
The ATX–LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
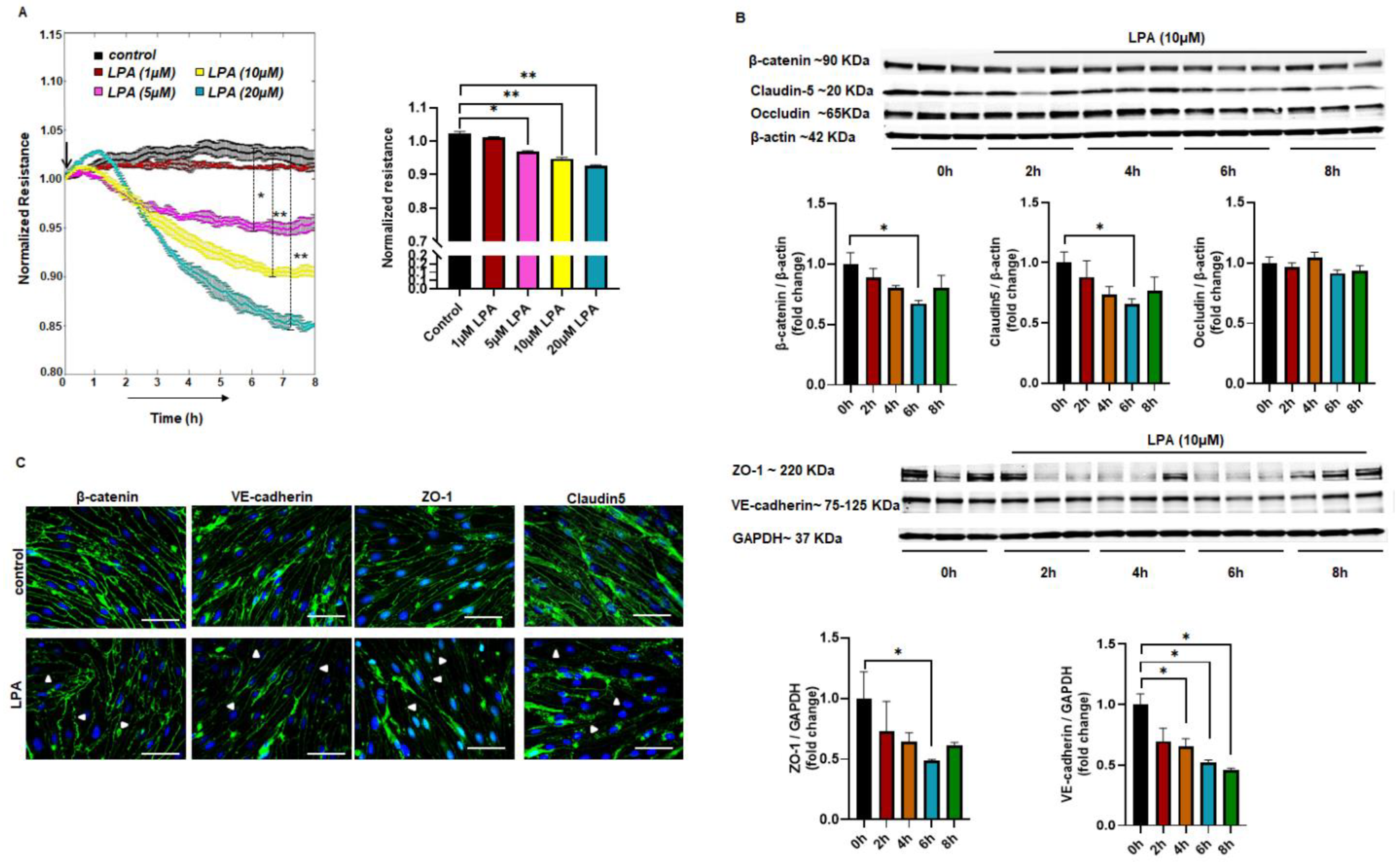
2.1. LPA-Mediated Endothelial Permeability In Vitro
2.2. Altered Expression of Junctional Proteins with LPA Treatment
2.3. LPA Increases Endothelial Permeability via the LPAR1–ROCK Pathway
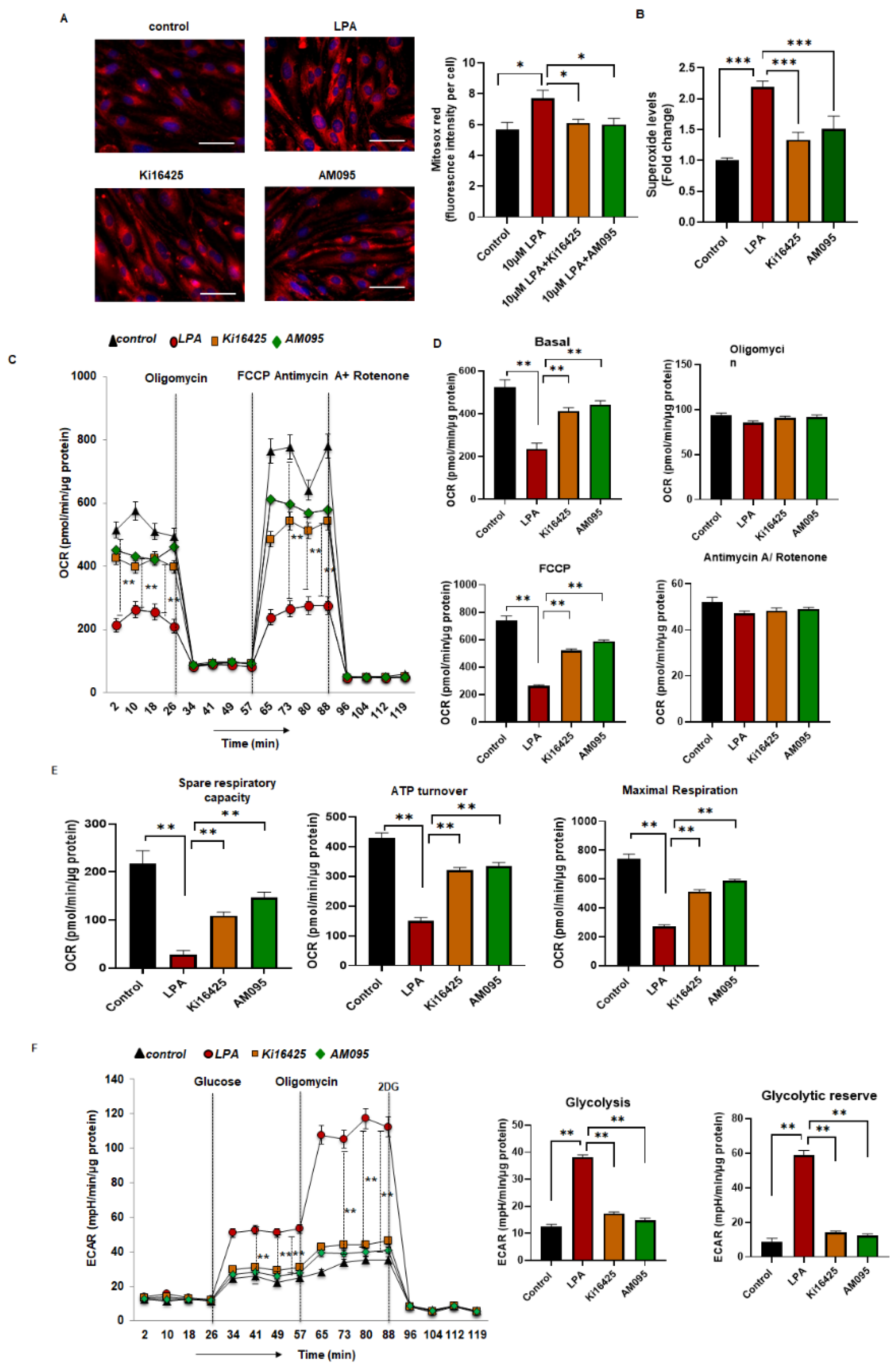
2.4. Increased Levels of Superoxide and Reduced Mitochondrial Bioenergetics in LPA-Treated MBMEC
2.5. Elevated Expression of ATX with Oxygen Glucose Deprivation–Reoxygenation (OGDR)
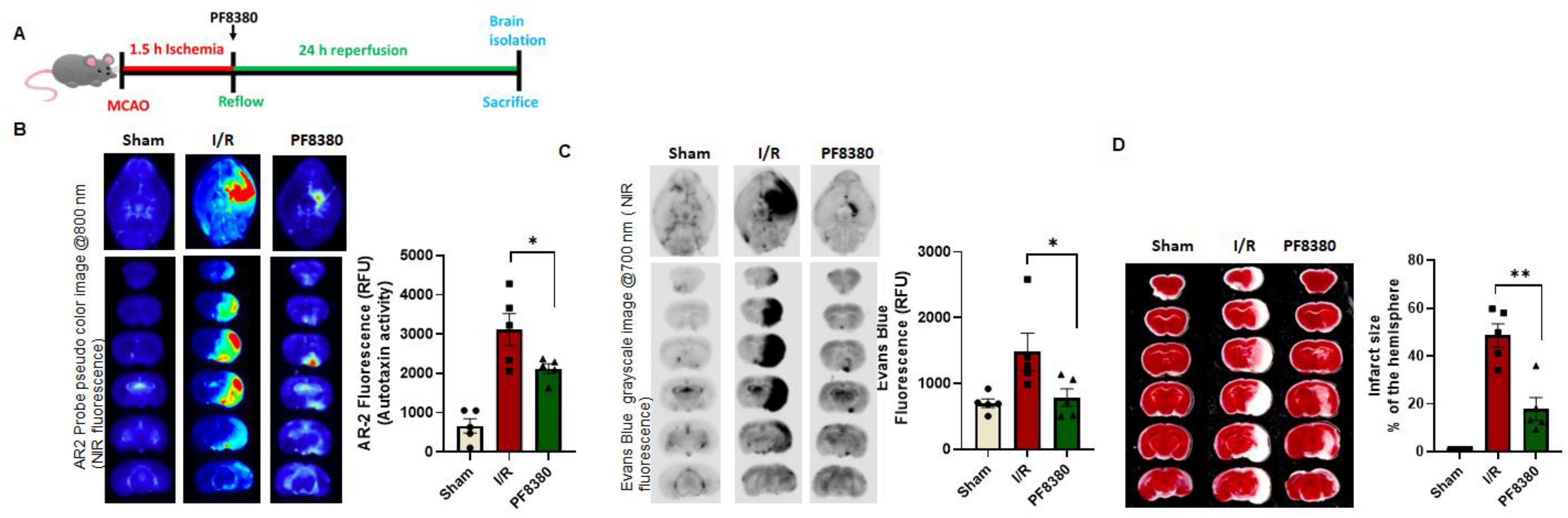
2.6. ATX Inhibitor Reduces Permeability in a Mouse Stroke Model
3. Discussion
4. Methods
4.1. Animal Models of Ischemic Stroke
4.2. Cell Culture
4.3. mRNA Expression of LPA Receptors
4.4. Superoxide Measurement
4.5. Mitochondrial Bioenergetics
4.6. Transendothelial Electrical Resistance (TEER)
4.7. Western Blot Analysis
4.8. Immunofluorescence Staining of Junctional Proteins
4.9. Mitosox Red Mitochondrial Superoxide Staining
4.10. OGDR (Oxygen Glucose Deprivation–Reoxygenation)
4.11. ATX Activity Assay and Evans Blue Permeability Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Zhao, Z.; Zlokovic, B.V. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J. Exp. Med. 2017, 214, 3151–3169. [Google Scholar] [CrossRef]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood–brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhu, L.; An, C.; Wang, R.; Yang, L.; Yu, W.; Li, P.; Gao, Y. The blood brain barrier in cerebral ischemic injury–Disruption and repair. Brain Hemorrhages 2020, 1, 34–53. [Google Scholar] [CrossRef]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef]
- Butler, J.; Heidari, P.; Blayney, S.; Hitomi, E.; Luby, M.; Leigh, R. Blood-brain barrier integrity of stroke patients presenting in an extended time window. BMC Neurol. 2020, 20, 54. [Google Scholar] [CrossRef]
- Bernardo-Castro, S.; Sousa, J.A.; Brás, A.; Cecília, C.; Rodrigues, B.; Almendra, L.; Machado, C.; Santo, G.; Silva, F.; Ferreira, L.; et al. Pathophysiology of Blood-Brain Barrier Permeability Throughout the Different Stages of Ischemic Stroke and Its Implication on Hemorrhagic Transformation and Recovery. Front. Neurol. 2020, 11, 594672. [Google Scholar] [CrossRef]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; Spohr, T.; Amaral, R.F.D.; Fonseca, A.; Garcia, C.; Mendes, F.A.; Freitas, C.; dosSantos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Fukushima, N.; Ishii, S.; Tsujiuchi, T.; Kagawa, N.; Katoh, K. Comparative analyses of lysophosphatidic acid receptor-mediated signaling. Cell. Mol. Life Sci. CMLS 2015, 72, 2377–2394. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; He, P.; Han, Y.; Dong, L.; Yun, C.C. Control of Intestinal Epithelial Permeability by Lysophosphatidic Acid Receptor 5. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1073–1092. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Watsky, M.A. LPA and S1P increase corneal epithelial and endothelial cell transcellular resistance. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1927–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Wei, J.; Li, S.; Suber, T.; Zhao, J. AM966, an Antagonist of Lysophosphatidic Acid Receptor 1, Increases Lung Microvascular Endothelial Permeability through Activation of Rho Signaling Pathway and Phosphorylation of VE-Cadherin. Mediat. Inflamm. 2017, 2017, 6893560. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Savant, S.; Toews, M.; Miller, D.W. Rapid and reversible enhancement of blood-brain barrier permeability using lysophosphatidic acid. J. Cereb. Blood Flow Metab. 2013, 33, 1944–1954. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef]
- Tanaka, M.; Okudaira, S.; Kishi, Y.; Ohkawa, R.; Iseki, S.; Ota, M.; Noji, S.; Yatomi, Y.; Aoki, J.; Arai, H. Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. J. Biol. Chem. 2006, 281, 25822–25830. [Google Scholar] [CrossRef] [Green Version]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.K.; Brindley, D.N. Role of the autotaxin–lysophosphatidate axis in the development of resistance to cancer therapy. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2020, 1865, 158716. [Google Scholar] [CrossRef] [PubMed]
- Orosa, B.; García, S.; Conde, C. The autotaxin-lysophosphatidic acid pathway in pathogenesis of rheumatoid arthritis. Eur. J. Pharmacol. 2015, 765, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Uojima, H.; Setsu, T.; Okubo, T.; Atsukawa, M.; Furuichi, Y.; Arase, Y.; Hidaka, H.; Tanaka, Y.; Nakazawa, T.; et al. Usefulness of autotaxin for the complications of liver cirrhosis. World J. Gastroenterol. 2020, 26, 97–108. [Google Scholar] [CrossRef]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.H.; Crack, P.J. Modulation of neuro-inflammation and vascular response by oxidative stress following cerebral ischemia-reperfusion injury. Curr. Med. Chem. 2008, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- DiNapoli, V.A.; Huber, J.D.; Houser, K.; Li, X.; Rosen, C.L. Early disruptions of the blood-brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol. Aging 2008, 29, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Merali, Z.; Huang, K.; Mikulis, D.; Silver, F.; Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE 2017, 12, e0171558. [Google Scholar] [CrossRef] [Green Version]
- Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Blood-Brain Barrier Protection as a Therapeutic Strategy for Acute Ischemic Stroke. AAPS J. 2017, 19, 957–972. [Google Scholar] [CrossRef]
- Ren, Y.; Guo, L.; Tang, X.; Apparsundaram, S.; Kitson, C.; Deguzman, J.; Fuentes, M.E.; Coyle, L.; Majmudar, R.; Allard, J.; et al. Comparing the differential effects of LPA on the barrier function of human pulmonary endothelial cells. Microvasc. Res. 2013, 85, 59–67. [Google Scholar] [CrossRef]
- Ueda, H.; Neyama, H.; Sasaki, K.; Miyama, C.; Iwamoto, R. Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol. Pain 2019, 5, 100020. [Google Scholar] [CrossRef]
- Sarker, M.H.; Hu, D.E.; Fraser, P.A. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation 2010, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Johnson, A.M.; Sladojevic, N.; Keep, R.F.; Andjelkovic, A.V. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann. N. Y. Acad. Sci. 2017, 1397, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix Metalloproteinase-Mediated Disruption of Tight Junction Proteins in Cerebral Vessels is Reversed by Synthetic Matrix Metalloproteinase Inhibitor in Focal Ischemia in Rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Wu, W.T.; Chen, C.-N.; Lin, C.I.; Chen, J.H.; Lee, H. Lysophospholipids enhance matrix metalloproteinase-2 expression in human endothelial cells. Endocrinology 2005, 146, 3387–3400. [Google Scholar] [CrossRef]
- Yu, Y.; Qin, J.; Liu, M.; Ruan, Q.; Li, Y.; Zhang, Z. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int. J. Mol. Med. 2014, 33, 661–669. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Chi, C.; Xu, Y.Q.; Wang, C.; Wang, T.Y.; Lv, D.; Li, X. Occludin endocytosis is involved in the disruption of the intestinal epithelial barrier in a mouse model of alcoholic steatohepatitis. J. Dig. Dis. 2019, 20, 476–485. [Google Scholar] [CrossRef]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vasc. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [Green Version]
- Xiang, H.; Lu, Y.; Shao, M.; Wu, T. Lysophosphatidic Acid Receptors: Biochemical and Clinical Implications in Different Diseases. J. Cancer 2020, 11, 3519–3535. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuw Amerongen, G.P.; Vermeer, M.A.; van Hinsbergh, V.W. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2000, 20, e127–e133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Han, J.; Song, S.Y.; Kim, W.S.; Shin, S.; Kim, J.H.; Ahn, H.; Jeong, J.H.; Hwang, S.J.; Sung, J.H. Lysophosphatidic acid increases the proliferation and migration of adipose-derived stem cells via the generation of reactive oxygen species. Mol. Med. Rep. 2015, 12, 5203–5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klomsiri, C.; Rogers, L.C.; Soito, L.; McCauley, A.K.; King, S.B.; Nelson, K.J.; Poole, L.B.; Daniel, L.W. Endosomal H2O2 production leads to localized cysteine sulfenic acid formation on proteins during lysophosphatidic acid-mediated cell signaling. Free Radic. Biol. Med. 2014, 71, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabowski, D.S.; Kadlec, A.O.; Ait-Aissa, K.; Hockenberry, J.C.; Pearson, P.J.; Beyer, A.M.; Gutterman, D.D. Lysophosphatidic acid acts on LPA1 receptor to increase H2 O2 during flow-induced dilation in human adipose arterioles. Br. J. Pharmacol. 2018, 175, 4266–4280. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, K.; Nzirorera, C.; Cowie, A.M.; Varghese, G.P.; Trivedi, P.; Eichmann, T.O.; Biswas, D.; Touaibia, M.; Morris, A.J.; Aidinis, V.; et al. Autotaxin-LPA signaling contributes to obesity-induced insulin resistance in muscle and impairs mitochondrial metabolism. J. Lipid Res. 2018, 59, 1805–1817. [Google Scholar] [CrossRef] [Green Version]
- Chandra, M.; Escalante-Alcalde, D.; Bhuiyan, M.S.; Orr, A.W.; Kevil, C.; Morris, A.J.; Nam, H.; Dominic, P.; McCarthy, K.J.; Miriyala, S.; et al. Cardiac-specific inactivation of LPP3 in mice leads to myocardial dysfunction and heart failure. Redox. Biol. 2018, 14, 261–271. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Nakada, M.; Demuth, T.; Rosensteel, T.; Reavie, L.B.; Berens, M.E. Autotaxin: A secreted autocrine/paracrine factor that promotes glioma invasion. J. Neuro Oncol. 2008, 86, 297–309. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef] [Green Version]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Heyman, S.N.; Leibowitz, D.; Mor-Yosef Levi, I.; Liberman, A.; Eisenkraft, A.; Alcalai, R.; Khamaisi, M.; Rosenberger, C. Adaptive response to hypoxia and remote ischaemia pre-conditioning: A new hypoxia-inducible factors era in clinical medicine. Acta Physiol. 2016, 216, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Madan, D.; Ferguson, C.G.; Lee, W.Y.; Prestwich, G.D.; Testa, C.A. Non-invasive imaging of tumors by monitoring autotaxin activity using an enzyme-activated near-infrared fluorogenic substrate. PLoS ONE 2013, 8, e79065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A Novel Autotaxin Inhibitor Reduces Lysophosphatidic Acid Levels in Plasma and the Site of Inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.W.; Lim, W.; Jo, D.; Kim, S.; Park, J.T.; Min, J.J.; Hyun, H.; Kim, H.S. Low-Dose Evans Blue Dye for Near-Infrared Fluorescence Imaging in Photothrombotic Stroke Model. Int. J. Med. Sci. 2018, 15, 696–702. [Google Scholar] [CrossRef] [Green Version]
- Van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradère, J.P.; Pettit, T.R.; Wakelam, M.J.O.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattarai, S.; Sharma, S.; Subedi, U.; Ara, H.; Shum, A.; Milena, M.; Bhuiyan, M.S.; Kidambi, S.; Sun, H.; Miriyala, S.; et al. The ATX–LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. Int. J. Mol. Sci. 2022, 23, 4138. https://doi.org/10.3390/ijms23084138
Bhattarai S, Sharma S, Subedi U, Ara H, Shum A, Milena M, Bhuiyan MS, Kidambi S, Sun H, Miriyala S, et al. The ATX–LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. International Journal of Molecular Sciences. 2022; 23(8):4138. https://doi.org/10.3390/ijms23084138
Chicago/Turabian StyleBhattarai, Susmita, Sudha Sharma, Utsab Subedi, Hosne Ara, Alika Shum, Murov Milena, Md. Shenuarin Bhuiyan, Srivatsan Kidambi, Hong Sun, Sumitra Miriyala, and et al. 2022. "The ATX–LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion" International Journal of Molecular Sciences 23, no. 8: 4138. https://doi.org/10.3390/ijms23084138