1. Introduction
Chimeric antigen receptor (CAR)-expressing T cell (CAR-T) therapy targeting CD19 has been introduced into clinic settings for the treatment of refractory B cell lymphomas [
1,
2]. While its novelty and impressive efficacy have thrust CAR-T into the scientific limelight, its serious side effects, including cytokine release syndrome and on-target off-tumour effects, represent a major drawback [
3,
4]. Consequently, a comprehensive CAR design framework and effective CAR-T cell control methods for minimising side effects are of the utmost necessity. However, information and evidence regarding the relationship between CAR structure and CAR-T cell activity remain scarce. Thus, research into the fine tuning of CAR-T cell activity is lagging behind.
The CAR protein consists of an antigen-recognising domain (ARD), a hinge domain (HD), a transmembrane domain (TMD) and an intracellular signal transduction domain (STD). Single-chain variable fragments (scFvs) and various components from immune-related signalling proteins have been used to construct CARs. We previously used a variety of CAR components to construct several CAR structural variants in an attempt to acquire insight into the relationship between CAR structure and CAR-T cell activity [
5,
6,
7]. However, these efforts were made through activity correlation analyses based on alterations in the primary structure (amino acid sequence) of the CAR protein. To date, analyses of the effects of post-translational modifications on CAR-T cell activity are lacking. Within their original proteins, these borrowed components undergo post-translational modifications, such as disulphide bonding and glycosylation, which are known to play an important role in their function [
8,
9,
10,
11,
12,
13]. It is therefore quite likely that these moieties also undergo post-translational modification as part of the CAR construct, which would in turn affect CAR expression or CAR-T cell function. However, the link between CAR post-translational modification and CAR-T cell activity remains elusive, as CAR design has been largely heuristic in nature.
Thus, in this study, we used first-generation CARs consisting of a mouse vascular endothelial growth factor receptor 2 (VEGFR2)-specific scFv tandemly linked to a CD28- or CD8α-derived HD/TMD and a CD3ζ-derived STD in order to investigate CAR post-translational modifications and their effects on CAR-T cell function.
3. Discussion
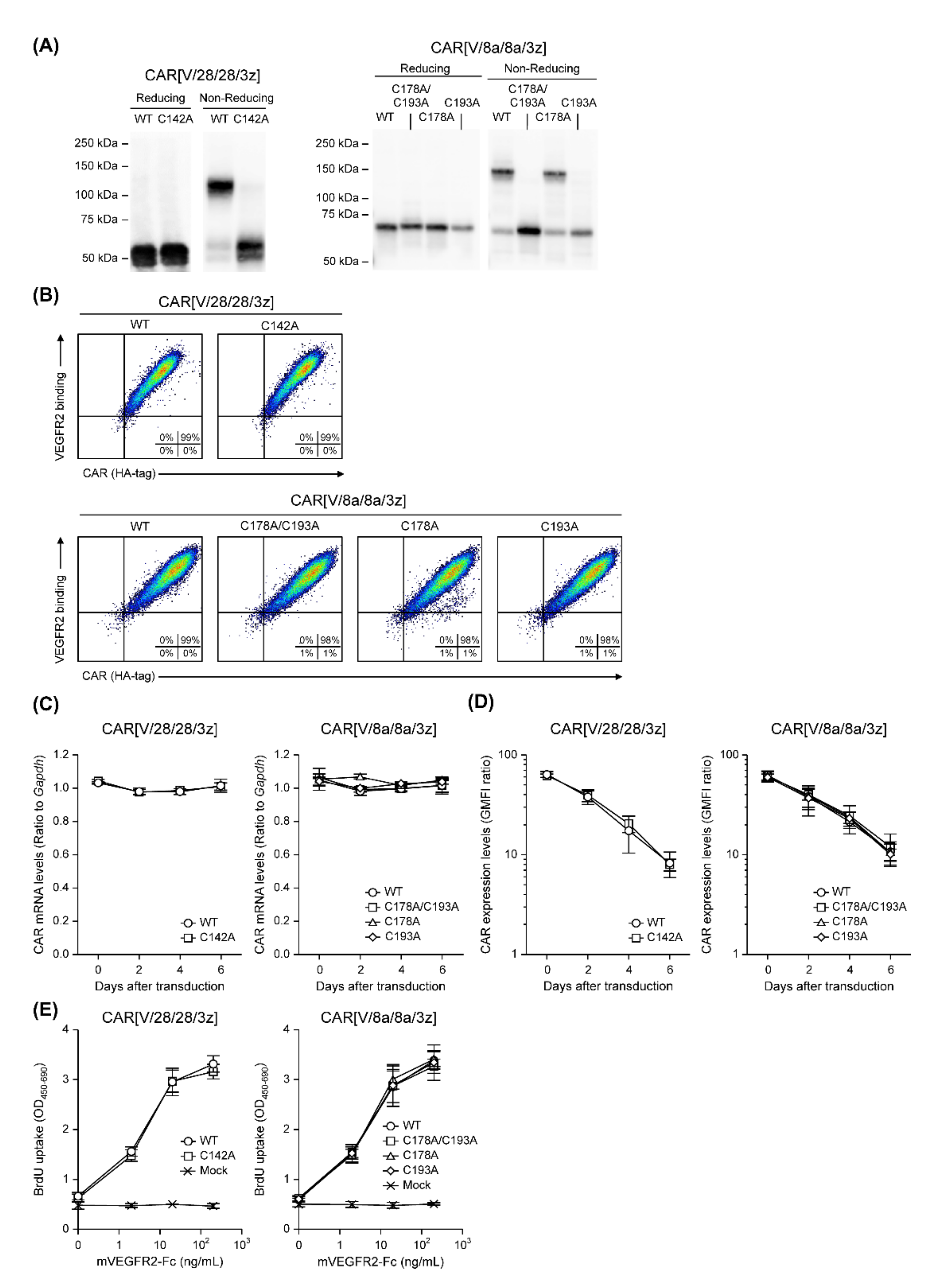
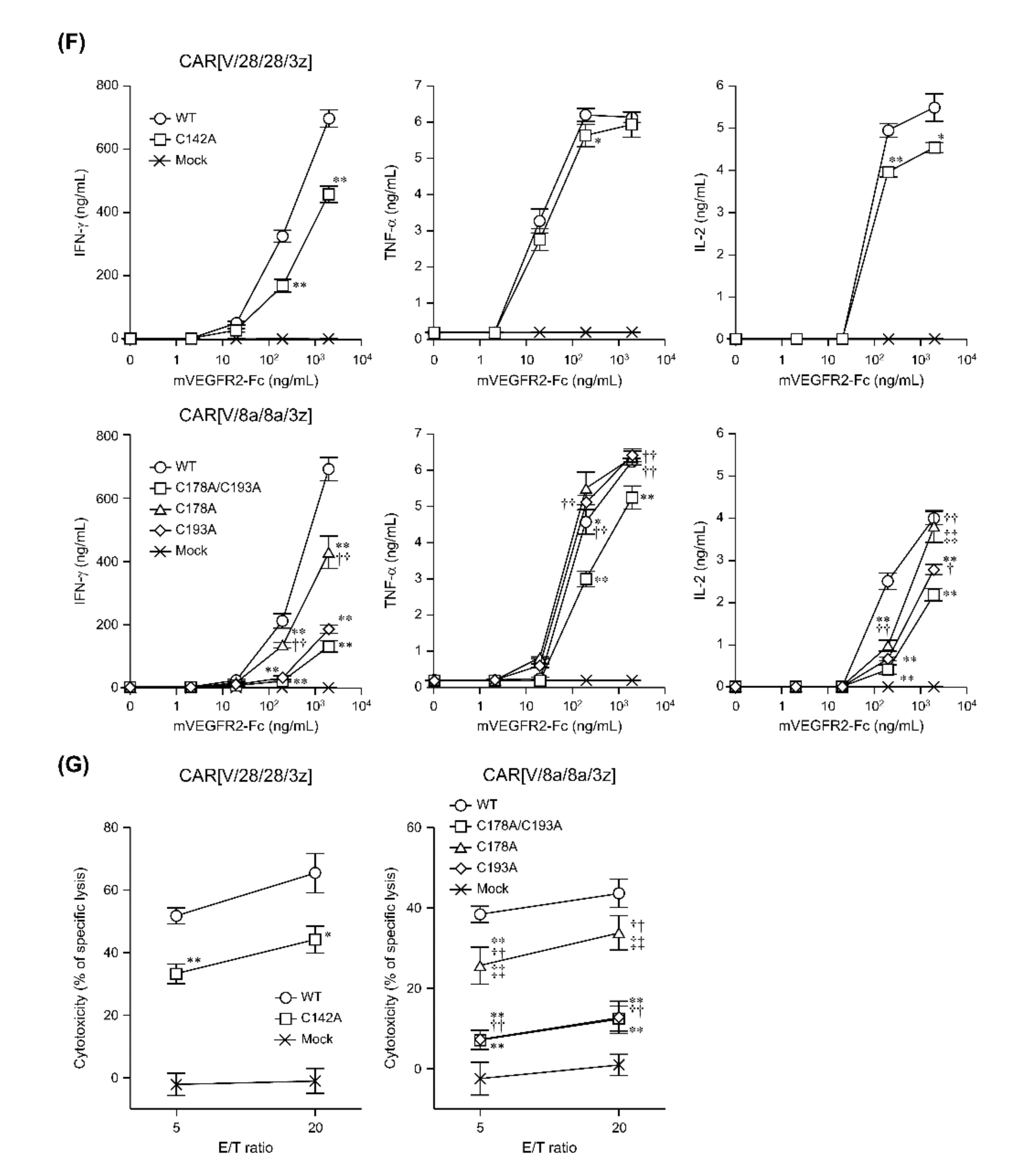
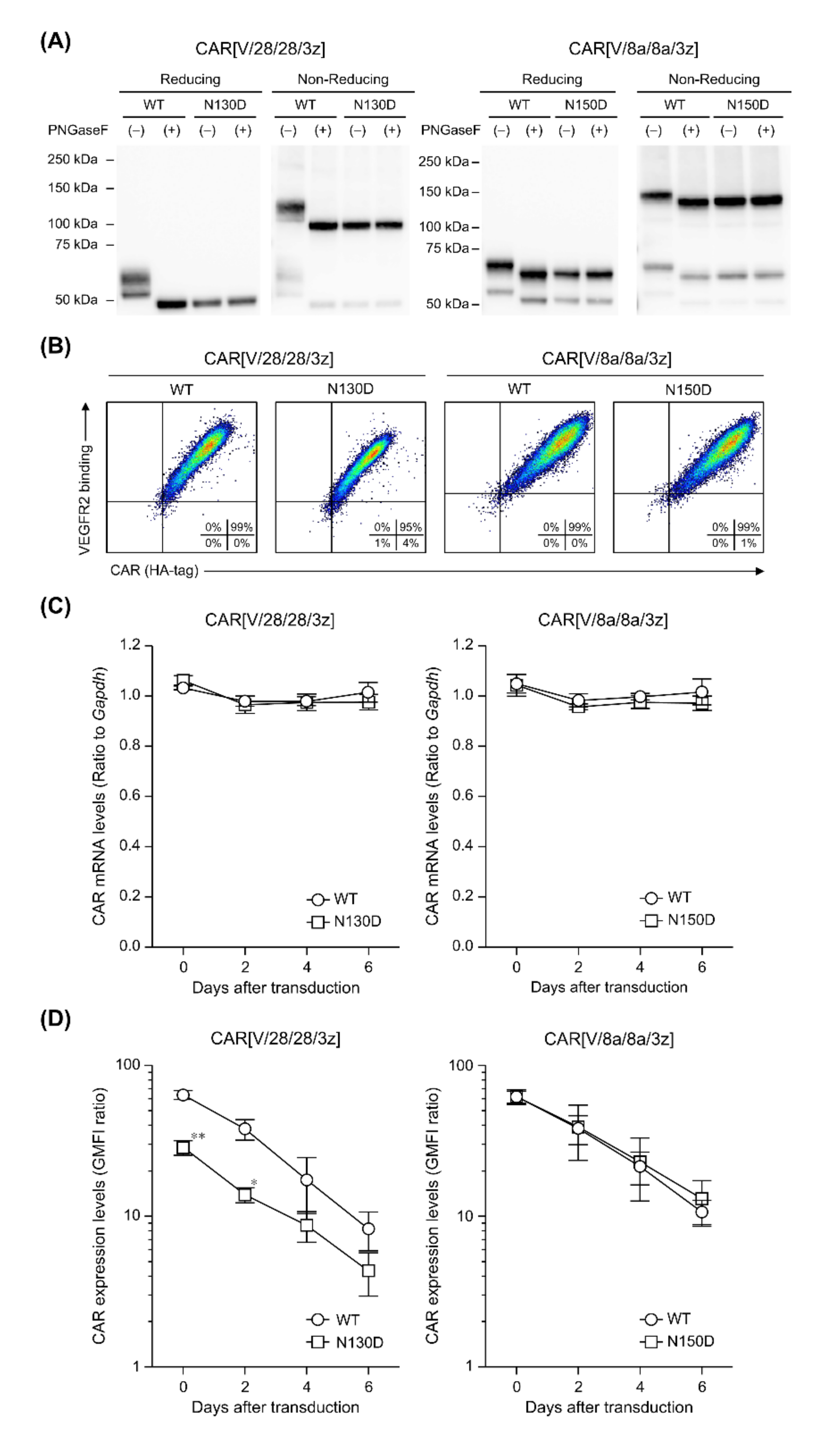
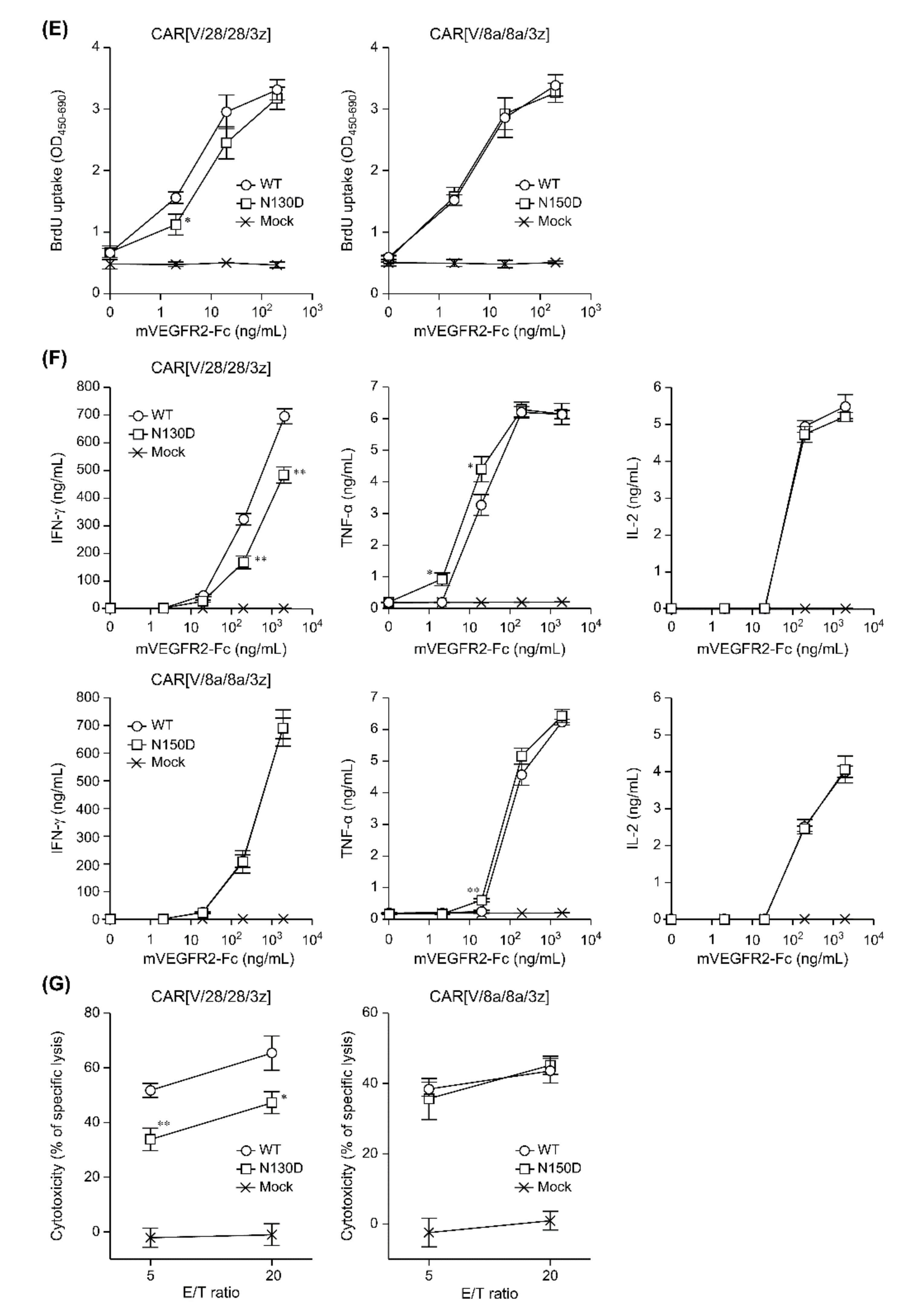
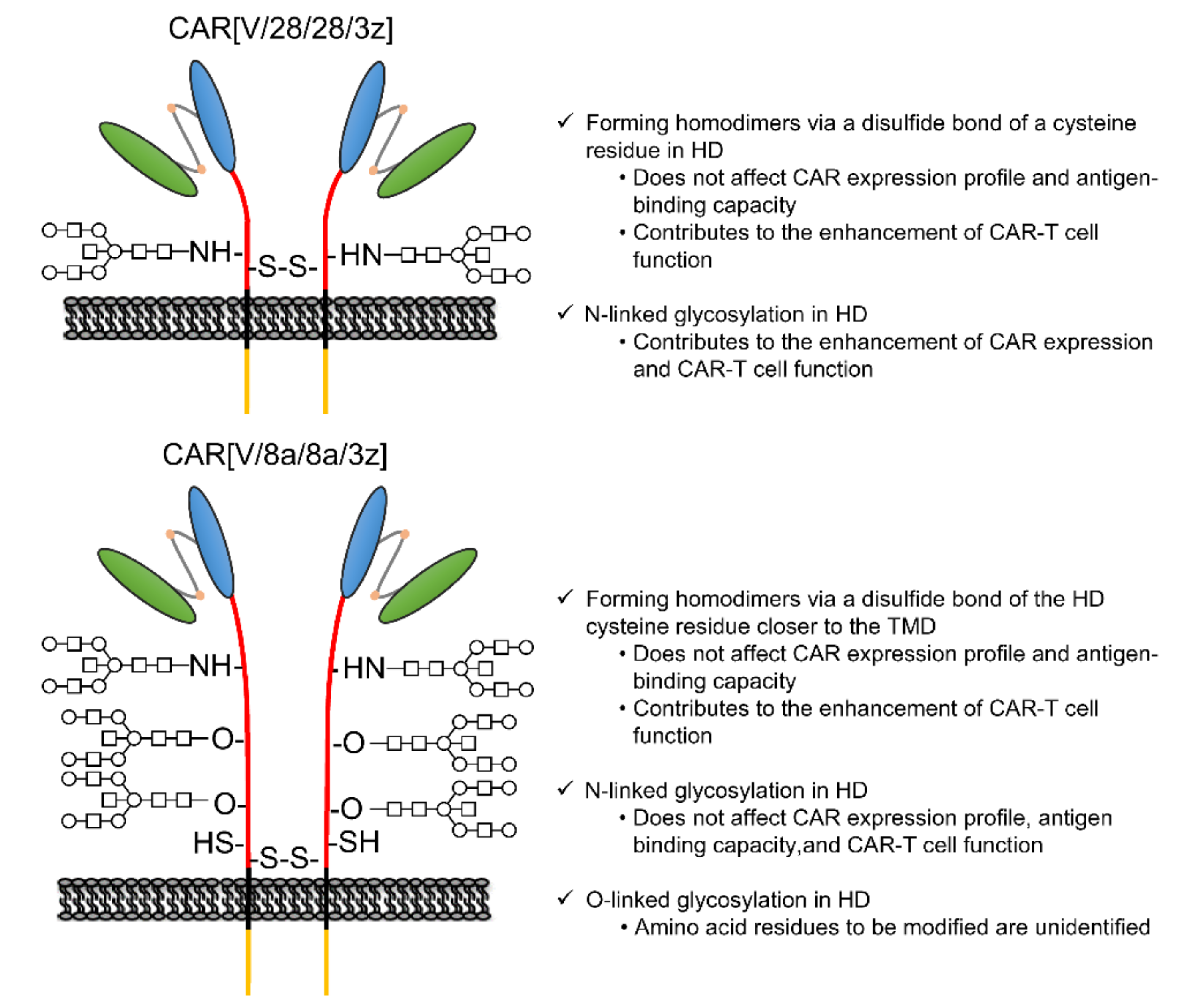
In this study, among the various components of the CAR protein, we focused on the HD in particular. Using two first-generation CARs, CAR[V/28/28/3z] and CAR[V/8a/8a/3z], as our models, we analysed the effects of disulphide bonding at cysteine residues and glycosylation on CAR-T cell activity. As summarised in
Figure 5, while homodimerisation of both CAR[V/28/28/3z] and CAR[V/8a/8a/3z] via HD cysteine residues did not affect CAR expression levels or avidity, it clearly contributed to antigen-specific CAR-T cell function. We are not the only group to report that selection of different HD/TMD components can enable the control of CAR-T cell activity without affecting its expression levels [
6,
14]. CAR is also known to form immune synapses on T cell membranes [
15]. Further, CARs with CD28-derived HD/TMD can exert rapid cytotoxic activity by forming such immune synapses more efficiently than CARs with CD8α-derived HD/TMD [
16]. Taken together with the current findings, we can surmise that CAR dimerisation via HD disulphide bonding can alter the activity of CAR-T cells without changing their expression levels. This is due to the effect on antigen-recognition-dependent clustering of CARs at the cell membrane and subsequent immune synapse formation.
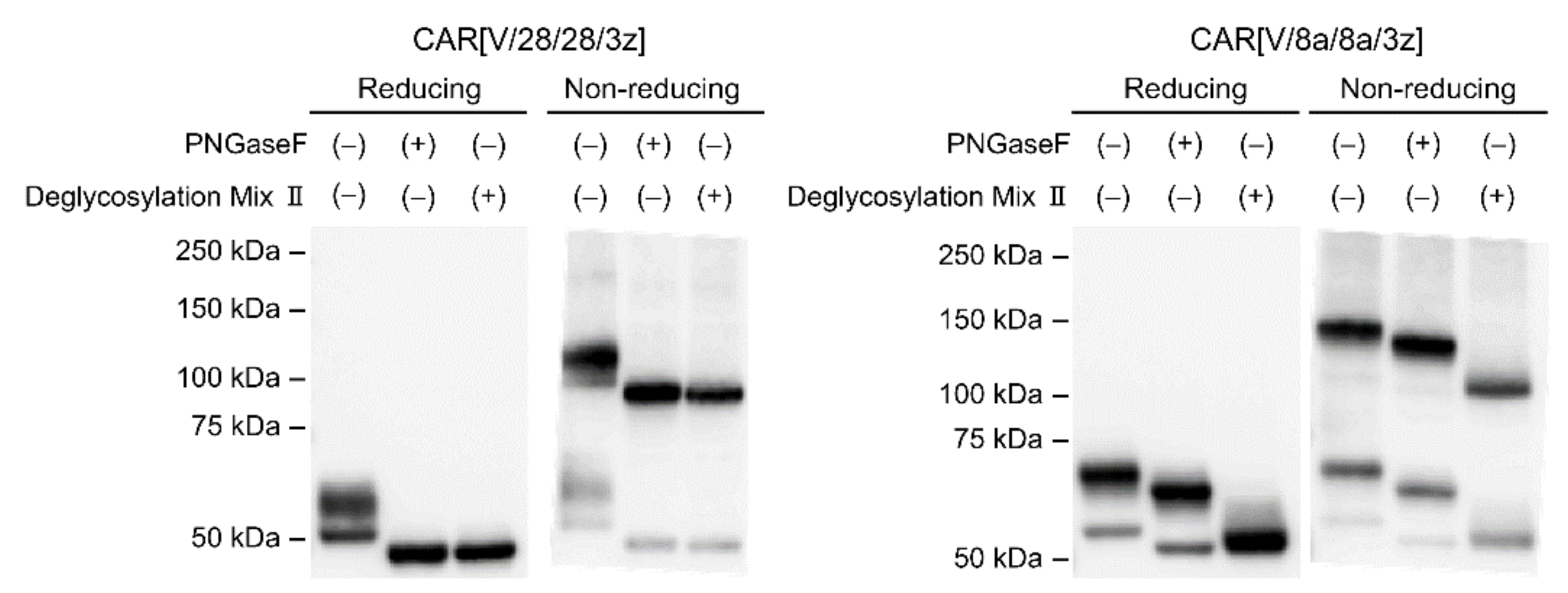
This study suggested that N-linked glycosylation in CD28-derived HD may contribute to the maintenance of CAR expression on the T cell membrane. We found that the glycosylation-enhanced maintenance of CAR expression affects CAR-T cell function. Herein, Western blotting analysis was performed only immediately after the completion of the gene transfer operation (day 0). It is unclear whether the decrease in CAR expression intensity following the removal of N-linked glycosylation was due to changes in the efficiency of CAR transfer to the cell surface or due to changes in CAR turnover. In the future, we will analyse the intracellular localisation and behaviour of CAR[V/28/28/3z] and CAR[V/28/28/3z](N130D) over time. In this manner, we would like to clarify the role of N-linked glycosylation in the CD28-derived HD. On the other hand, N-linked glycosylation in the HD of CAR[V/8a/8a/3z] had a small effect on the maintenance of CAR expression. Thus, it was not considered to be of importance for CAR-T cell function. The replacement/removal of N-linked glycosylation sites in CD8α-derived HD may represent one approach for improving CAR-T cell homogeneity as a drug formulation. Although it was found that CAR[V/8a/8a/3z] also undergoes O-linked glycosylation, we were unable to identify the amino acid residues at which these modifications occur. Regarding O-linked glycosylation of the original CD8α molecule, it has been reported that glycosylation status changes depending on the T cell activation state and differentiation stage [
17]. Although the previous report [
18] did not discuss O-linked glycosylation of CAR, it nonetheless demonstrates that even if one O-linked glycosylation site is deleted, another is modified. Therefore, it is possible that the same phenomenon takes place in CAR[V/8a/8a/3z]. In the future, we plan to identify the O-linked glycosylation site in the CD8α-derived HD by using the alanine mutagenesis screening technique or the mass spectrometric approach and to determine its effect on CAR-T cell function. There has been no report on the relationship between the presence or absence of the glycosylation of CAR and CAR-T cell function. Although we have not yet been able to investigate O-linked glycosylation in detail, we think these results are an important stepping stone to presenting its significance, the regulation of glycosylation for optimisation of CAR function and the homogenisation of CAR-T cell quality. In addition, this study was evaluated in a mouse model. The effects of the HD region on CAR-T cells have not been evaluated in either mice and humans, and the results in mice may not be applicable to humans, but we think that the evaluation in humans is also an issue for the future.
It has also been reported that the positional relationship between the CAR extracellular domain and the epitope affects CAR-T cell function [
19,
20,
21]. It is therefore necessary to consider the length of the HD, both in terms of the number of amino acids that comprise it and its folded, three-dimensional structure. It is important to note that the current findings were only obtained from two types of model CARs, both of which have HDs that adopt a random coil structure. In the future, we will prepare more CAR variants with various numbers and combinations of secondary structural units (α-helix and β-strand) in their HDs. In this way, we plan to comprehensively analyse the effects of HD length and flexibility on CAR-T cell activity.
4. Materials and Methods
4.1. Cell Lines
Human Plat-E cells were obtained from Cell Biolabs (San Diego, CA, USA) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA), 1 µg/mL puromycin (Merck, Darmstadt, Germany) and 10 µg/mL blasticidin (FUJIFILM Wako Pure Chemical, Osaka, Japan). Murine EL4 cells were obtained from the Cell Resource Center for Biomedical Research, Institute of Development, Aging, and Cancer, Tohoku University (Sendai, Japan). These were cultured in complete RPMI 1640 (cRPMI) medium (FUJIFILM Wako Pure Chemical, Osaka, Japan) supplemented with 10% FBS, Antibiotic-Antimycotic Mixed Stock Solution (100×) 5 mL (10,000 U/mL penicillin, 10,000 µg/mL streptomycin, 25 µg/mL amphotericin B, Nacalai Tesque, Kyoto, Japan) and 50 µM 2-mercaptoethanol (2-ME, Merck, Darmstadt, Germany). Murine-VEGFR2-expressing EL4 (VEGFR2+ EL4) cells were generated via Rv transduction (containing the VEGFR2 gene and a puromycin resistance cassette) and were grown in cRPMI medium, supplemented with 5 µg/mL puromycin. All cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.
4.2. Mice
Female C57BL/6 mice (6 weeks old) were purchased from SLC (Hamamatsu, Japan) and were maintained in the experimental animal facility at Osaka University.
4.3. Construction of CAR Structural Variants
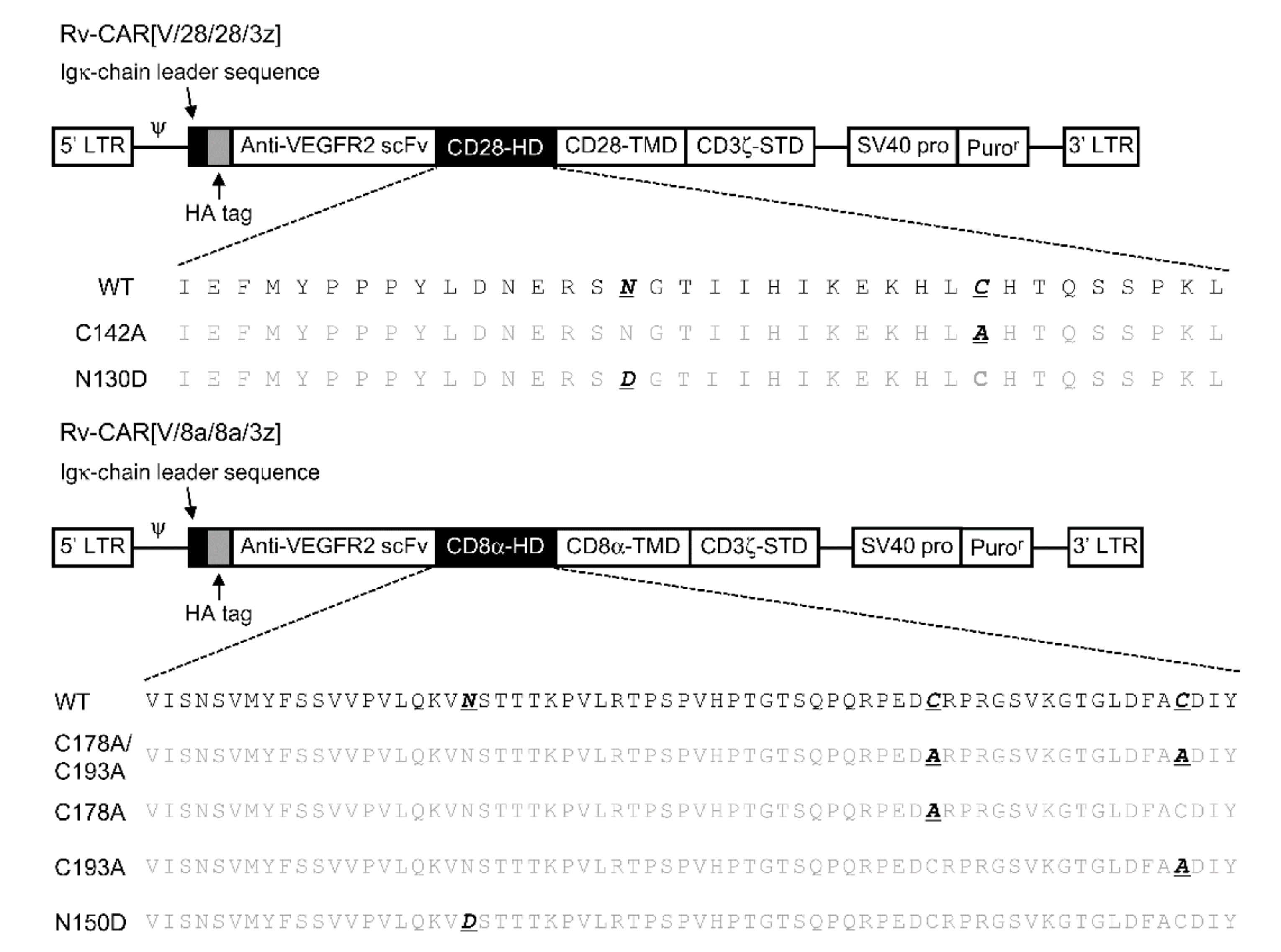
The basic structure of CAR followed the previous report [
7]. The notation and structure of various structurally modified CARs and the amino acid sequence of the HD part are summarised in
Figure 2. An HA-tag was incorporated into the N-terminus of CAR. We took two CARs consisting of a mouse-VEGFR-specific scFv tandemly linked to a CD28- or CD8α-derived HD/TMD and a CD3ζ-derived STD as our basic structural units. We then constructed six structural variants of these CARs by substituting cysteines in the HD with alanine and by substituting various amino acids in a region described as a potential glycosylation site in the literature [
13] as well as in protein databases. In the CD28-derived HD, we substituted one cysteine residue with one alanine and one asparagine that is an N-linked glycosylation site with aspartate. In the CD8α-derived HD, we substituted either one, the other, or both cysteine residues with alanine, in addition to substituting one asparagine, an N-linked glycosylation site, with aspartate.
4.4. Production of CAR-T Cells
Murine CAR-T cells were produced as previously described [
22]. Briefly, a murine leukaemia virus-derived pMXs-Puro Retroviral Vector (Cell Biolabs, San Diego, CA, USA) was used as a plasmid for Rv production. The Rv packaging CAR gene was produced by transfecting Plat-E cells with the pMXs-Puro/CAR. The culture supernatant of Plat-E cells obtained 48 h later was filtered through a 0.45 μm filter and used as an Rv solution for gene transfer. Murine T cells were activated by incubation with an anti-CD3ε mAb (clone 145-2C11, Bioxcell, West Lebanon, NH, USA) and anti-CD28 mAb (clone 37.51, Bioxcell, West Lebanon, NH, USA) and then transduced with Rv-bound Retronectin (Takara Bio, Kusatsu, Japan) under anti-CD3ε/CD28 mAbs stimulation. The gene-transduced cells were cultured in cRPMI medium, supplemented with 5 μg/mL puromycin. We defined the end of the 24 h culture as the end of the gene transfer operation on the 0th day (day 0).
4.5. Reverse Transcription and Quantitative Polymerase Chain Reaction
mRNA was extracted from genetically modified T cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA). Complementary DNA (cDNA) was prepared through a reverse transcription reaction using Super Script III Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) after DNase treatment. The CAR gene-encoded mRNA was detected via the TaqMan Gene Expression Assay (Thermo Fisher Scientific, Waltham, MA, USA) with primers and probes that bind to the mouse-VEGFR2-specific scFv. We also detected mRNA encoded by the GAPDH gene as an endogenous control. The expression level of each gene was measured on a CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA).
4.6. Flow Cytometry (FCM) Analysis for Surface Expression and Antigen-Binding Capacity of CAR
Using a staining buffer (PBS supplemented with 2% FBS and 0.05% NaN
3), 5 × 10
5 T cells were first treated with a TruStain fcX (anti-mouse CD16/32) Antibody (clone 93; BioLegend, San Diego, CA, USA). Staining was then performed using the Zombie Aqua Fixable Viability Kit (BioLegend, San Diego, CA, USA), an eFluor450-conjugated anti-mouse CD4 mAb (clone GK1.5; Thermo Fisher Scientific, Waltham, MA, USA) and a PE-Cy7-conjugated anti-mouse CD8 mAb (clone 53-6.7; Thermo Fisher Scientific, Waltham, MA, USA). Since we incorporated an HA-tag at the CAR N-terminus, we used APC-conjugated anti-HA mAb (clone GG8-1F3.3.1, Miltenyi Biotec, Bergisch Gladbach, Germany) and APC-conjugated mouse IgG1κ Isotype Control (clone P3.6.2.8.1, Thermo Fisher Scientific, Waltham, MA, USA) to detect CAR expression. The geometric mean fluorescence intensity (GMFI) of live cells, lymphocytes and CD8-positive fractions was measured via FCM analysis using anti-HA-Tag antibody or isotype control. The GMFI ratio was calculated from the following formula, and this value was quantified as the CAR expression intensity.
To analyse the avidity of our CARs for mouse VEGFR2, we used a staining buffer (PBS supplemented with 2% FBS and 0.05% NaN
3) and first treated 5 × 10
5 T cells with TruStain fcX (anti-mouse CD16/32) Antibody (clone 93; BioLegend, San Diego, CA, USA). Then, the CAR-antigen reaction was carried out using recombinant mouse VEGFR2-Fc Chimera (BioLegend, San Diego, CA, USA). As a control, we subjected a group of cells to the same procedures without addition of the antigen. Excess antigen was removed by washing with staining buffer, and staining was performed by adding Zombie Aqua Fixable Viability Kit, eFlu-or450-conjugated anti-mouse CD4 mAb and PE-Cy7-conjugated anti-mouse CD8 mAb. The recombinant mouse VEGFR2-Fc chimera we used contained a His-tag. Therefore, we used Alexa Fluor-conjugated anti-His-tag mAb (clone OGHis, MBL) to detect antigens bound to the CAR. The GMFI of live cells, lymphocytes and CD8-positive fractions with or without antigen was measured via FCM analysis. The GMFI ratio was calculated via the following formula and quantified as the antigen-binding capacity of the CAR:
BD FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA) was used for FCM analysis.
4.7. Protein Extraction from CAR-T Cells and Enzymatic Deglycosylation
Total protein was extracted from CAR-T cells immediately after the gene transfer operation (day 0) using cell lysis buffer (50 mM Tris-HCl (pH 7.4) supplemented with 150 mM NaCl, 1 mM EDTA, 5% glycerol, 1% Triton-X). Thereafter, protein levels were quantified via the Lowry method using a DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA).
A sample from which N-linked glycans had been removed was prepared by treating the protein extract sample from CAR-T cells with PNGaseF (New England Biolabs, Ipswich, MA, USA). In addition, a sample from which both N-linked and O-linked glycans had been removed was prepared via treatment with Deglycosylation Mix II (New England Biolabs, Ipswich, MA, USA).
4.8. Western Blotting
Lysate sample was mixed with Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA, USA) containing 20% 2-mercaptoethanol or not, and heat-denatured. Next, lysate samples were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis with 7.5% SuperSep Ace (FUJIFILM Wako Pure Chemical, Osaka, Japan) and transferred to a polyvinylidene difluoride membrane (GE Healthcare, Menlo Park, CA, USA) using a Trans-Blot SD Cell (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was blocked in 4% Block Ace (KAC Co., Kyoto, Japan), and then reacted with HA-Tag Rabbit mAb (clone C29F4, Cell Signaling Technology, Danvers, MA, USA) in tris-buffered saline containing 0.05% Tween-20 and 1% bovine serum albumin. The membrane was then washed and reacted with anti-rabbit IgG HRP-linked antibody (Cell Signaling Technology, Danvers, MA, USA). Immunoreactive proteins on the membrane were detected using ImmunoStar Zeta (FUJIFILM Wako Pure Chemical, Osaka, Japan) and ImageQuant LAS4010 (GE Healthcare, Menlo Park, CA, USA).
4.9. BrdU Proliferation Assay and Cytokine-Enzyme-Linked Immunosorbent Assay (ELISA)
Four days after Rv transduction, CAR-T cells were cultured for 24 h on a plate coated with VEGFR2-Fc (2–2000 ng/mL). Proliferation activity was measured via BrdU uptake ELISA (Sigma-Aldrich, St. Louis, MO, USA). IFN-γ, TNF-α and IL-2 levels in the supernatants were determined using the OptiEIA™ ELISA Set (BD Biosciences, Franklin Lakes, NJ, USA).
4.10. Cytotoxicity Assay
EL4 cells were stained with Violet Proliferation Dye 450 (BD Biosciences, Franklin Lakes, NJ, USA), and EL4 cells expressing mVEGFR2 were stained with Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific, Waltham, MA, USA). These target cells were co-cultured in batches of 1 × 10
5 cells for 18 h with enough CAR-T cells to match the effector/target ratio of each well. Four days after the end of the gene transfer operation, CAR-T cells were used for this experiment. Count Bright Absolute Counting Beads (Thermo Fisher Scientific, Waltham, MA, USA) were added to unify the analysis volume of all samples, and 7-AAD Viability Staining Solution (BioLegend, San Diego, CA, USA) was added to stain dead cells. FCM analysis was then performed and terminated when 1000 Count Bright Absolute Counting Beads were detected in each sample. The ratio (R) of the number of mVEGFR2-expressing EL4 cells to the number of living EL4 cells was calculated for each well, and the cytotoxic activity was calculated from the following formula.
control well: seeded only with target cells; test well: seeded with both effector and target cells.
4.11. Statistical Analysis
All experimental data are presented as the mean ± SD. Statistical significance was evaluated via Welch’s t-test or Tukey’s test.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






