
GABAA Receptor-Stabilizing Protein Ubqln1 Affects Hyperexcitability and Epileptogenesis after Traumatic Brain Injury and in a Model of In Vitro Epilepsy in Mice

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
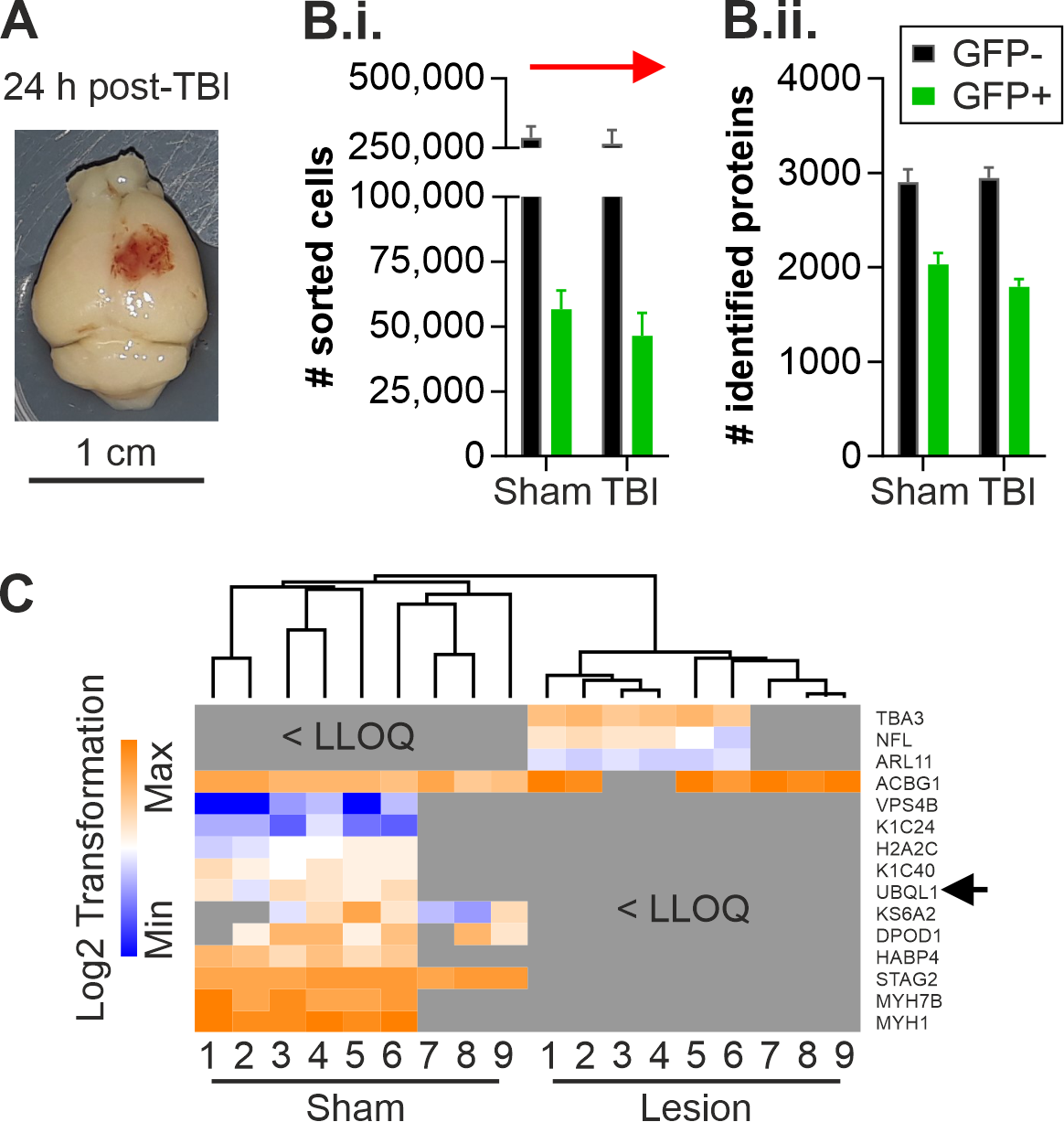
2.1. Altered Protein Expression of Ubqln1 in Cortical GABAergic Interneurons Isolated from the Contralateral Cortex 24 h Post-TBI
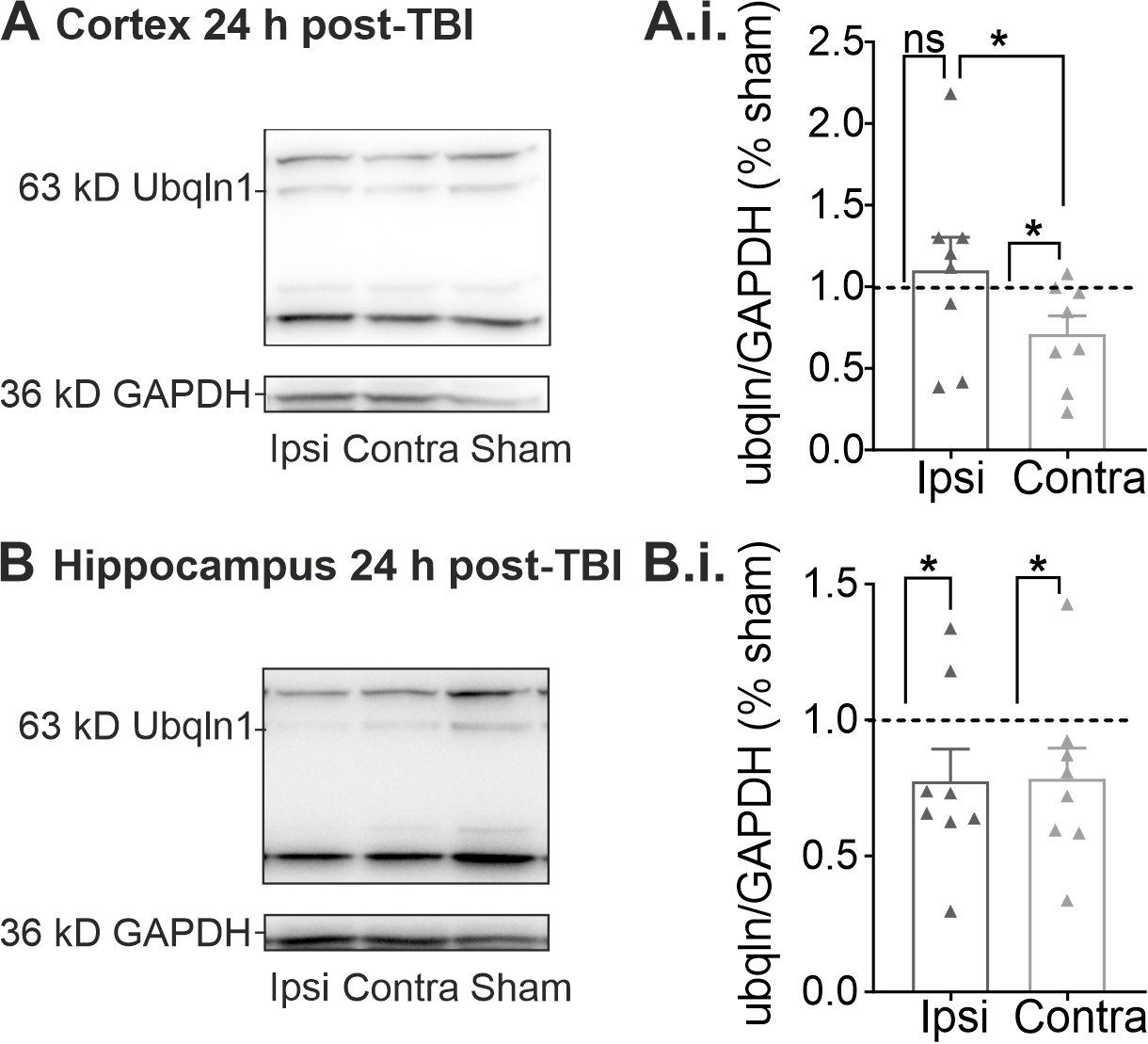
2.2. Regulation of Ubqln1 Expression in Cortex and Hippocampus 24 h Post-TBI
2.3. Properties of Pharmacologically Induced Epileptiform Activity in Hippocampal Brain Slices during Multi-Electrode Array (MEA) Recordings In Vitro
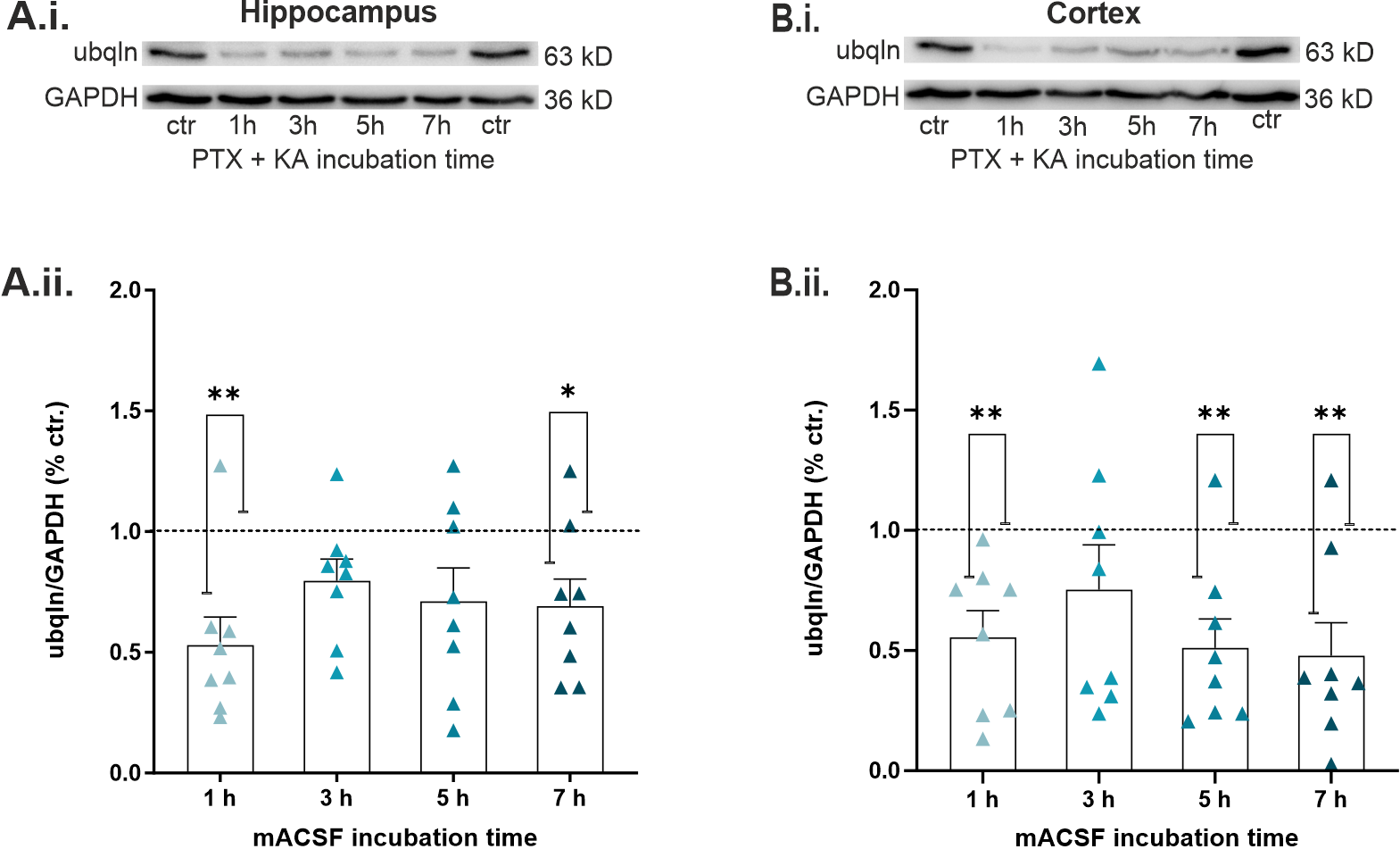
2.4. Epileptiform Activity Impaired Ubqln1 Expression in Hippocampal and Cortical Slices
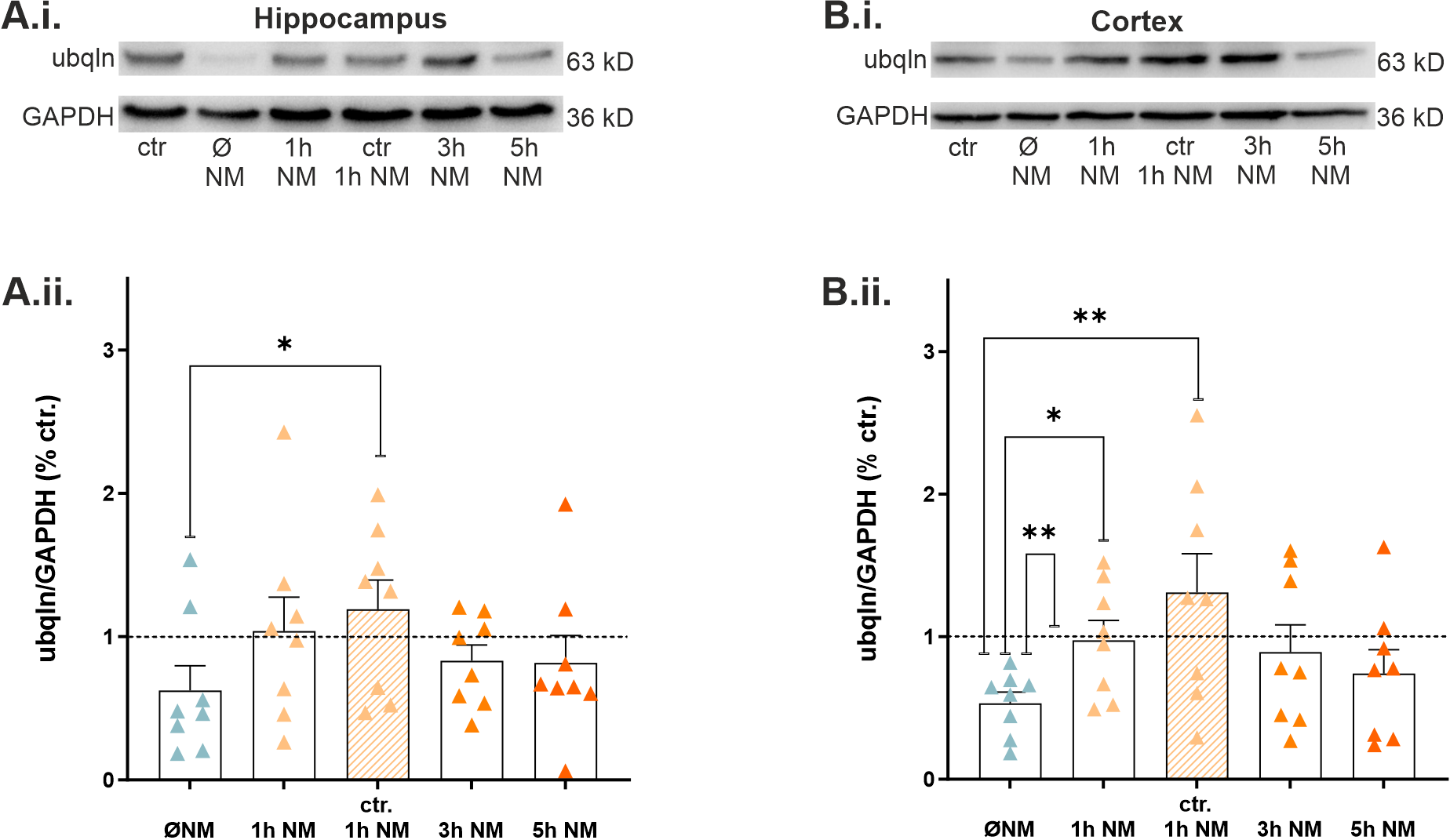
2.5. Nialamide, a Non-Selective Monoamine Oxidase Inhibitor, Rescued Expression of Ubqln1 in Hippocampal and Cortical Slices
2.6. The MAO Inhibitor Nialamide Alleviated Epileptiform Discharges and the Peak Amplitudes in Slices during MEA Recordings
3. Discussion
4. Materials and Methods
4.1. Animals and Ethical Approval
4.2. Controlled Cortical Impact
4.3. Electrophysiology In Vitro
4.3.1. Preparation of Brain Slices
4.3.2. In Vitro Epilepsy Model
4.3.3. Multi-Electrode Array Recordings
4.4. Western Blots
4.5. Fluorescence-Activated Cell Sorting (FACS)
4.6. Liquid Chromatography Mass Spectrometry Analysis and Label-Free Quantification Analysis
4.7. Statistical Evaluation
4.8. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | acetonitrile |
| ACSF | artificial cerebrospinal fluid |
| BCA | bicinchoninic acid assay |
| CaCl2·H2O | calcium chloride monohydrate |
| CA1 | cornu ammonis subfield CA1 |
| CCI | controlled cortical impact |
| CO2 | carbon dioxide |
| Ctr. | Control |
| D | dopamine |
| DIA | data-independent acquisition |
| DMSO | dimethyl sulfoxide |
| DPBS | Dulbecco’s phosphate buffered saline |
| DTT | dithiothreitol |
| ECL | enhanced chemiluminescence |
| EDTA | ethylenediaminetetraacetic acid |
| ER | endoplasmic reticulum |
| EtOH | ethanol |
| FA | formic acid |
| FDR | false discovery rate |
| GABA | gamma aminobutyric acid |
| GAD67 | glutamate decarboxylase 67 kD |
| GAPDH | glyceraldehyde 3-phosphatase dehydrogenase |
| GFP | green fluorescent protein |
| HRP | horseradish peroxidase |
| IAA | iodoacetamide |
| IMS | ion-mobility separation |
| I/R injury | ischemia/reperfusion injury |
| KA | kainic acid/ kainate |
| KCl | potassium chloride |
| LC-MS | liquid chromatography mass spectrometry |
| LDS | lithium dodecyl sulfate |
| mACSF | modified artificial cerebrospinal fluid |
| MAO | monoamine oxidase |
| MEA | multi-electrode array |
| MgSO4·7H2O | magnesium sulfate heptahydrate |
| N | number |
| N2 | nitrogen |
| NaCl | sodium chloride |
| NaHCO3 | sodium hydrogen carbonate |
| NaH2PO4·H2O | sodium phosphate monobasic monohydrate |
| NFL | neurofilament |
| NM | nialamide |
| Norm. | normalized |
| Ns | statistically not significant |
| O2 | oxygen |
| PFA | paraformaldehyde |
| PLGS | proteinlynx global server |
| PTE | posttraumatic epilepsy |
| PTX | picrotoxin |
| PTZ | pentylenetetrazole |
| PVDF | polyvinylidene difluoride |
| SD | spreading depolarization |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| SEM | standard error of mean |
| SLE | seizure-like event |
| TBI | traumatic brain injury |
| TBS-T | mixture of Tris-buffered saline with polysorbate 20 (tween 20) |
| TiN | titanium nitride |
| ubqln1 | ubiquilin-1/plic-1 |
| UPS | ubiquitin proteasome system |
| 5-HT | serotonin |
References
- Lucke-Wold, B.P.; Nguyen, L.; Turner, R.C.; Logsdon, A.F.; Chen, Y.W.; Smith, K.E.; Huber, J.D.; Matsumoto, R.; Rosen, C.L.; Tucker, E.S.; et al. Traumatic brain injury and epilepsy: Underlying mechanisms leading to seizure. Seizure 2015, 33, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fordington, S.; Manford, M. A review of seizures and epilepsy following traumatic brain injury. J. Neurol. 2020, 267, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, D.H. Epilepsy after head injury: An overview. Epilepsia 2009, 50 (Suppl. 2), 4–9. [Google Scholar] [CrossRef]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.J. Review: Animal models of acquired epilepsy: Insights into mechanisms of human epileptogenesis. Neuropathol. Appl. Neurobiol. 2018, 44, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, F.; Zapfe, W.P.K.; Draguhn, A.; Gutiérrez, R. Early Appearance and Spread of Fast Ripples in the Hippocampus in a Model of Cortical Traumatic Brain Injury. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 9034–9046. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Pedersen, M.G.; Pedersen, C.B.; Sidenius, P.; Olsen, J.; Vestergaard, M. Long-term risk of epilepsy after traumatic brain injury in children and young adults: A population-based cohort study. Lancet 2009, 373, 1105–1110. [Google Scholar] [CrossRef]
- Briggs, S.W.; Galanopoulou, A.S. Altered GABA signaling in early life epilepsies. Neural Plast. 2011, 2011, 527605. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, R.M.; Giza, C.C.; Rotenberg, A. Glutamate and GABA imbalance following traumatic brain injury. Curr. Neurol. Neurosci. Rep. 2015, 15, 27. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Z.; Gu, J.; Zhang, Y.; Wang, W.; Shen, H.; Chen, G.; Wang, X. Plic-1, a new target in repressing epileptic seizure by regulation of GABAAR function in patients and a rat model of epilepsy. Clin. Sci. 2015, 129, 1207–1223. [Google Scholar] [CrossRef]
- Köhling, R. Prolonged seizures: What are the mechanisms that predispose or cease to be protective? A review of animal data. Epileptic Disord. Int. Epilepsy J. Videotape 2014, 16, S23–S36. [Google Scholar] [CrossRef] [PubMed]
- Dugladze, T.; Maziashvili, N.; Börgers, C.; Gurgenidze, S.; Häussler, U.; Winkelmann, A.; Haas, C.A.; Meier, J.C.; Vida, I.; Kopell, N.J.; et al. GABA(B) autoreceptor-mediated cell type-specific reduction of inhibition in epileptic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 15073–15078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantu, D.; Walker, K.; Andresen, L.; Taylor-Weiner, A.; Hampton, D.; Tesco, G.; Dulla, C.G. Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb. Cortex 2015, 25, 2306–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrosci, B.; Mittmann, T. Functional consequences of the disturbances in the GABA-mediated inhibition induced by injuries in the cerebral cortex. Neural Plast. 2011, 2011, 614329. [Google Scholar] [CrossRef] [PubMed]
- Imbrosci, B.; Wang, Y.; Arckens, L.; Mittmann, T. Neuronal mechanisms underlying transhemispheric diaschisis following focal cortical injuries. Brain Struct. Funct. 2015, 220, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.S.; Obara, K.; Muramatsu, K. Diaschisis. Neurol. Res. 1993, 15, 362–366. [Google Scholar] [CrossRef]
- Le Prieult, F.; Thal, S.C.; Engelhard, K.; Imbrosci, B.; Mittmann, T. Acute Cortical Transhemispheric Diaschisis after Unilateral Traumatic Brain Injury. J. Neurotrauma 2017, 34, 1097–1110. [Google Scholar] [CrossRef]
- Kleijnen, M.F.; Shih, A.H.; Zhou, P.; Kumar, S.; Soccio, R.E.; Kedersha, N.L.; Gill, G.; Howley, P.M. The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 2000, 6, 409–419. [Google Scholar] [CrossRef]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef] [Green Version]
- Upadhya, S.C.; Hegde, A.N. A potential proteasome-interacting motif within the ubiquitin-like domain of parkin and other proteins. Trends Biochem. Sci. 2003, 28, 280–283. [Google Scholar] [CrossRef]
- Ko, H.S.; Uehara, T.; Tsuruma, K.; Nomura, Y. Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 2004, 566, 110–114. [Google Scholar] [PubMed]
- Jansen, A.H.; Reits, E.A.; Hol, E.M. The ubiquitin proteasome system in glia and its role in neurodegenerative diseases. Front. Mol. Neurosci. 2014, 7, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saliba, R.S.; Pangalos, M.; Moss, S.J. The ubiquitin-like protein Plic-1 enhances the membrane insertion of GABAA receptors by increasing their stability within the endoplasmic reticulum. J. Biol. Chem. 2008, 283, 18538–18544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, F.K.; Kittler, J.T.; Muller, E.; Thomas, P.; Uren, J.M.; Merlo, D.; Wisden, W.; Triller, A.; Smart, T.G.; Moss, S.J. GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat. Neurosci. 2001, 4, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Natunen, T.; Takalo, M.; Kemppainen, S.; Leskelä, S.; Marttinen, M.; Kurkinen, K.M.A.; Pursiheimo, J.P.; Sarajärvi, T.; Viswanathan, J.; Gabbouj, S.; et al. Relationship between ubiquilin-1 and BACE1 in human Alzheimer’s disease and APdE9 transgenic mouse brain and cell-based models. Neurobiol. Dis. 2016, 85, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Saunders, A.J. An emerging role for Ubiquilin 1 in regulating protein quality control system and in disease pathogenesis. Discov. Med. 2009, 8, 18–22. [Google Scholar]
- Safren, N.; Chang, L.; Dziki, K.M.; Monteiro, M.J. Signature changes in ubiquilin expression in the R6/2 mouse model of Huntington’s disease. Brain Res. 2015, 1597, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Liu, Y.; Tu, X.; Ren, X.; Zhao, W.; Liu, J.; Zhang, L.; Chen, W.; Zhang, P.; Wang, W.; et al. Decreased expression of ubiquilin-1 following neonatal hypoxia-ischemic brain injury in mice. Mol. Med. Rep. 2019, 19, 4597–4602. [Google Scholar] [CrossRef]
- Liu, Y.; Feng, S.; Subedi, K.; Wang, H. Attenuation of Ischemic Stroke-Caused Brain Injury by a Monoamine Oxidase Inhibitor Involves Improved Proteostasis and Reduced Neuroinflammation. Mol. Neurobiol. 2020, 57, 937–948. [Google Scholar] [CrossRef]
- Ihbe, N.; Le Prieult, F.; Wang, Q.; Distler, U.; Sielaff, M.; Tenzer, S.; Thal, S.C.; Mittmann, T. Adaptive Mechanisms of Somatostatin-Positive Interneurons after Traumatic Brain Injury through a Switch of α Subunits in L-Type Voltage-Gated Calcium Channels. Cereb. Cortex 2022, 32, 1093–1109. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gao, H.M.; Hong, J.S. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: Role of neuroinflammation. Environ. Health Perspect. 2003, 111, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.C.; Cheney, R.E. Myosins in cell junctions. Bioarchitecture 2012, 2, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridler, T.; Matthews, P.; Phillips, K.G.; Randall, A.D.; Brown, J.T. Initiation and slow propagation of epileptiform activity from ventral to dorsal medial entorhinal cortex is constrained by an inhibitory gradient. J. Physiol. 2018, 596, 2251–2266. [Google Scholar] [CrossRef] [Green Version]
- Gaiottino, J.; Norgren, N.; Dobson, R.; Topping, J.; Nissim, A.; Malaspina, A.; Bestwick, J.P.; Monsch, A.U.; Regeniter, A.; Lindberg, R.L.; et al. Increased neurofilament light chain blood levels in neurodegenerative neurological diseases. PLoS ONE 2013, 8, e75091. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, Z.; Lv, X.; Wu, Q.; Yan, J.; Mao, G.; Xing, W. Neurofilament light chain level in traumatic brain injury: A system review and meta-analysis. Medicine 2020, 99, e22363. [Google Scholar] [CrossRef]
- Shahim, P.; Politis, A.; van der Merwe, A.; Moore, B.; Chou, Y.Y.; Pham, D.L.; Butman, J.A.; Diaz-Arrastia, R.; Gill, J.M.; Brody, D.L.; et al. Neurofilament light as a biomarker in traumatic brain injury. Neurology 2020, 95, e610–e622. [Google Scholar] [CrossRef]
- Garland, P.; Morton, M.; Zolnourian, A.; Durnford, A.; Gaastra, B.; Toombs, J.; Heslegrave, A.J.; More, J.; Zetterberg, H.; Bulters, D.O.; et al. Neurofilament light predicts neurological outcome after subarachnoid haemorrhage. Brain J. Neurol. 2021, 144, 761–768. [Google Scholar] [CrossRef]
- Carron, S.F.; Yan, E.B.; Allitt, B.J.; Rajan, R. Immediate and Medium-term Changes in Cortical and Hippocampal Inhibitory Neuronal Populations after Diffuse TBI. Neuroscience 2018, 388, 152–170. [Google Scholar] [CrossRef]
- Schwarzer, C.; Tsunashima, K.; Wanzenböck, C.; Fuchs, K.; Sieghart, W.; Sperk, G. GABA(A) receptor subunits in the rat hippocampus II: Altered distribution in kainic acid-induced temporal lobe epilepsy. Neuroscience 1997, 80, 1001–1017. [Google Scholar] [CrossRef]
- Figueiredo, T.; Harbert, C.L.; Pidoplichko, V.; Almeida-Suhett, C.P.; Rossetti, K.; Braga, M.F.M.; Marini, A.M. The Recovery of GABAergic Function in the Hippocampus CA1 Region After mTBI. Mol. Neurobiol. 2020, 57, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Suhett, C.P.; Prager, E.M.; Pidoplichko, V.; Figueiredo, T.H.; Marini, A.M.; Li, Z.; Eiden, L.E.; Braga, M.F. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 2015, 273, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Ochoa, M.A.; Chiprés-Tinajero, G.A.; González-Domínguez, N.P.; Medina-Ceja, L. Causal relationship of CA3 back-projection to the dentate gyrus and its role in CA1 fast ripple generation. BMC Neurosci. 2021, 22, 37. [Google Scholar] [CrossRef]
- Gibbs, J.W., 3rd; Shumate, M.D.; Coulter, D.A. Differential epilepsy-associated alterations in postsynaptic GABA(A) receptor function in dentate granule and CA1 neurons. J. Neurophysiol. 1997, 77, 1924–1938. [Google Scholar] [CrossRef] [Green Version]
- Shulman, K.I.; Herrmann, N.; Walker, S.E. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs 2013, 27, 789–797. [Google Scholar] [CrossRef]
- Kaludercic, N.; Carpi, A.; Menabò, R.; Di Lisa, F.; Paolocci, N. Monoamine oxidases (MAO) in the pathogenesis of heart failure and ischemia/reperfusion injury. Biochim. Biophys. Acta 2011, 1813, 1323–1332. [Google Scholar] [CrossRef] [Green Version]
- Baker, G.; Matveychuk, D.; MacKenzie, E.M.; Holt, A.; Wang, Y.; Kar, S. Attenuation of the effects of oxidative stress by the MAO-inhibiting antidepressant and carbonyl scavenger phenelzine. Chem. Biol. Interact. 2019, 304, 139–147. [Google Scholar] [CrossRef]
- Aguiar, C.C.; Almeida, A.B.; Araújo, P.V.; de Abreu, R.N.; Chaves, E.M.; do Vale, O.C.; Macêdo, D.S.; Woods, D.J.; Fonteles, M.M.; Vasconcelos, S.M. Oxidative stress and epilepsy: Literature review. Oxid. Med. Cell. Longev. 2012, 2012, 795259. [Google Scholar] [CrossRef] [Green Version]
- Cebak, J.E.; Singh, I.N.; Hill, R.L.; Wang, J.A.; Hall, E.D. Phenelzine Protects Brain Mitochondrial Function In Vitro and In Vivo following Traumatic Brain Injury by Scavenging the Reactive Carbonyls 4-Hydroxynonenal and Acrolein Leading to Cortical Histological Neuroprotection. J. Neurotrauma 2017, 34, 1302–1317. [Google Scholar] [CrossRef] [Green Version]
- Culpepper, L. The use of monoamine oxidase inhibitors in primary care. J. Clin. Psychiatry 2012, 73 (Suppl. 1), 37–41. [Google Scholar] [CrossRef] [PubMed]
- Gillman, P.K.; Feinberg, S.S.; Fochtmann, L.J. Revitalizing monoamine oxidase inhibitors: A call for action. CNS Spectr. 2020, 25, 452–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, A.M.; Starr, M.S. Dopaminergic modulation of pilocarpine-induced motor seizures in the rat: The role of hippocampal D2 receptors. Neuroscience 1993, 53, 425–431. [Google Scholar] [CrossRef]
- Clinckers, R.; Smolders, I.; Meurs, A.; Ebinger, G.; Michotte, Y. Anticonvulsant action of hippocampal dopamine and serotonin is independently mediated by D and 5-HT receptors. J. Neurochem. 2004, 89, 834–843. [Google Scholar] [CrossRef]
- Bagdy, G.; Kecskemeti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef]
- Foreman, B.; Lee, H.; Okonkwo, D.O.; Strong, A.J.; Pahl, C.; Shutter, L.A.; Dreier, J.P.; Ngwenya, L.B.; Hartings, J.A. The Relationship between Seizures and Spreading Depolarizations in Patients with Severe Traumatic Brain Injury. Neurocrit. Care 2022, 1–18. [Google Scholar] [CrossRef]
- Cozzolino, O.; Marchese, M.; Trovato, F.; Pracucci, E.; Ratto, G.M.; Buzzi, M.G.; Sicca, F.; Santorelli, F.M. Understanding Spreading Depression from Headache to Sudden Unexpected Death. Front. Neurol. 2018, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Hartings, J.A.; Andaluz, N.; Bullock, M.R.; Hinzman, J.M.; Mathern, B.; Pahl, C.; Puccio, A.; Shutter, L.A.; Strong, A.J.; Vagal, A.; et al. Prognostic Value of Spreading Depolarizations in Patients With Severe Traumatic Brain Injury. JAMA Neurol. 2020, 77, 489–499. [Google Scholar] [CrossRef]
- Tamamaki, N.; Yanagawa, Y.; Tomioka, R.; Miyazaki, J.; Obata, K.; Kaneko, T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 2003, 467, 60–79. [Google Scholar] [CrossRef]
- Huusko, N.; Romer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkanen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef]
- Olsen, R.W. Allosteric ligands and their binding sites define γ-aminobutyric acid (GABA) type A receptor subtypes. Adv. Pharmacol. 2015, 73, 167–202. [Google Scholar] [PubMed]
- Butler, J.L.; Paulsen, O. Hippocampal network oscillations—Recent insights from in vitro experiments. Curr. Opin. Neurobiol. 2015, 31, 40–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, F.E.; Sutula, T.P. Epileptogenesis in the dentate gyrus: A critical perspective. Prog. Brain Res. 2007, 163, 755–773. [Google Scholar] [PubMed]
- Sielaff, M.; Kuharev, J.; Bohn, T.; Hahlbrock, J.; Bopp, T.; Tenzer, S.; Distler, U. Evaluation of FASP, SP3, and iST Protocols for Proteomic Sample Preparation in the Low Microgram Range. J. Proteome Res. 2017, 16, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Kuharev, J.; Navarro, P.; Levin, Y.; Schild, H.; Tenzer, S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2014, 11, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Kuharev, J.; Navarro, P.; Tenzer, S. Label-free quantification in ion mobility-enhanced data-independent acquisition proteomics. Nat. Protoc. 2016, 11, 795–812. [Google Scholar] [CrossRef]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition* S. Mol. Cell. Proteom. 2006, 5, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Nemes, A.; Najm, I.M.; Gale, J.T.; Ying, Z.; Johnson, M.; Gonzalez-Martinez, J. Underlying Cortical Dysplasia as Risk Factor for Traumatic Epilepsy: An Animal Study. J. Neurotrauma 2016, 33, 1883–1891. [Google Scholar] [CrossRef]
- Sakakura, K.; Fujimoto, A.; Arai, Y.; Ichikawa, N.; Sato, K.; Baba, S.; Inenaga, C.; Matsumura, A.; Ishikawa, E.; Enoki, H.; et al. Posttraumatic epilepsy may be a state in which underlying epileptogenicity involves focal cortical dysplasia. Epilepsy Behav. 2021, 114, 107352. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kürten, T.; Ihbe, N.; Ueberbach, T.; Distler, U.; Sielaff, M.; Tenzer, S.; Mittmann, T. GABAA Receptor-Stabilizing Protein Ubqln1 Affects Hyperexcitability and Epileptogenesis after Traumatic Brain Injury and in a Model of In Vitro Epilepsy in Mice. Int. J. Mol. Sci. 2022, 23, 3902. https://doi.org/10.3390/ijms23073902
Kürten T, Ihbe N, Ueberbach T, Distler U, Sielaff M, Tenzer S, Mittmann T. GABAA Receptor-Stabilizing Protein Ubqln1 Affects Hyperexcitability and Epileptogenesis after Traumatic Brain Injury and in a Model of In Vitro Epilepsy in Mice. International Journal of Molecular Sciences. 2022; 23(7):3902. https://doi.org/10.3390/ijms23073902
Chicago/Turabian StyleKürten, Tabea, Natascha Ihbe, Timo Ueberbach, Ute Distler, Malte Sielaff, Stefan Tenzer, and Thomas Mittmann. 2022. "GABAA Receptor-Stabilizing Protein Ubqln1 Affects Hyperexcitability and Epileptogenesis after Traumatic Brain Injury and in a Model of In Vitro Epilepsy in Mice" International Journal of Molecular Sciences 23, no. 7: 3902. https://doi.org/10.3390/ijms23073902
APA StyleKürten, T., Ihbe, N., Ueberbach, T., Distler, U., Sielaff, M., Tenzer, S., & Mittmann, T. (2022). GABAA Receptor-Stabilizing Protein Ubqln1 Affects Hyperexcitability and Epileptogenesis after Traumatic Brain Injury and in a Model of In Vitro Epilepsy in Mice. International Journal of Molecular Sciences, 23(7), 3902. https://doi.org/10.3390/ijms23073902