
LPS Response Is Impaired by Urban Fine Particulate Matter

 , ,
, , 
 and
and
Abstract
:1. Introduction
2. Results
2.1. Exposure and PM2.5 Characterization
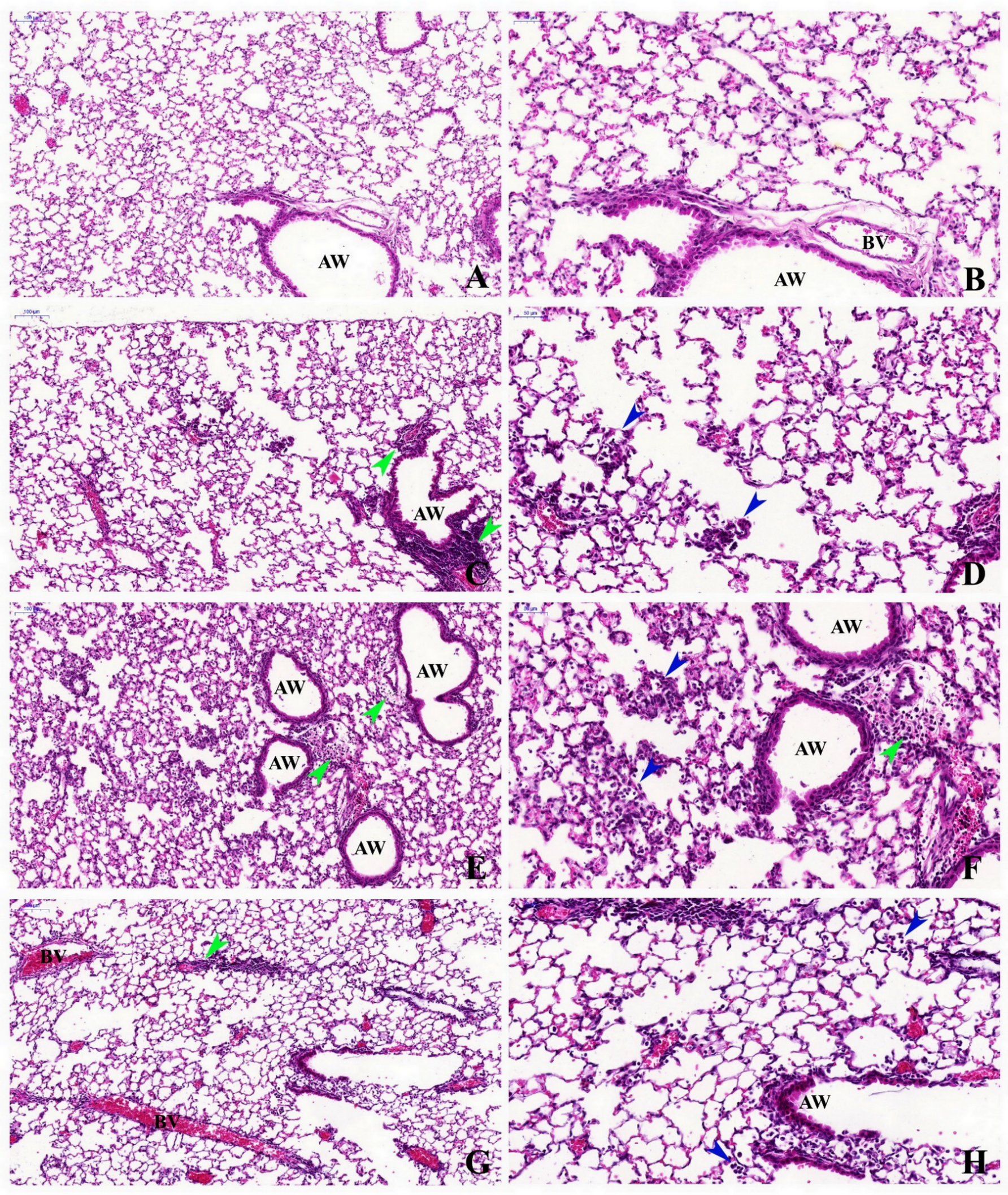
2.2. Histopathological Description
2.3. Stereological Analysis
2.4. Inflammatory Cells Assessment
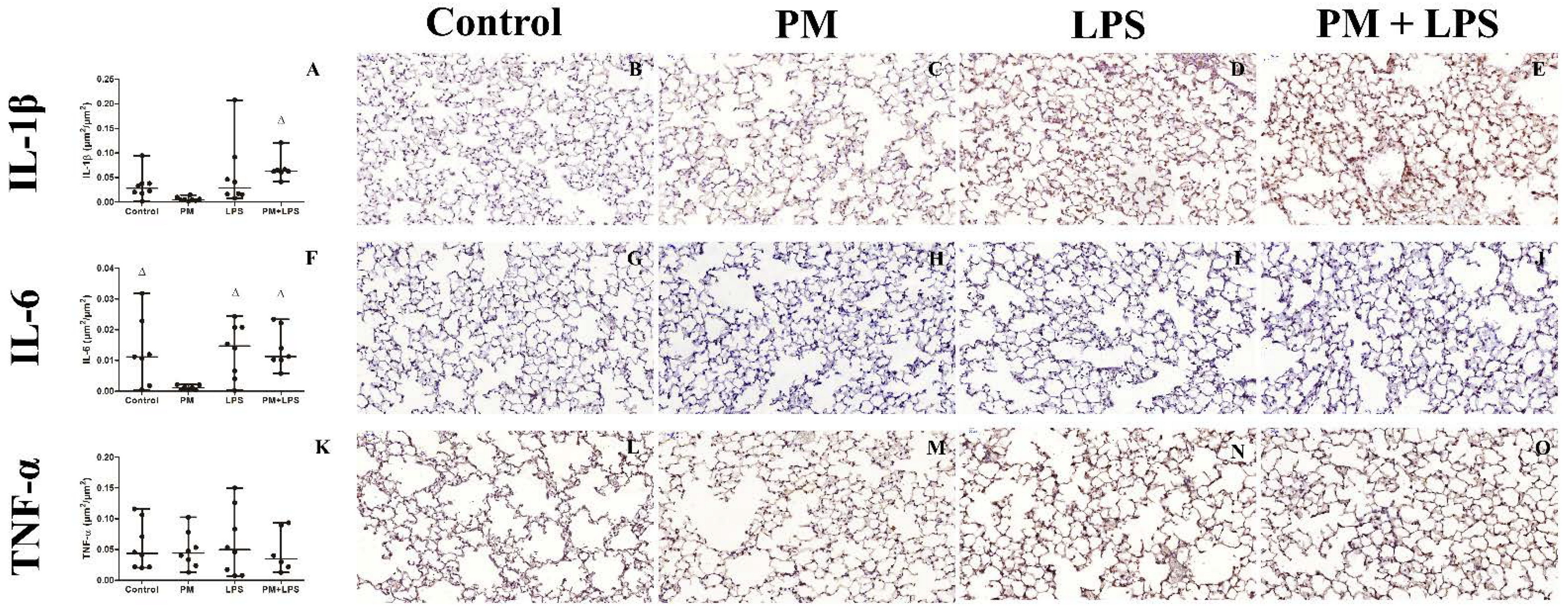
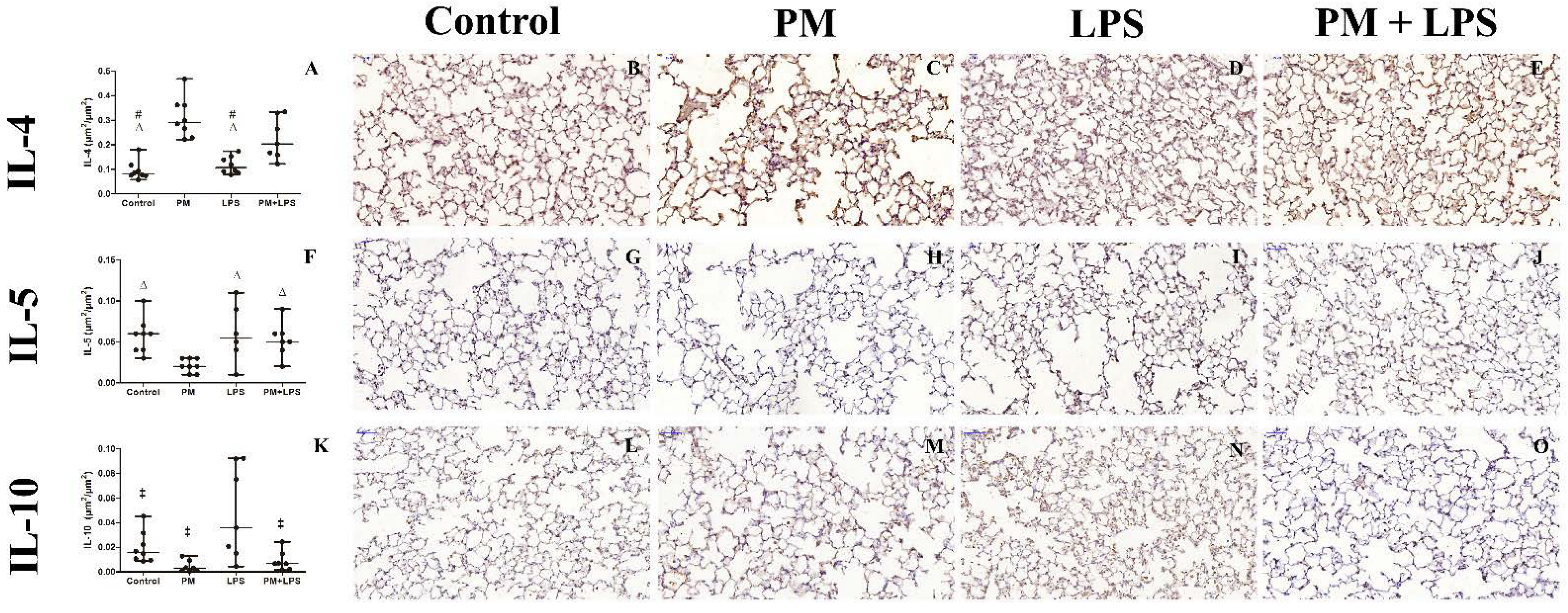
2.5. Inflammatory Cytokines
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. PM2.5 Elementary Characterization
4.3. Blood and Bronchoalveolar Lavage Fluid (BALF) Analysis
4.4. Stereological Analysis and Descriptive Analysis
4.5. Molecular Analysis
4.6. Immunohistochemically Inflammation Assessment
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schraufnagel, D.E.; Balmes, J.R.; Cowl, C.T.; De Matteis, S.; Jung, S.H.; Mortimer, K.; Perez-Padilla, R.; Rice, M.B.; Riojas-Rodriguez, H.; Sood, A.; et al. Air Pollution and Noncommunicable Diseases: A Review by the Forum of International Respiratory Societies’ Environmental Committee, Part 1: The Damaging Effects of Air Pollution. Chest 2019, 155, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Global Air Quality Guidelines. Particulate Matter (PM2.5 and PM10), Ozone, Nitrogen Dioxide, Sulfur Dioxide, and Carbon Monoxide. Available online: https://www.who.int/publications/i/item/9789240034228?ua=1 (accessed on 27 November 2021).
- Forbes, L.J.; Kapetanakis, V.; Rudnicka, A.R.; Cook, D.G.; Bush, T.; Stedman, J.R.; Whincup, P.H.; Strachan, D.P.; Anderson, H.R. Chronic exposure to outdoor air pollution and lung function in adults. Thorax 2009, 64, 657–663. [Google Scholar] [CrossRef] [Green Version]
- Reilly, J.P.; Zhao, Z.; Shashaty, M.G.S.; Koyama, T.; Christie, J.D.; Lanken, P.N.; Wang, C.; Balmes, J.R.; Matthay, M.A.; Calfee, C.S.; et al. Low to Moderate Air Pollutant Exposure and Acute Respiratory Distress Syndrome after Severe Trauma. Am. J. Respir. Crit. Care Med. 2019, 199, 62–70. [Google Scholar] [CrossRef]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Pérez, R.D.; Taborda, N.A.; Gómez, D.M.; Narvaez, J.F.; Porras, J.; Hernandez, J.C. Inflammatory effects of particulate matter air pollution. Environ. Sci. Pollut. Res. 2020, 27, 42390–42404. [Google Scholar] [CrossRef] [PubMed]
- Gałuszka, A.; Stec, M.; Węglarczyk, K.; Kluczewska, A.; Siedlar, M.; Baran, J. Transition Metal Containing Particulate Matter Promotes Th1 and Th17 Inflammatory Response by Monocyte Activation in Organic and Inorganic Compounds Dependent Manner. Int. J. Environ. Res. Public Health 2020, 17, 1227. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef]
- Ebbensgaard, A.; Mordhorst, H.; Aarestrup, F.M.; Hansen, E.B. The Role of Outer Membrane Proteins and Lipopolysaccharides for the Sensitivity of Escherichia coli to Antimicrobial Peptides. Front. Microbiol. 2018, 9, 2153. [Google Scholar] [CrossRef] [Green Version]
- Klein, G.; Müller-Loennies, S.; Lindner, B.; Kobylak, N.; Brade, H.; Raina, S. Molecular and structural basis of inner core lip-opolysaccharide alterations in Escherichia coli: Incorporation of glucuronic acid and phosphoethanolamine in the heptose re-gion. J. Biol. Chem. 2013, 288, 8111–8127. [Google Scholar] [CrossRef] [Green Version]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef]
- Yanagisawa, R.; Takano, H.; Inoue, K.; Ichinose, T.; Sadakane, K.; Yoshino, S.; Yamaki, K.; Kumagai, Y.; Uchiyama, K.; Yoshikawa, T.; et al. Enhancement of acute lung injury related to bacterial endotoxin by components of diesel exhaust particles. Thorax 2003, 58, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, H.; Yanagisawa, R.; Ichinose, T.; Sadakane, K.; Yoshino, S.; Yoshikawa, T.; Morita, M. Diesel exhaust particles enhance lung injury related to bacterial endotoxin through expression of proinflammatory cytokines, chemokines, and intercellular adhesion molecule-1. Am. J. Respir. Crit. Care Med. 2002, 165, 1329–1335. [Google Scholar] [CrossRef]
- Kim, D.I.; Song, M.K.; Lee, K. Diesel Exhaust Particulates Enhances Susceptibility of LPS-Induced Acute Lung Injury through Upregulation of the IL-17 Cytokine-Derived TGF-β1/Collagen I Expression and Activation of NLRP3 Inflammasome Signaling in Mice. Biomolecules 2021, 11, 67. [Google Scholar] [CrossRef]
- Andrade, M.D.; de Miranda, R.M.; Fornaro, A.; Kerr, A.; Oyama, B.; de Andre, P.A.; Saldiva, P. Vehicle emissions and PM(2.5) mass concentrations in six Brazilian cities. Air Qual. Atmos. Health 2012, 5, 79–88. [Google Scholar] [CrossRef] [Green Version]
- de Miranda, R.M.; de Fatima Andrade, M.; Fornaro, A.; Astolfo, R.; de Andre, P.A.; Saldiva, P. Urban air pollution: A representative survey of PM(2.5) mass concentrations in six Brazilian cities. Air Qual. Atmos. Health 2012, 5, 63–77. [Google Scholar] [CrossRef] [Green Version]
- de Barros Mendes Lopes, T.; Groth, E.E.; Veras, M.; Furuya, T.K.; de Souza Xavier Costa, N.; Ribeiro Júnior, G.; Lopes, F.D.; de Almeida, F.M.; Cardoso, W.V.; Saldiva, P.H.N.; et al. Pre- and postnatal exposure of mice to concentrated urban PM2.5 decreases the number of alveoli and leads to altered lung function at an early stage of life. Environ. Pollut. 2018, 241, 511–520. [Google Scholar] [CrossRef]
- Yoshizaki, K.; Fuziwara, C.S.; Brito, J.M.; Santos, T.M.N.; Kimura, E.T.; Correia, A.T.; Amato-Lourenco, L.F.; Vasconcellos, P.; Silva, L.F.; Brentani, M.M.; et al. The effects of urban particulate matter on the nasal epithelium by gender: An experimental study in mice. Environ. Pollut. 2016, 213, 359–369. [Google Scholar] [CrossRef]
- de Souza Xavier Costa, N.; Ribeiro Júnior, G.; Dos Santos Alemany, A.A.; Belotti, L.; Schalch, A.S.; Cavalcante, M.F.; Ribeiro, S.; Veras, M.M.; Kallás, E.G.; Saldiva, P.H.N.; et al. Air pollution impairs recovery and tissue remodeling in a murine model of acute lung injury. Sci. Rep. 2020, 10, 15314. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Frevert, C.W.; Martin, T.R. Animal models of acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L379–L399. [Google Scholar] [CrossRef] [Green Version]
- Park, B.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, X.; Dou, M.; He, H.; Ju, M.; Ji, S.; Zhou, J.; Chen, C.; Zhang, D.; Miao, C.; et al. Particulate matter disrupts airway epithelial barrier via oxidative stress to promote Pseudomonas aeruginosa infection. J. Thorac. Dis. 2019, 11, 2617–2627. [Google Scholar] [CrossRef]
- Sahuquillo-Arce, J.M.; Ibáñez-Martínez, E.; Hernández-Cabezas, A.; Ruiz-Gaitán, A.; Falomir-Salcedo, P.; Menéndez, R.; López-Hontangas, J.L. Influence of environmental conditions and pollution on the incidence of Streptococcus pneumoniae infections. ERJ Open Res. 2017, 3, 00014–02017. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.J.; Lin, H.C.; Yang, Y.F.; Chen, C.Y.; Ling, M.P.; Chen, S.C.; Chen, W.Y.; You, S.H.; Lu, T.H.; Liao, C.M. Association Between Ambient Air Pollution and Elevated Risk of Tuberculosis Development. Infect. Drug Resist. 2019, 12, 3835–3847. [Google Scholar] [CrossRef] [Green Version]
- Domingo, J.L.; Rovira, J. Effects of air pollutants on the transmission and severity of respiratory viral infections. Environ. Res. 2020, 187, 109650. [Google Scholar] [CrossRef]
- Renwick, L.C.; Brown, D.; Clouter, A.; Donaldson, K. Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types. Occup. Environ. Med. 2004, 61, 442–447. [Google Scholar] [CrossRef]
- Becker, S.; Soukup, J.M.; Sioutas, C.; Cassee, F.R. Response of human alveolar macrophages to ultrafine, fine, and coarse urban air pollution particles. Exp. Lung Res. 2003, 29, 29–44. [Google Scholar] [CrossRef]
- Miyata, R.; van Eeden, S.F. The innate and adaptive immune response induced by alveolar macrophages exposed to ambient particulate matter. Toxicol. Appl. Pharmacol. 2011, 257, 209–226. [Google Scholar] [CrossRef]
- Yang, J.; Kim, E.K.; Park, H.J.; McDowell, A.; Kim, Y.K. The impact of bacteria-derived ultrafine dust particles on pulmonary diseases. Exp. Mol. Med. 2020, 52, 338–347. [Google Scholar] [CrossRef]
- Gurczynski, S.J.; Moore, B.B. IL-17 in the lung: The good, the bad, and the ugly. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L6–L16. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.G.; Santos, J.M.B.; Foster, R.; Rossi, M.; Luna Junior, L.A.; Katekaru, C.M.; de Sá, M.C.; Jonckheere, A.C.; Almeida, F.M.; Amaral, J.B.; et al. The Effect of Particulate Matter Exposure on the Inflammatory Airway Response of Street Runners and Sedentary People. Atmosphere 2020, 11, 43. [Google Scholar] [CrossRef] [Green Version]
- van Voorhis, M.; Knopp, S.; Julliard, W.; Fechner, J.H.; Zhang, X.; Schauer, J.J.; Mezrich, J.D. Exposure to atmospheric particulate matter enhances Th17 polarization through the aryl hydrocarbon receptor. PLoS ONE 2013, 8, e82545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.; Zhong, X.; Zhang, J.; Lao, Q.; He, Z.; Bai, J. The expression of Foxp3 and ROR gamma t in lung tissues from normal smokers and chronic obstructive pulmonary disease patients. Int. Immunopharmacol. 2011, 11, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, K.; McDonald-Hyman, C.; Noth, E.M.; Pratt, B.; Hammond, S.K.; Balmes, J.; Tager, I. Ambient air pollution impairs regulatory T-cell function in asthma. J. Allergy Clin. Immunol. 2010, 126, 845–852. [Google Scholar] [CrossRef]
- Nomura, F.; Akashi, S.; Sakao, Y.; Sato, S.; Kawai, T.; Matsumoto, M.; Nakanishi, K.; Kimoto, M.; Miyake, K.; Takeda, K.; et al. Cutting edge: Endotoxin tolerance in mouse peritoneal macrophages correlates with down-regulation of surface toll-like receptor 4 expression. J. Immunol. 2000, 164, 3476–3479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Beltrán, S.; Torres, M.; Arellano, M.; Juárez, E. Human macrophages chronically exposed to LPS can be reactivated by stimulation with MDP to acquire an antimicrobial phenotype. Cell Immunol. 2017, 315, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.P.; Burton, D.S.; Nagel, D.J.; Bhattacharya, S.; Falsey, A.R.; Georas, S.N.; Hopke, P.K.; Johnston, C.J.; Kottmann, R.M.; Litonjua, A.A.; et al. The effect of air pollution on the transcriptomics of the immune response to respiratory infection. Sci. Rep. 2021, 11, 19436. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.F.; Kumar, P.; Freitas, E.D.; Ynoue, R.Y.; Martins, J.; Martins, L.D.; Nogueira, T.; Perez-Martinez, P.; Miranda, R.M.; Albuquerque, T.; et al. Air quality in the megacity of São Paulo: Evolution over the last 30 years and future perspectives. Atmos. Environ. 2017, 159, 66–82. [Google Scholar] [CrossRef] [Green Version]
- CETESB. Qualidade do ar no Estado de São Paulo 2020. Available online: https://cetesb.sp.gov.br/ar/wp-content/uploads/sites/28/2021/05/Relatorio-de-Qualidade-do-Ar-no-Estado-de-Sao-Paulo-2020.pdf (accessed on 27 November 2021).
- Phalen, R.F.; Oldham, M.J.; Wolff, R.K. The relevance of animal models for aerosol studies. J. Aerosol Med. Pulm. Drug Deliv. 2008, 21, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Sioutas, C.; Koutrakis, P.; Burton, R.M. A technique to expose animals to concentrated fine ambient aerosols. Environ. Health Perspect. 1995, 103, 172–177. [Google Scholar] [CrossRef]
- de Souza Xavier Costa, N.; Ribeiro Júnior, G.; Dos Santos Alemany, A.A.; Belotti, L.; Zati, D.H.; Frota Cavalcante, M.; Matera Veras, M.; Ribeiro, S.; Kallás, E.G.; Nascimento Saldiva, P.H.; et al. Early and late pulmonary effects of nebulized LPS in mice: An acute lung injury model. PLoS ONE 2017, 12, e0185474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, C.C.; Hyde, D.M.; Ochs, M.; Weibel, E.R. An official research policy statement of the American Thoracic Society/European Respiratory Society: Standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 2010, 181, 394–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyerholz, D.K.; Sieren, J.C.; Beck, A.P.; Flaherty, H.A. Approaches to evaluate lung inflammation in translational research. Vet. Pathol. 2018, 55, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Control | PM | LPS | PM+LPS | LPS Nebulization | PM2.5 Exposure | Interaction |
|---|---|---|---|---|---|---|---|
| Initial Body Weight (g) | 22 (3) | 23 (2) | 22 (2) | 24 (4) | ns | ns | ns |
| Final Body weight (g) | 24 (2) bc | 26.5 (3.5) bc | 21.5 (2) | 20 (4) | p ≤ 0.0001 | ns | p = 0.042 |
| Peribronchial inflammation score | 0 (1) bc | 1 (1) bc | 1.5 (1) | 1.5 (1) | p ≤ 0.0001 | ns | ns |
| Alveolar inflammatory infiltrate score | 0 (1) b | 0 (1) b | 2 (1) | 1 (1) | p ≤ 0.0001 | ns | ns |
| Lung volume (mm3) | 431.2 (145) | 486.1 (172) | 462.6 (55) | 470.4 (94) | ns | ns | ns |
| Lung volume per body weight ratio (mm3/g) | 18.8 (4.7) | 17.5 (8.4) | 20.9 (3.3) | 22.7 (3) a | p = 0.02 | ns | ns |
| Vv parenchyma (%) | 78.1 (10) | 78.5 (6) | 81.6 (8) | 77.6 (8) | ns | ns | ns |
| Vv septa (%) | 28.8 (7.4) | 32.8 (4.7) | 34.3 (3.3) | 34.5 (7.7) a | p = 0.031 | ns | ns |
| Vv alveolar airspace (%) | 49.3 (2.6) | 46.9 (5.8) c | 48.6 (6.1) | 44.3 (5.8) | ns | p = 0.01 | ns |
| Vv non-parenchyma (%) | 21.9 (15) | 21.5 (6) | 18.4 (8) | 22.4 (8) | ns | ns | ns |
| Vt parenchyma (mm3) | 332.9 (116.8) | 392.2 (162.6) | 368.4 (48.8) | 369.2 (50.9) | ns | ns | ns |
| Vt septa (mm3) | 125.4 (31.2) | 163.5 (61.1) a | 181.7 (35.2) | 129.9 (70.9) | ns | ns | p = 0.002 |
| Vt alveolar airspace (mm3) | 199.4 (73.9) | 230.2 (110) c | 264.9 (71.7) | 183.6 (63.2) | ns | ns | p = 0.004 |
| Vt non-parenchyma (mm3) | 80.4 (65.5) | 105.9 (47.9) | 76.5 (40.15) | 102 (41.3) | ns | ns | ns |
| Sv septa (mm−1) | 238.9 (53.6) | 211 (47.1) | 199.8 (27.8) a | 204.1 (16.9) a | p = 0.01 | ns | ns |
| St septa (103 mm2) | 94.6 (56.9) | 113.1 (52) | 89.8 (23.9) | 92.8 (23.6) | ns | ns | ns |
| Septal thickness (µm) | 2.6 (1) | 3.1 (1.2) | 3.7 (1.3) a | 3.3 (1.7) a | p = 0.004 | ns | ns |
| Control | PM | LPS | PM+LPS | LPS Nebulization | PM2.5 Exposure | Interaction | |
|---|---|---|---|---|---|---|---|
| KC | |||||||
| Serum (pg/mL) | 3.8 (1.3) b | 3.7 (2.4) b | 16.8 (9.7) | 7.5 (3) b | p ≤ 0.0001 | p = 0.007 | p = 0.013 |
| BALF (pg/mL) | 0.6 (2.5) bd | 0.9 (5.7) bd | 103.6 (24.3) | 60.7 (74.2) b | p ≤ 0.0001 | p = 0.03 | p = 0.018 |
| IL-1β | |||||||
| Serum (pg/mL) | >LD | >LD | 0 (0.04) | >LD | ns | ns | ns |
| BALF (pg/mL) | >LD bc | >LD bd | 23.7 (15.1) | 12.9 (7.5) | p ≤ 0.0001 | ns | ns |
| Tissue (µm2/µm2) | 0.02 (0.02) | 0.005 (0.005) d | 0.03 (0.06) | 0.06 (0.006) | p = 0.005 | ns | ns |
| IL-6 | |||||||
| Serum (pg/mL) | 0.1 (0.3) | 0.1 (0.2) b | 9.3 (7.9) | 7.7 (13.2) | p = 0.002 | ns | ns |
| BALF (pg/mL) | >LD bd | 0.01 (0.3) bd | 800.8 (613.5) | 325.3 (231.2) b | p ≤ 0.0001 | p ≤ 0.0001 | p ≤ 0.0001 |
| Tissue (µm2/µm2) | 0.01 (0.02) c | 0.001 (0.002) | 0.02 (0.02) c | 0.01 (0.01) c | p = 0.035 | ns | p = 0.044 |
| TNF | |||||||
| Serum (pg/mL) | 0.8 (1.2) b | 0.9 (1.7) b | 5.1 (3.4) | 2.3 (0.8) b | p = 0.001 | ns | p = 0.009 |
| BALF (pg/mL) | 0.01 (0.4) b | 0.1 (1) b | 2822.1 (1310.3) | 486.5 (379.8) b | p ≤ 0.0001 | p ≤ 0.0001 | p ≤ 0.0001 |
| Tissue (µm2/µm2) | 0.04 (0.07) | 0.04 (0.05) | 0.05 (0.1) | 0.03 (0.07) | ns | ns | ns |
| IL-4 | |||||||
| Serum (pg/mL) | >LD | >LD | >LD | >LD | ns | ns | ns |
| BALF (pg/mL) | >LD | >LD | >LD | >LD | ns | ns | ns |
| Tissue (µm2/µm2) | 0.08 (0.03) cd | 0.3 (0.1) | 0.1 (0.06) cd | 0.2 (0.2) | ns | p ≤ 0.0001 | p = 0.027 |
| IL-5 | |||||||
| Serum (pg/mL) | 0.3 (0.5) | 0.8 (0.8) | >LD c | >LD c | p ≤ 0.0001 | ns | ns |
| BALF (pg/mL) | >LD | >LD | >LD | >LD | ns | ns | ns |
| Tissue (µm2/µm2) | 0.05 (0.06) c | 0.02 (0.01) | 0.06 (0.06) c | 0.05 (0.02) | ns | p = 0.015 | ns |
| IL-10 | |||||||
| Serum (pg/mL) | 0.4 (1.5) | 0.1 (0.7) | 1.1 (2.3) | 0 (0.1) | ns | ns | ns |
| BALF (pg/mL) | 0 (0.9) | 0 (0.3) | 1.1 (2.9) | 0 (0.4) | ns | ns | ns |
| Tissue (µm2/µm2) | 0.015 (0.02) | 0.003 (0.008) b | 0.035 (0.08) | 0.007 (0.01) b | p = 0.04 | p = 0.001 | ns |
| IL-17A | |||||||
| Serum (pg/mL) | 1.7 (0.4) | 1.8 (0.4) | 1.5 (0.5) | 1.6 (0.1) | ns | ns | ns |
| BALF (pg/mL) | 1.2 (0.1) b | 1.2 (0.2) b | 6.5 (9.3) | 3.9 (3.1) | p = 0.009 | ns | ns |
| Tissue (µm2/µm2) | 0.01 (0.01) c | 0.1 (0.08) c | 0.03 (0.01) | 0.04 (0.03) c | ns | p ≤ 0.0001 | p = 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza Xavier Costa, N.; Ribeiro Júnior, G.; dos Santos Alemany, A.A.; Belotti, L.; Frota Cavalcante, M.; Ribeiro, S.; Matera Veras, M.; Kallás, E.G.; Saldiva, P.H.N.; Dolhnikoff, M.; et al. LPS Response Is Impaired by Urban Fine Particulate Matter. Int. J. Mol. Sci. 2022, 23, 3913. https://doi.org/10.3390/ijms23073913
de Souza Xavier Costa N, Ribeiro Júnior G, dos Santos Alemany AA, Belotti L, Frota Cavalcante M, Ribeiro S, Matera Veras M, Kallás EG, Saldiva PHN, Dolhnikoff M, et al. LPS Response Is Impaired by Urban Fine Particulate Matter. International Journal of Molecular Sciences. 2022; 23(7):3913. https://doi.org/10.3390/ijms23073913
Chicago/Turabian Stylede Souza Xavier Costa, Natália, Gabriel Ribeiro Júnior, Adair Aparecida dos Santos Alemany, Luciano Belotti, Marcela Frota Cavalcante, Susan Ribeiro, Mariana Matera Veras, Esper Georges Kallás, Paulo Hilário Nascimento Saldiva, Marisa Dolhnikoff, and et al. 2022. "LPS Response Is Impaired by Urban Fine Particulate Matter" International Journal of Molecular Sciences 23, no. 7: 3913. https://doi.org/10.3390/ijms23073913
APA Stylede Souza Xavier Costa, N., Ribeiro Júnior, G., dos Santos Alemany, A. A., Belotti, L., Frota Cavalcante, M., Ribeiro, S., Matera Veras, M., Kallás, E. G., Saldiva, P. H. N., Dolhnikoff, M., & Ferraz da Silva, L. F. (2022). LPS Response Is Impaired by Urban Fine Particulate Matter. International Journal of Molecular Sciences, 23(7), 3913. https://doi.org/10.3390/ijms23073913