, 62, 243–250. [Google Scholar] [CrossRef]
- Campbell, D.B.; Sutcliffe, J.S.; Ebert, P.J.; Militerni, R.; Bravaccio, C.; Trillo, S.; Elia, M.; Schneider, C.; Melmed, R.; Sacco, R.; et al. A genetic variant that disrupts MET transcription is associated with autism. Proc. Natl. Acad. Sci. USA 2006, 103, 16834–16839. [Google Scholar] [CrossRef] [Green Version]
- Sousa, I.; Clark, T.G.; Toma, C.; Kobayashi, K.; Choma, M.; Holt, R.; Sykes, N.H.; Lamb, J.A.; Bailey, A.J.; Battaglia, A.; et al. MET and autism susceptibility: Family and case–control studies. Eur. J. Hum. Genet. 2008, 17, 749–758. [Google Scholar] [CrossRef]
- Thanseem, I.; Nakamura, K.; Miyachi, T.; Toyota, T.; Yamada, S.; Tsujii, M.; Tsuchiya, K.J.; Anitha, A.; Iwayama, Y.; Yamada, K.; et al. Further evidence for the role of MET in autism susceptibility. Neurosci. Res. 2010, 68, 137–141. [Google Scholar] [CrossRef]
- Sun, H.; Lesche, R.; Li, D.-M.; Liliental, J.; Zhang, H.; Gao, J.; Gavrilova, N.; Mueller, B.; Liu, X.; Wu, H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 6199–6204. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilot, A.; Frazier, T.W.; Eng, C. Balancing Proliferation and Connectivity in PTEN-associated Autism Spectrum Disorder. Neurotherapeutics 2015, 12, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.A.; Pastore, M.; Prior, T.; Herman, G.E.; McBride, K.L. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet. Med. 2009, 11, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarn, N.; Jaini, R.; Thacker, S.; Lee, H.; Dutta, R.; Eng, C. Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol. Psychiatry 2020, 26, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.-H.; Zhu, X.; Zhang, J.; Knoop, L.L.; Tharp, R.; Smeyne, R.J.; Eberhart, C.G.; Burger, P.C.; Baker, S.J. Pten regulates neuronal soma size: A mouse model of Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 404–411. [Google Scholar] [CrossRef]
- Nolan, S.O.; Jefferson, T.S.; Reynolds, C.D.; Smith, G.D.; Holley, A.J.; Hodges, S.L.; Lugo, J.N. Neuronal deletion of phosphatase and tensin homolog results in cerebellar motor learning dysfunction and alterations in intracellular signaling. NeuroReport 2019, 30, 556–561. [Google Scholar] [CrossRef]
- Cupolillo, D.; Hoxha, E.; Faralli, A.; De Luca, A.; Rossi, F.; Tempia, F.; Carulli, D. Autistic-Like Traits and Cerebellar Dysfunction in Purkinje Cell PTEN Knock-Out Mice. Neuropsychopharmacology 2015, 41, 1457–1466. [Google Scholar] [CrossRef] [Green Version]
- Sadakata, T.; Mizoguchi, A.; Sato, Y.; Katoh-Semba, R.; Fukuda, M.; Mikoshiba, K.; Furuichi, T. The Secretory Granule-Associated Protein CAPS2 Regulates Neurotrophin Release and Cell Survival. J. Neurosci. 2004, 24, 43–52. [Google Scholar] [CrossRef]
- Bonora, E.; Graziano, C.; Minopoli, F.; Bacchelli, E.; Magini, P.; Diquigiovanni, C.; Lomartire, S.; Bianco, F.; Vargiolu, M.; Parchi, P.; et al. Maternally inherited genetic variants of CADPS 2 are present in Autism Spectrum Disorders and Intellectual Disability patients. EMBO Mol. Med. 2014, 6, 795–809. [Google Scholar] [CrossRef]
- Grabowski, P.A.P.; Bello, A.F.; Rodrigues, D.L.; Forbeci, M.J.; Motter, V.; Raskin, S. Deletion Involving the 7q31-32 Band at the CADPS2 Gene Locus in a Patient with Autism Spectrum Disorder and Recurrent Psychotic Syndrome Triggered by Stress. Case Rep. Psychiatry 2017, 2017, 4254152. [Google Scholar] [CrossRef] [Green Version]
- Sadakata, T.; Kakegawa, W.; Mizoguchi, A.; Washida, M.; Katoh-Semba, R.; Shutoh, F.; Okamoto, T.; Nakashima, H.; Kimura, K.; Tanaka, M.; et al. Impaired Cerebellar Development and Function in Mice Lacking CAPS2, a Protein Involved in Neurotrophin Release. J. Neurosci. 2007, 27, 2472–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, E.H.; Courchesne, R.Y.; Cox, N.J.; Lord, C.; Gonen, D.; Guter, S.J.; Lincoln, A.; Nix, K.; Haas, R.; Leventhal, B.L.; et al. Linkage-Disequilibrium Mapping of Autistic Disorder, with 15q11-13 Markers. Am. J. Hum. Genet. 1998, 62, 1077–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homanics, G.E.; DeLorey, T.M.; Firestone, L.L.; Quinlan, J.J.; Handforth, A.; Harrison, N.L.; Krasowski, M.D.; Rick, C.E.M.; Korpi, E.R.; Mäkelä, R.; et al. Mice devoid of γ-aminobutyrate type A receptor β3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc. Natl. Acad. Sci. USA 1997, 94, 4143–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser-McCaw, B.; Hecht, F.; Cadien, J.D.; Moore, B.C. Fragile X-linked mental retardation. Am. J. Med. Genet. 1980, 7, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Meloni, I.; Bruttini, M.; Longo, I.; Mari, F.; Rizzolio, F.; D’Adamo, P.; Denvriendt, K.; Fryns, J.-P.; Toniolo, D.; Renieri, A. A Mutation in the Rett Syndrome Gene, MECP2, Causes X-Linked Mental Retardation and Progressive Spasticity in Males. Am. J. Hum. Genet. 2000, 67, 982–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, A.; Smith, M.; Yates, J.R. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat. Genet. 1994, 6, 193–196. [Google Scholar] [CrossRef]
- Kalsner, L.; Chamberlain, S.J. Prader-Willi, Angelman, and 15q11-q13 Duplication Syndromes. Pediatr. Clin. N. Am. 2015, 62, 587–606. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, R.J.; Berry-Kravis, E.; Hazlett, H.C.; Bailey, D.B., Jr.; Moine, H.; Kooy, R.F.; Tassone, F.; Gantois, I.; Sonenberg, N.; Mandel, J.L.; et al. Fragile X syndrome. Nat. Rev. Dis. Primers 2017, 3, 17065. [Google Scholar] [CrossRef]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef] [Green Version]
- Boyle, L.; Kaufmann, W.E. The behavioral phenotype of FMR1 mutations. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154C, 469–476. [Google Scholar] [CrossRef]
- Gothelf, D.; Furfaro, J.A.; Hoeft, F.; Eckert, M.A.; Hall, S.S.; O’Hara, R.; Erba, H.W.; Bs, J.R.; Bs, K.M.H.; Ms, S.P.; et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann. Neurol. 2008, 63, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostofsky, S.H.; Mazzocco, M.M.; Aakalu, G.; Warsofsky, I.S.; Denckla, M.B.; Reiss, A.L. Decreased cerebellar posterior vermis size in fragile X syndrome: Correlation with neurocognitive performance. Neurology 1998, 50, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sabaratnam, M. Pathological and neuropathological findings in two males with fragile-X syndrome. J. Intellect. Disabil. Res. 2000, 44 Pt 1, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Navarro, C.S.; Hunsaker, M.R.; Maezawa, I.; Shuler, J.F.; Tassone, F.; Delany, M.; Au, J.W.; Berman, R.F.; Jin, L.-W.; et al. Neuropathologic features in the hippocampus and cerebellum of three older men with fragile X syndrome. Mol. Autism 2011, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.S.; Adams, P.E.; Nguyen, D.; Brunberg, J.A.; Tassone, F.; Zhang, W.; Koldewyn, K.; Rivera, S.M.; Grigsby, J.; Zhang, L.; et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS). Neurology 2007, 69, 851–859. [Google Scholar] [CrossRef]
- Koekkoek, S.; Yamaguchi, K.; Milojkovic, B.; Dortland, B.; Ruigrok, T.; Maex, R.; De Graaf, W.; Smit, A.; VanderWerf, F.; Bakker, C.; et al. Deletion of FMR1 in Purkinje Cells Enhances Parallel Fiber LTD, Enlarges Spines, and Attenuates Cerebellar Eyelid Conditioning in Fragile X Syndrome. Neuron 2005, 47, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Tobia, M.J.; Woodruff-Pak, D.S. Delay eyeblink classical conditioning is impaired in Fragile X syndrome. Behav. Neurosci. 2009, 123, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Sunamura, N.; Iwashita, S.; Enomoto, K.; Kadoshima, T.; Isono, F. Loss of the fragile X mental retardation protein causes aberrant differentiation in human neural progenitor cells. Sci. Rep. 2018, 8, 11585. [Google Scholar] [CrossRef] [Green Version]
- Pacey, L.K.K.; Guan, S.; Tharmalingam, S.; Thomsen, C.; Hampson, D.R. Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav. 2015, 5, e00400. [Google Scholar] [CrossRef] [Green Version]
- Krasovska, V.; Doering, L.C. Regulation of IL-6 Secretion by Astrocytes via TLR4 in the Fragile X Mouse Model. Front. Mol. Neurosci. 2018, 11, 272. [Google Scholar] [CrossRef]
- Bernardet, M.; Crusio, W. Fmr1KO Mice as a Possible Model of Autistic Features. Sci. World J. 2006, 6, 1164–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ey, E.; Leblond, C.S.; Bourgeron, T. Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011, 4, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Ellegood, J.; Pacey, L.K.; Hampson, D.R.; Lerch, J.P.; Henkelman, R.M. Anatomical phenotyping in a mouse model of fragile X syndrome with magnetic resonance imaging. NeuroImage 2010, 53, 1023–1029. [Google Scholar] [CrossRef]
- Rett, A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966, 116, 723–726. [Google Scholar] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef]
- Percy, A.K. Rett Syndrome: Exploring the autism link. Arch. Neurol. 2011, 68, 985–989. [Google Scholar] [CrossRef] [Green Version]
- Glaze, D.G.; Frost, J.D.; Zoghbi, H.Y.; Percy, A.K. Rett’s Syndrome. Correlation of electroencephalographic characteristics with clinical staging. Arch. Neurol. 1987, 44, 1053–1056. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of BDNF Transcription Involves Calcium-Dependent Phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA Methylation-Related Chromatin Remodeling in Activity-Dependent Bdnf Gene Regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theoharides, T.C.; Athanassiou, M.; Panagiotidou, S.; Doyle, R. Dysregulated brain immunity and neurotrophin signaling in Rett syndrome and autism spectrum disorders. J. Neuroimmunol. 2014, 279, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Maezawa, I.; Calafiore, M.; Wulff, H.; Jin, L.-W. Does microglial dysfunction play a role in autism and Rett syndrome? Neuron Glia Biol. 2011, 7, 85–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maezawa, I.; Jin, L.-W. Rett Syndrome Microglia Damage Dendrites and Synapses by the Elevated Release of Glutamate. J. Neurosci. 2010, 30, 5346–5356. [Google Scholar] [CrossRef] [Green Version]
- Oldfors, A.; Sourander, P.; Armstrong, D.L.; Percy, A.K.; Witt-Engerström, I.; Hagberg, B.A. Rett syndrome: Cerebellar pathology. Pediatr. Neurol. 1990, 6, 310–314. [Google Scholar] [CrossRef]
- Murakami, J.W.; Courchesne, E.; Haas, R.H.; Press, G.; Yeung-Courchesne, R. Cerebellar and cerebral abnormalities in Rett syndrome: A quantitative MR analysis. Am. J. Roentgenol. 1992, 159, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Belichenko, N.P.; Belichenko, P.V.; Li, H.H.; Mobley, W.C.; Francke, U. Comparative study of brain morphology inMecp2mutant mouse models of Rett syndrome. J. Comp. Neurol. 2008, 508, 184–195. [Google Scholar] [CrossRef]
- Achilly, N.P.; He, L.-J.; Kim, O.; Ohmae, S.; Wojaczynski, G.J.; Lin, T.; Sillitoe, R.V.; Medina, J.F.; Zoghbi, H.Y. Deleting Mecp2 from the cerebellum rather than its neuronal subtypes causes a delay in motor learning in mice. eLife 2021, 10, e64833. [Google Scholar] [CrossRef]
- Li, W.; Pozzo-Miller, L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014, 76 Pt C, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Chang, Q.; Khare, G.; Dani, V.; Nelson, S.; Jaenisch, R. The Disease Progression of Mecp2 Mutant Mice Is Affected by the Level of BDNF Expression. Neuron 2006, 49, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhala, R.; Korhonen, L.; Mikelsaar, M.; Lindholm, D.; Riikonen, R. Neurotrophic factors in cerebrospinal fluid and serum of patients with Rett syndrome. J. Child Neurol. 1998, 13, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, R. Neurotrophic factors in the pathogenesis of Rett syndrome. J. Child Neurol. 2003, 18, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Abuhatzira, L.; Makedonski, K.; Kaufman, Y.; Razin, A.; Shemer, R. MeCP2 Deficiency in the Brain Decreases BDNF Levels by REST/CoREST-Mediated Repression and Increases TRKB Production. Epigenetics 2007, 2, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Curatolo, P.; Moavero, R.; de Vries, P.J. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- de Vries, P.J.; Whittemore, V.H.; Leclezio, L.; Byars, A.W.; Dunn, D.; Ess, K.C.; Hook, D.; King, B.H.; Sahin, M.; Jansen, A. Tuberous Sclerosis Associated Neuropsychiatric Disorders (TAND) and the TAND Checklist. Pediatr. Neurol. 2015, 52, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Gipson, T.T.; Gerner, G.; Wilson, M.A.; Blue, M.E.; Johnston, M.V. Potential for treatment of severe autism in tuberous sclerosis complex. World J. Clin. Pediatr. 2013, 2, 16–25. [Google Scholar] [CrossRef]
- Smalley, S.L. Autism and tuberous sclerosis. J. Autism Dev. Disord. 1998, 28, 407–414. [Google Scholar] [CrossRef]
- Jeste, S.S.; Sahin, M.; Bolton, P.; Ploubidis, G.B.; Humphrey, A. Characterization of Autism in Young Children with Tuberous Sclerosis Complex. J. Child Neurol. 2008, 23, 520–525. [Google Scholar] [CrossRef]
- Martí-Bonmatí, L.; Menor, F.; Dosdá, R. Tuberous Sclerosis: Differences between Cerebral and Cerebellar Cortical Tubers in a Pediatric Population. Am. J. Neuroradiol. 2000, 21, 557–560. [Google Scholar]
- Ertan, G.; Arulrajah, S.; Tekes, A.; Jordan, L.; Huisman, T. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J. Neuroradiol. 2010, 37, 231–238. [Google Scholar] [CrossRef]
- Menor, F.; Marti-Bonmati, L.; Mulas, F.; Poyatos, C.; Cortina, H. Neuroimaging in tuberous sclerosis: A clinicoradiological evaluation in pediatric patients. Pediatr. Radiol. 1992, 22, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.; Hagiwara, M.; Katz, J.; Roth, J.; Devinsky, O.; Weiner, H.; Milla, S. MRI Characterization and Longitudinal Study of Focal Cerebellar Lesions in a Young Tuberous Sclerosis Cohort. Am. J. Neuroradiol. 2013, 34, 655–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boer, K.; Troost, D.; Jansen, F.; Nellist, M.; Ouweland, A.M.V.D.; Geurts, J.J.; Spliet, W.G.; Crino, P.; Aronica, E. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 2008, 28, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.M.; Egelhoff, J.C.; McKellop, J.M.; Franz, D.N. Autism and the cerebellum: Evidence from tuberous sclerosis. J. Autism Dev. Disord. 2000, 30, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.; Sahin, M. Cerebellar Development and Autism Spectrum Disorder in Tuberous Sclerosis Complex. J. Child Neurol. 2015, 30, 1954–1962. [Google Scholar] [CrossRef] [Green Version]
- Ito, N.; Rubin, G.M. Gigas, a Drosophila Homolog of Tuberous Sclerosis Gene Product-2, Regulates the Cell Cycle. Cell 1999, 96, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Lipton, J.O.; Sahin, M. The Neurology of mTOR. Neuron 2014, 84, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Miloloza, A.; Rosner, M.; Nellist, M.; Halley, D.; Bernaschek, G.; Hengstschläger, M. The TSC1 gene product, hamartin, negatively regulates cell proliferation. Hum. Mol. Genet. 2000, 9, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Manning, B.D. The TSC1–TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Sosunov, A.A.; Wu, X.; Weiner, H.L.; Mikell, C.B.; Goodman, R.R.; Crino, P.D.; McKhann, G.M. Tuberous sclerosis: A primary pathology of astrocytes? Epilepsia 2008, 49 (Suppl. 2), 53–62. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, M.; Baybis, M.; Newman, D.; Kolson, D.L.; Chen, W.; McKhann, G.; Gutmann, D.; Crino, P.B. Expression of ICAM-1, TNF-α, NFκB, and MAP kinase in tubers of the tuberous sclerosis complex. Neurobiol. Dis. 2003, 14, 279–290. [Google Scholar] [CrossRef]
- Boer, K.; Jansen, F.; Nellist, M.; Redeker, S.; Ouweland, A.V.D.; Spliet, W.; van Nieuwenhuizen, O.; Troost, D.; Crino, P.; Aronica, E. Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res. 2008, 78, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Boer, K.; Crino, P.B.; Gorter, J.A.; Nellist, M.; Jansen, F.E.; Spliet, W.G.; Van Rijen, P.C.; Wittink, F.R.; Breit, T.M.; Troost, D.; et al. Gene Expression Analysis of Tuberous Sclerosis Complex Cortical Tubers Reveals Increased Expression of Adhesion and Inflammatory Factors. Brain Pathol. 2010, 20, 704–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, D.H.; Zhang, Y.; Hasbani, M.J.; Goldberg, M.; Plank, T.L.; Henske, E.P. Expression of the tuberous sclerosis complex gene products, hamartin and tuberin, in central nervous system tissues. Acta Neuropathol. 2000, 99, 223–230. [Google Scholar] [CrossRef]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Kloth, A.D.; Badura, A.; Li, A.; Cherskov, A.; Connolly, S.G.; Giovannucci, A.; Bangash, M.A.; Grasselli, G.; Peñagarikano, O.; Piochon, C.; et al. Cerebellar associative sensory learning defects in five mouse autism models. eLife 2015, 4, e06085. [Google Scholar] [CrossRef]
- Clayton-Smith, J. Angelman syndrome: A review of the clinical and genetic aspects. J. Med. Genet. 2003, 40, 87–95. [Google Scholar] [CrossRef]
- Reith, R.M.; Way, S.; McKenna, J.; Haines, K.; Gambello, M.J. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol. Dis. 2011, 43, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Buiting, K.; Williams, C.; Horsthemke, K.B.B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef]
- Fang, P.; Lev-Lehman, E.; Tsai, T.-F.; Matsuura, T.; Benton, C.S.; Sutcliffe, J.S.; Christian, S.L.; Kubota, T.; Halley, D.J.; Meijers-Heijboer, H.; et al. The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet. 1999, 8, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Berrios, J.; Newbern, J.; Snider, W.D.; Philpot, B.D.; Hahn, K.; Zylka, M.J. An Autism-Linked Mutation Disables Phosphorylation Control of UBE3A. Cell 2015, 162, 795–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogart, A.; Wu, D.; LaSalle, J.M.; Schanen, N.C. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 2010, 38, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trillingsgaard, A.; Østergaard, J.R. Autism in Angelman Syndrome: An exploration of comorbidity. Autism 2004, 8, 163–174. [Google Scholar] [CrossRef]
- Peters, S.U.; Horowitz, L.; Barbieri-Welge, R.; Taylor, J.L.; Hundley, R.J. Longitudinal follow-up of autism spectrum features and sensory behaviors in Angelman syndrome by deletion class. J. Child Psychol. Psychiatry 2011, 53, 152–159. [Google Scholar] [CrossRef]
- Moreno-De-Luca, D.; Sanders, S.J.; Willsey, A.J.; Mulle, J.G.; Lowe, J.K.; Geschwind, D.H.; State, M.W.; Martin, C.L.; Ledbetter, D.H. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol. Psychiatry 2013, 18, 1090–1095. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, U.; Sutcliffe, J.S.; Cattanach, B.M.; Beechey, C.V.; Armstrong, D.; Eichele, G.; Beaudet, A.L. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 1997, 17, 75–78. [Google Scholar] [CrossRef]
- Dindot, S.V.; Antalffy, B.A.; Bhattacharjee, M.B.; Beaudet, A.L. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2007, 17, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Rougeulle, C.; Glatt, H.; Lalande, M. The Angelman syndrome candidate gene, UBE3AIE6-AP, is imprinted in brain. Nat. Genet. 1997, 17, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kishino, T.; Li, E.; Webber, H.; Dikkes, P.; Holmes, G.L.; Wagstaff, J. Neurobehavioral and Electroencephalographic Abnormalities in Ube3aMaternal-Deficient Mice. Neurobiol. Dis. 2002, 9, 149–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.-H.; Armstrong, D.; Albrecht, U.; Atkins, C.; Noebels, J.; Eichele, G.; Sweatt, J.D.; Beaudet, A.L. Mutation of the Angelman Ubiquitin Ligase in Mice Causes Increased Cytoplasmic p53 and Deficits of Contextual Learning and Long-Term Potentiation. Neuron 1998, 21, 799–811. [Google Scholar] [CrossRef] [Green Version]
- Cheron, G.; Servais, L.; Wagstaff, J.; Dan, B. Fast cerebellar oscillation associated with ataxia in a mouse model of angelman syndrome. Neuroscience 2005, 130, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Egawa, K.; Kitagawa, K.; Inoue, K.; Takayama, M.; Takayama, C.; Saitoh, S.; Kishino, T.; Kitagawa, M.; Fukuda, A. Decreased Tonic Inhibition in Cerebellar Granule Cells Causes Motor Dysfunction in a Mouse Model of Angelman Syndrome. Sci. Transl. Med. 2012, 4, 163ra157. [Google Scholar] [CrossRef]
- Battaglia, A. The inv dup (15) or idic (15) syndrome (Tetrasomy 15q). Orphanet J. Rare Dis. 2008, 3, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urraca, N.; Cleary, J.; Brewer, V.; Pivnick, E.K.; McVicar, K.; Thibert, R.L.; Schanen, N.C.; Esmer, C.; Lamport, D.; Reiter, L.T. The Interstitial Duplication 15q11.2-q13 Syndrome Includes Autism, Mild Facial Anomalies and a Characteristic EEG Signature. Autism Res. 2013, 6, 268–279. [Google Scholar] [CrossRef] [Green Version]
- Distefano, C.; Gulsrud, A.; Huberty, S.; Kasari, C.; Cook, E.; Reiter, L.T.; Thibert, R.; Jeste, S.S. Identification of a distinct developmental and behavioral profile in children with Dup15q syndrome. J. Neurodev. Disord. 2016, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Depienne, C.; Moreno-De-Luca, D.; Heron, D.; Bouteiller, D.; Gennetier, A.; Delorme, R.; Chaste, P.; Siffroi, J.-P.; Chantot-Bastaraud, S.; Benyahia, B. Screening for Genomic Rearrangements and Methylation Abnormalities of the 15q11-q13 Region in Autism Spectrum Disorders. Biol. Psychiatry 2009, 66, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.; Scherer, S. Copy-number variations associated with neuropsychiatric conditions. Nature 2008, 455, 919–923. [Google Scholar] [CrossRef]
- Piochon, C.; Kloth, A.D.; Grasselli, G.; Titley, H.; Nakayama, H.; Hashimoto, K.; Wan, V.; Simmons, D.; Eissa, T.; Nakatani, J.; et al. Cerebellar plasticity and motor learning deficits in a copy-number variation mouse model of autism. Nat. Commun. 2014, 5, 5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatani, J.; Tamada, K.; Hatanaka, F.; Ise, S.; Ohta, H.; Inoue, K.; Tomonaga, S.; Watanabe, Y.; Chung, Y.J.; Banerjee, R.; et al. Abnormal Behavior in a Chromosome- Engineered Mouse Model for Human 15q11-13 Duplication Seen in Autism. Cell 2009, 137, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soda, T.; Mapelli, L.; Locatelli, F.; Botta, L.; Goldfarb, M.; Prestori, F.; D’Angelo, E. Hyper-excitability and hyper-plasticity disrupt cerebellar signal transfer in the IB2 KO mouse model of autism. J. Neurosci. 2019, 39, 2383–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giza, J.; Urbanski, M.; Prestori, F.; Bandyopadhyay, B.; Yam, A.; Friedrich, V.; Kelley, K.; D’Angelo, E.; Goldfarb, M. Be-havioral and cerebellar transmission deficits in mice lacking the autism-linked gene islet brain-2. J. Neurosci. 2010, 30, 14805–14816. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, K.M.; Ramachandran, D.; Patel, V.C.; Shetty, A.C.; Cutler, D.J.; Zwick, M.E. Identification of rare X-linked neuroligin variants by massively parallel sequencing in males with autism spectrum disorder. Mol. Autism 2012, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Berkel, S.; Marshall, C.R.; Weiss, B.; Howe, J.; Roeth, R.; Moog, U.; Endris, V.; Roberts, W.; Szatmari, P.; Pinto, D.; et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010, 42, 489–491. [Google Scholar] [CrossRef]
- Sato, D.; Lionel, A.C.; Leblond, C.S.; Prasad, A.; Pinto, D.; Walker, S.; O’Connor, I.; Russell, C.; Drmic, I.E.; Hamdan, F.F.; et al. SHANK1 Deletions in Males with Autism Spectrum Disorder. Am. J. Hum. Genet. 2012, 90, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Phelan, M.; Rogers, R.; Saul, R.; Stapleton, G.; Sweet, K.; McDermid, H.; Shaw, S.; Claytor, J.; Willis, J.; Kelly, D. 22q13 deletion syndrome. Am. J. Med. Genet. 2001, 101, 91–99. [Google Scholar] [CrossRef]
- Leblond, C.S.; Nava, C.; Polge, A.; Gauthier, J.; Huguet, G.; Lumbroso, S.; Giuliano, F.; Stordeur, C.; Depienne, C.; Mouzat, K.; et al. Meta-analysis of SHANK Mutations in Autism Spectrum Disorders: A Gradient of Severity in Cognitive Impairments. PLoS Genet. 2014, 10, e1004580. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.M.; Betancur, C.; Boeckers, T.M.; Bockmann, J.; Chaste, P.; Fauchereau, F.; Nygren, G.; Rastam, M.; Gillberg, I.C.; Anckarsäter, H.; et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007, 39, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Moessner, R.; Marshall, C.R.; Sutcliffe, J.S.; Skaug, J.; Pinto, D.; Vincent, J.; Zwaigenbaum, L.; Fernandez, B.; Roberts, W.; Szatmari, P.; et al. Contribution of SHANK3 Mutations to Autism Spectrum Disorder. Am. J. Hum. Genet. 2007, 81, 1289–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, J.; Spiegelman, D.; Piton, A.; Lafrenière, R.G.; Laurent, S.; St-Onge, J.; Lapointe, L.; Hamdan, F.F.; Cossette, P.; Mottron, L.; et al. Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2009, 150B, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Kim, E. The Shank family of scaffold proteins. J. Cell Sci. 2000, 113 Pt 11, 1851–1856. [Google Scholar] [CrossRef]
- Grabrucker, A.M.; Knight, M.J.; Proepper, C.; Bockmann, J.; Joubert, M.; Rowan, M.; Nienhaus, G.U.; Garner, C.C.; Bowie, J.U.; Kreutz, M.R.; et al. Concerted action of zinc and ProSAP/Shank in synaptogenesis and synapse maturation. EMBO J. 2011, 30, 569–581. [Google Scholar] [CrossRef] [Green Version]
- Zitzer, H.; Hönck, H.-H.; Bächner, D.; Richter, D.; Kreienkamp, H.-J. Somatostatin Receptor Interacting Protein Defines a Novel Family of Multidomain Proteins Present in Human and Rodent Brain. J. Biol. Chem. 1999, 274, 32997–33001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckers, T.M.; Kreutz, M.R.; Winter, C.; Zuschratter, W.; Smalla, K.-H.; Sanmarti-Vila, L.; Wex, H.; Langnaese, K.; Bockmann, J.; Garner, C.C.; et al. Proline-Rich Synapse-Associated Protein-1/Cortactin Binding Protein 1 (ProSAP1/CortBP1) Is a PDZ-Domain Protein Highly Enriched in the Postsynaptic Density. J. Neurosci. 1999, 19, 6506–6518. [Google Scholar] [CrossRef]
- Boeckers, T.M.; Winter, C.; Smalla, K.-H.; Kreutz, M.R.; Bockmann, J.; Seidenbecher, C.; Garner, C.; Gundelfinger, E.D. Proline-Rich Synapse-Associated Proteins ProSAP1 and ProSAP2 Interact with Synaptic Proteins of the SAPAP/GKAP Family. Biochem. Biophys. Res. Commun. 1999, 264, 247–252. [Google Scholar] [CrossRef]
- Lim, S.; Naisbitt, S.; Yoon, J.; Hwang, J.-I.; Suh, P.-G.; Sheng, M.; Kim, E. Characterization of the Shank Family of Synaptic Proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem. 1999, 274, 29510–29518. [Google Scholar] [CrossRef] [Green Version]
- Böckers, T.M.; Segger-Junius, M.; Iglauer, P.; Bockmann, J.; Gundelfinger, E.D.; Kreutz, M.R.; Richter, D.; Kindler, S.; Kreienkamp, H.-J. Differential expression and dendritic transcript localization of Shank family members: Identification of a dendritic targeting element in the 3′ untranslated region of Shank1 mRNA. Mol. Cell. Neurosci. 2004, 26, 182–190. [Google Scholar] [CrossRef]
- Hung, A.Y.; Futai, K.; Sala, C.; Valtschanoff, J.G.; Ryu, J.; Woodworth, M.A.; Kidd, F.L.; Sung, C.C.; Miyakawa, T.; Bear, M.F.; et al. Smaller Dendritic Spines, Weaker Synaptic Transmission, but Enhanced Spatial Learning in Mice Lacking Shank1. J. Neurosci. 2008, 28, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Silverman, J.L.; Turner, S.M.; Barkan, C.L.; Tolu, S.S.; Saxena, R.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Sociability and motor functions in Shank1 mutant mice. Brain Res. 2011, 1380, 120–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wöhr, M.; Roullet, F.I.; Hung, A.Y.; Sheng, M.; Crawley, J.N. Communication Impairments in Mice Lacking Shank1: Reduced Levels of Ultrasonic Vocalizations and Scent Marking Behavior. PLoS ONE 2011, 6, e20631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungur, A.; Vörckel, K.J.; Schwarting, R.K.; Wöhr, M. Repetitive behaviors in the Shank1 knockout mouse model for autism spectrum disorder: Developmental aspects and effects of social context. J. Neurosci. Methods 2014, 234, 92–100. [Google Scholar] [CrossRef]
- Peter, S.; Brinke, M.M.T.; Stedehouder, J.; Reinelt, C.M.; Wu, B.; Zhou, H.; Zhou, K.; Boele, H.-J.; Kushner, S.A.; Lee, M.G.; et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 2016, 7, 12627. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Lee, D.; Cho, Y.S.; Chung, C.; Yoo, Y.-E.; Kim, J.; Lee, J.; Kim, W.; Kim, H.; Bae, Y.C.; et al. Cerebellar Shank2 Regulates Excitatory Synapse Density, Motor Coordination, and Specific Repetitive and Anxiety-Like Behaviors. J. Neurosci. 2016, 36, 12129–12143. [Google Scholar] [CrossRef] [Green Version]
- Stoodley, C.J. Distinct regions of the cerebellum show gray matter decreases in autism, ADHD, and developmental dyslexia. Front. Syst. Neurosci. 2014, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Bangash, M.A.; Park, J.M.; Melnikova, T.; Wang, D.; Jeon, S.K.; Lee, D.; Syeda, S.; Kim, J.; Kouser, M.; Schwartz, J.; et al. RETRACTED: Enhanced Polyubiquitination of Shank3 and NMDA Receptor in a Mouse Model of Autism. Cell 2011, 145, 758–772. [Google Scholar] [CrossRef] [Green Version]
- Peça, J.; Feliciano, C.; Ting, J.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; McCoy, P.A.; Rodriguiz, R.M.; Pan, Y.; Je, H.S.; Roberts, A.C.; Kim, C.J.; Berrios, J.; Colvin, J.S.; Bousquet-Moore, D.; et al. Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet. 2011, 20, 3093–3108. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.M.; Perroy, J.; Loll, F.; Perrais, D.; Fagni, L.; Bourgeron, T.; Montcouquiol, M.; Sans, N. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 2012, 17, 71–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouser, M.; Speed, H.; Dewey, C.M.; Reimers, J.M.; Widman, A.J.; Gupta, N.; Liu, S.; Jaramillo, T.C.; Bangash, M.; Xiao, B.; et al. Loss of Predominant Shank3 Isoforms Results in Hippocampus-Dependent Impairments in Behavior and Synaptic Transmission. J. Neurosci. 2013, 33, 18448–18468. [Google Scholar] [CrossRef] [PubMed]
- Duffney, L.J.; Zhong, P.; Wei, J.; Matas, E.; Cheng, J.; Qin, L.; Ma, K.; Dietz, D.; Kajiwara, Y.; Buxbaum, J.; et al. Autism-like Deficits in Shank3-Deficient Mice Are Rescued by Targeting Actin Regulators. Cell Rep. 2015, 11, 1400–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matas, E.; Maisterrena, A.; Thabault, M.; Balado, E.; Francheteau, M.; Balbous, A.; Galvan, L.; Jaber, M. Major motor and gait deficits with sexual dimorphism in a Shank3 mutant mouse model. Mol. Autism 2021, 12, 2. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nature 2008, 455, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A Neuroligin-3 Mutation Implicated in Autism Increases Inhibitory Synaptic Transmission in Mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gill-berg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef] [Green Version]
- Chadman, K.K.; Gong, S.; Scattoni, M.L.; Boltuck, S.E.; Gandhy, S.U.; Heintz, N.; Crawley, J.N. Minimal aberrant behavioral phenotypes of neuroligin-3 R451C knockin mice. Autism Res. 2008, 1, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Radyushkin, K.; Hammerschmidt, K.; Boretius, S.; Varoqueaux, F.; El-Kordi, A.; Ronnenberg, A.; Winter, D.; Frahm, J.; Fischer, J.; Brose, N.; et al. Neuroligin-3-deficient mice: Model of a monogenic heritable form of autism with an olfactory deficit. Genes Brain Behav. 2009, 8, 416–425. [Google Scholar] [CrossRef]
- Rothwell, P.; Fuccillo, M.; Maxeiner, S.; Hayton, S.J.; Gokce, O.; Lim, B.; Fowler, S.C.; Malenka, R.C.; Südhof, T.C. Autism-Associated Neuroligin-3 Mutations Commonly Impair Striatal Circuits to Boost Repetitive Behaviors. Cell 2014, 158, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Lai, E.S.K.; Nakayama, H.; Miyazaki, T.; Nakazawa, T.; Tabuchi, K.; Hashimoto, K.; Watanabe, M.; Kano, M. An Autism-Associated Neuroligin-3 Mutation Affects Developmental Synapse Elimination in the Cerebellum. Front. Neural Circuits 2021, 15, 676891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, L.Y.; Liu, X.; Maxeiner, S.; Lee, S.-J.; Gokce, O.; Südhof, T.C. Neuroligins Sculpt Cerebellar Purkinje-Cell Circuits by Differential Control of Distinct Classes of Synapses. Neuron 2015, 87, 781–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudouin, S.J.; Gaudias, J.; Gerharz, S.; Hatstatt, L.; Zhou, K.; Punnakkal, P.; Tanaka, K.F.; Spooren, W.; Hen, R.; De Zeeuw, C.I.; et al. Shared Synaptic Pathophysiology in Syndromic and Nonsyndromic Rodent Models of Autism. Science 2012, 338, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahaye, A.; Toutain, A.; Aboura, A.; Dupont, C.; Tabet, A.; Benzacken, B.; Elion, J.; Verloes, A.; Pipiras, E.; Drunat, S. Chromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3. Eur. J. Med. Genet. 2009, 52, 328–332. [Google Scholar] [CrossRef]
- Negri, S.; Oberson, A.; Steinmann, M.; Sauser, C.; Nicod, P.; Waeber, G.; Schorderet, D.F.; Bonny, C. cDNA Cloning and Mapping of a Novel Islet-Brain/JNK-Interacting Protein. Genomics 2000, 64, 324–330. [Google Scholar] [CrossRef]
- Yasuda, J.; Whitmarsh, A.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP Group of Mitogen-Activated Protein Kinase Scaffold Proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef] [Green Version]
- Bonaglia, M.C.; Giorda, R.; Beri, S.; De Agostini, C.; Novara, F.; Fichera, M.; Grillo, L.; Galesi, O.; Vetro, A.; Ciccone, R.; et al. Molecular Mechanisms Generating and Stabilizing Terminal 22q13 Deletions in 44 Subjects with Phelan/McDermid Syndrome. PLoS Genet. 2011, 7, e1002173. [Google Scholar] [CrossRef]
- Soorya, L.; Kolevzon, A.; Zweifach, J.; Lim, T.; Dobry, Y.; Schwartz, L.; Frank, Y.; Wang, A.T.; Cai, G.; Parkhomenko, E.; et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 2013, 4, 18. [Google Scholar] [CrossRef] [Green Version]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Chen, C.-F.; Sharp, J.L.; Rollins, J.D.; Collins, J.S.; Rogers, R.C.; Phelan, K.; DuPont, B.R. 22q13.2q13.32 genomic regions associated with severity of speech delay, developmental delay, and physical features in Phelan–McDermid syndrome. Genet. Med. 2014, 16, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Monaghan, D.T.; Cotman, C.W. Distribution of N-methyl-D-aspartate-sensitive L-[3H]glutamate-binding sites in rat brain. J. Neurosci. 1985, 5, 2909–2919. [Google Scholar] [CrossRef]
- Markram, K.; Markram, H. The Intense World Theory—A Unifying Theory of the Neurobiology of Autism. Front. Hum. Neurosci. 2010, 4, 224. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, T.; Perrodin, C.; Markram, H. Hyper-connectivity and hyper-plasticity in the medial prefrontal cortex in the valproic acid animal model of autism. Front. Neural Circuits 2008, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.M.; Khatri, V.; Land, P.W.; Simons, D.J. Thalamocortical Angular Tuning Domains within Individual Barrels of Rat Somatosensory Cortex. J. Neurosci. 2003, 23, 9565–9574. [Google Scholar] [CrossRef] [Green Version]
- Mountcastle, V.B. Modality and Topographic Properties of Single Neurons of Cat’s Somatic Sensory Cortex. J. Neurophysiol. 1957, 20, 408–434. [Google Scholar] [CrossRef]
- Mountcastle, V.B. The columnar organization of the neocortex. Brain 1997, 120 Pt 4, 701–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mountcastle, V.B. Introduction. Computation in cortical columns. Cereb. Cortex 2003, 13, 2–4. [Google Scholar] [CrossRef]
- Casanova, M.F. Neuropathological and Genetic Findings in Autism: The Significance of a Putative Minicolumnopathy. Neuroscientist 2006, 12, 435–441. [Google Scholar] [CrossRef]
- Hutsler, J.J.; Casanova, M.F. Cortical construction in autism spectrum disorder: Columns, connectivity and the subplate. Neuropathol. Appl. Neurobiol. 2015, 42, 115–134. [Google Scholar] [CrossRef]
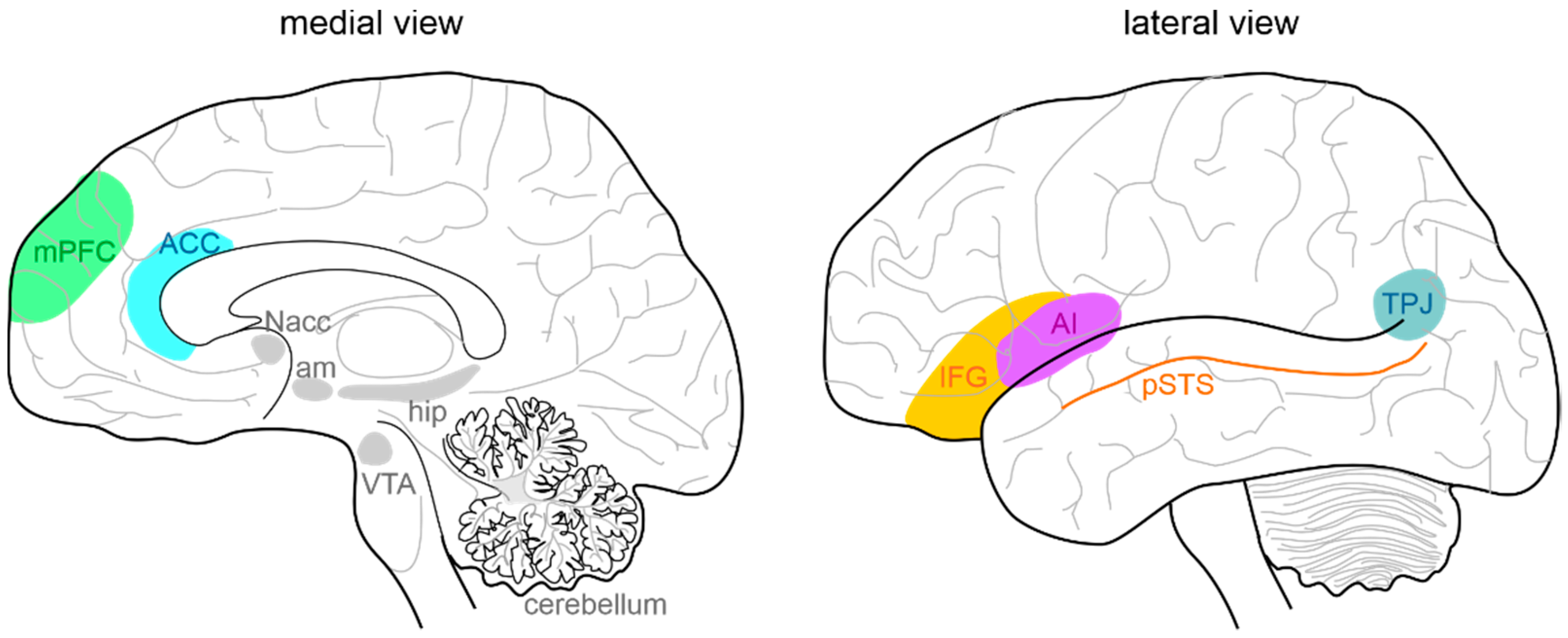

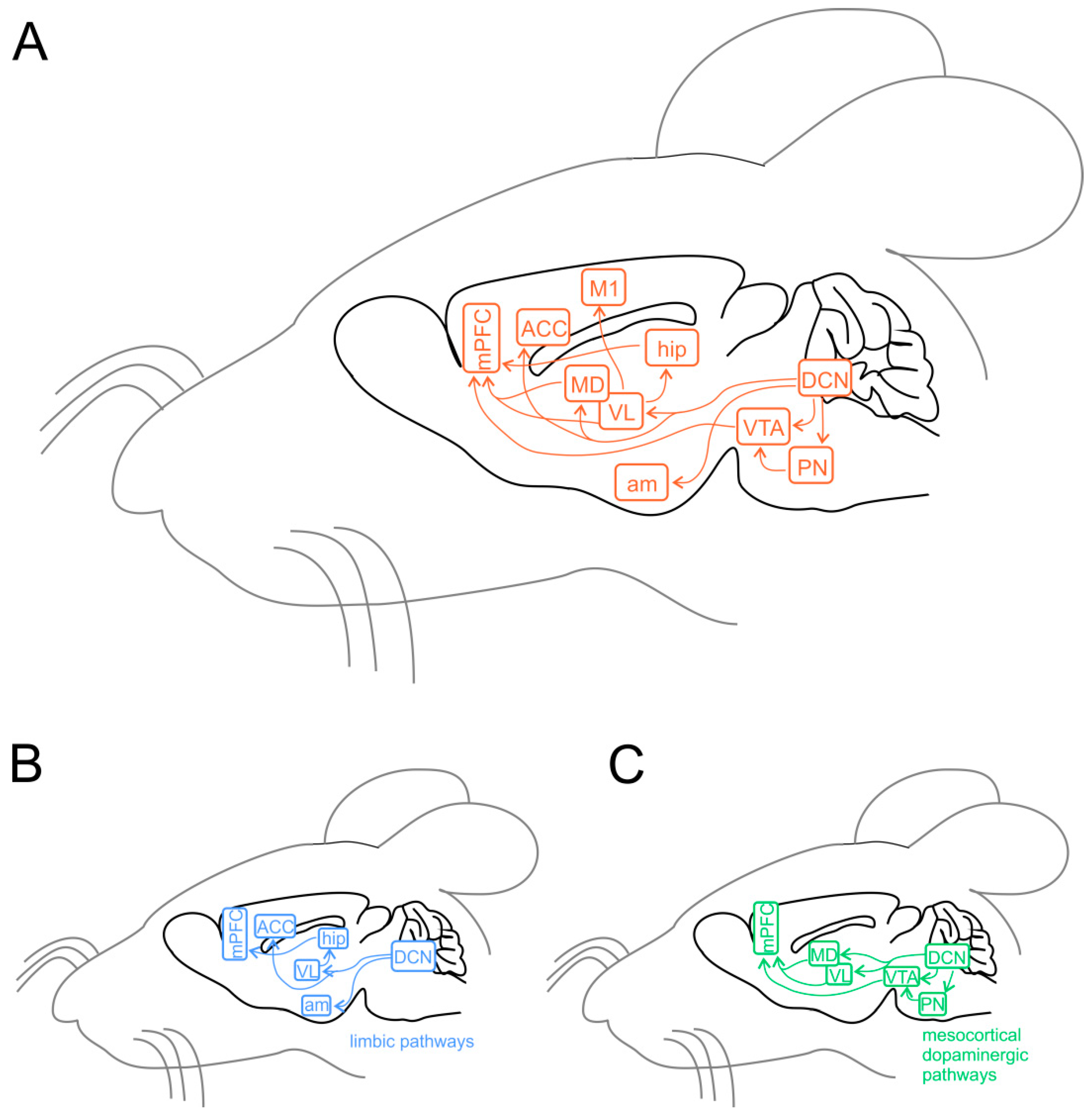



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Neurochemical Changes | Structural/Cellular Abnormalities | Functional Abnormalities | Behavioral Deficits |
|---|---|---|---|---|
| EN2-KO |
|
|
| |
| RORα-KO (staggerer) |
|
| ||
| FOXP2-KO |
|
|
| |
| Reelin-mutant (reeler) |
|
| ||
| MET-mutant |
|
| ||
| PTEN-KO (cerebellum) |
|
|
| |
| PTEN-KO (Purkinje cell) |
|
|
| |
| CAPDS2-KO |
|
|
| |
| GABRB3-KO |
|
|
| |
| FMR1-KO |
|
|
| |
| MeCP2-KO |
|
|
| ↓ |
| TSC1-KO TSC2-KO (Purkinje cell) |
|
|
| |
| UBE3A-KO |
|
| ||
| patDp/+ |
|
|
| |
| SHANK1-KO |
|
| ||
| SHANK2 e6/7-KO |
|
| ||
| SHANK2 e7-KO |
| |||
| SHANK2-KO (L7 Purkinje cell) |
| |||
| SHANK2-KO (Pcp2 Purkinje cell) |
| |||
| SHANK3-ΔC |
|
|
| |
| NLGN3-KO |
|
| ||
| NLGN3-R451C |
|
|
| |
| IB2-KO |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mapelli, L.; Soda, T.; D’Angelo, E.; Prestori, F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. Int. J. Mol. Sci. 2022, 23, 3894. https://doi.org/10.3390/ijms23073894
Mapelli L, Soda T, D’Angelo E, Prestori F. The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models. International Journal of Molecular Sciences. 2022; 23(7):3894. https://doi.org/10.3390/ijms23073894
Chicago/Turabian StyleMapelli, Lisa, Teresa Soda, Egidio D’Angelo, and Francesca Prestori. 2022. "The Cerebellar Involvement in Autism Spectrum Disorders: From the Social Brain to Mouse Models" International Journal of Molecular Sciences 23, no. 7: 3894. https://doi.org/10.3390/ijms23073894