
Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation


 ,
,  , , , , ,
, , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
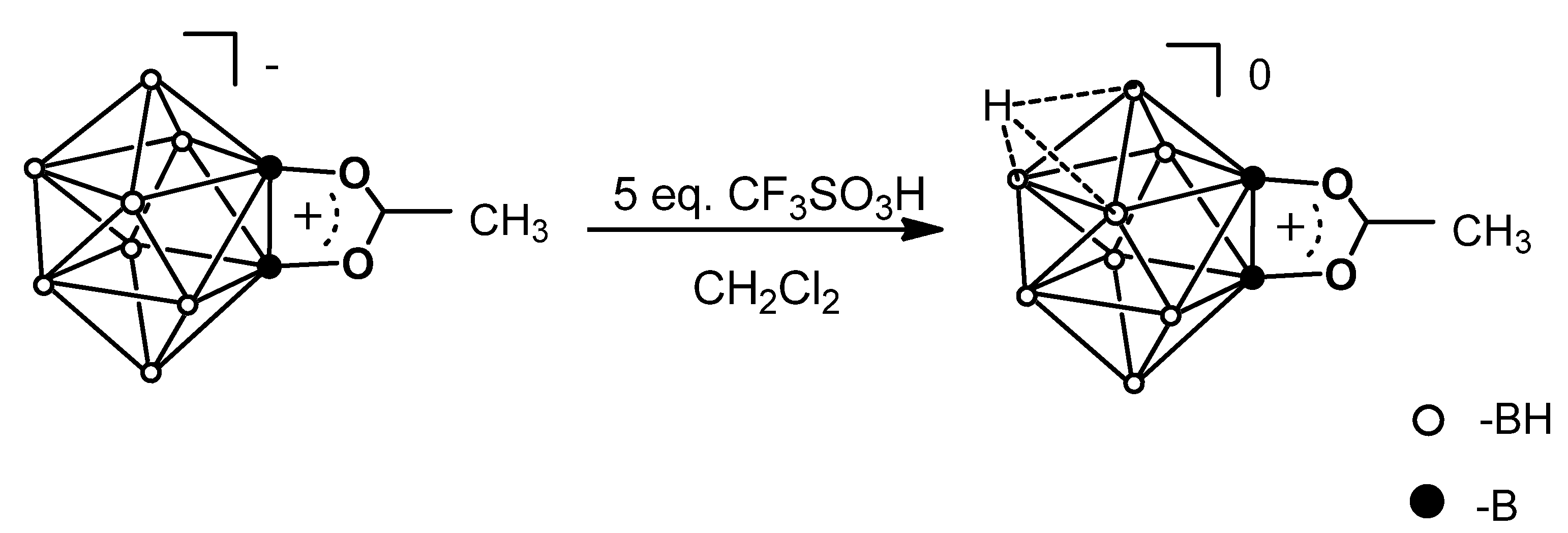
2.1. Experimental Protonation
2.2. Fukui Function
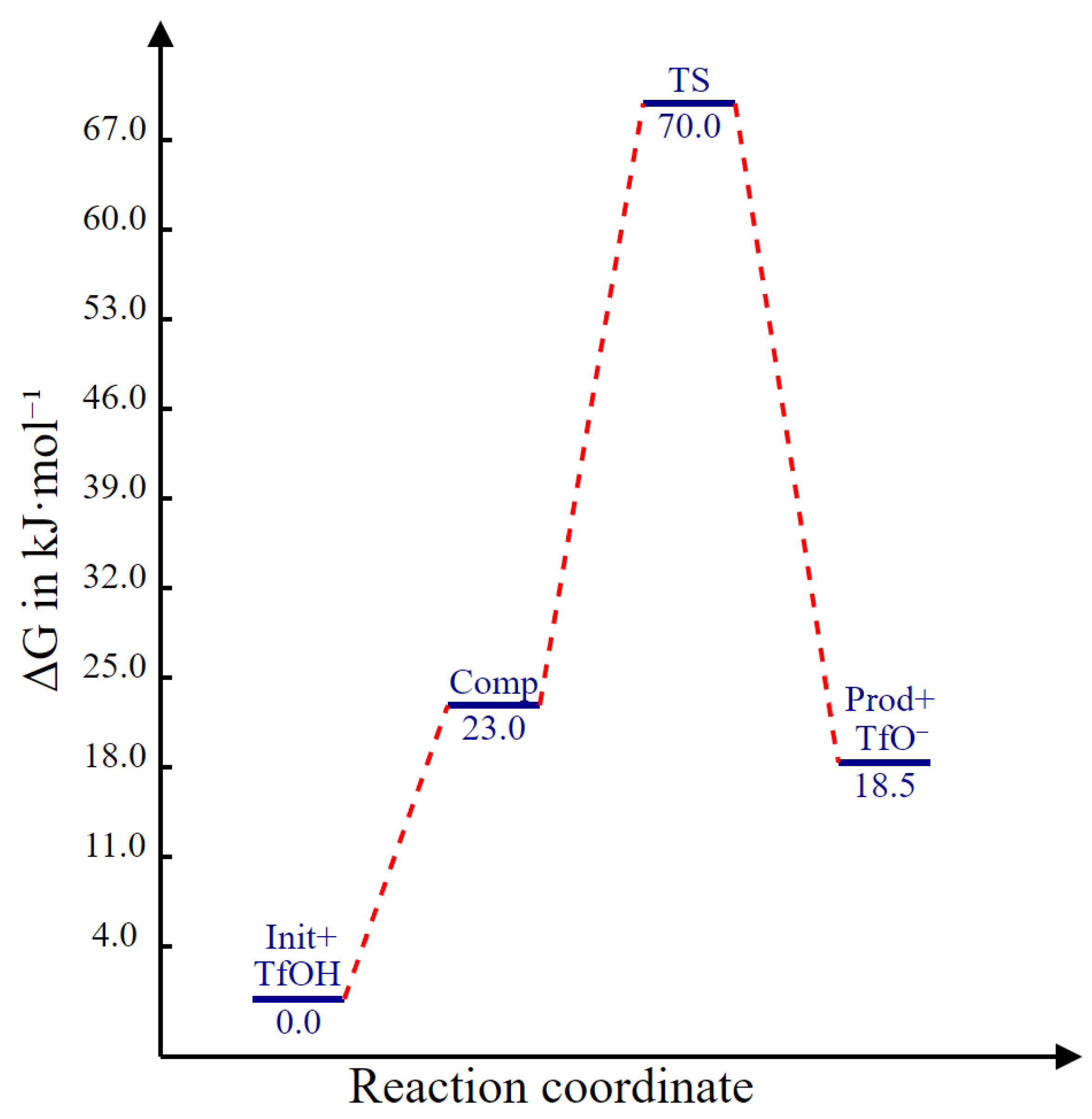
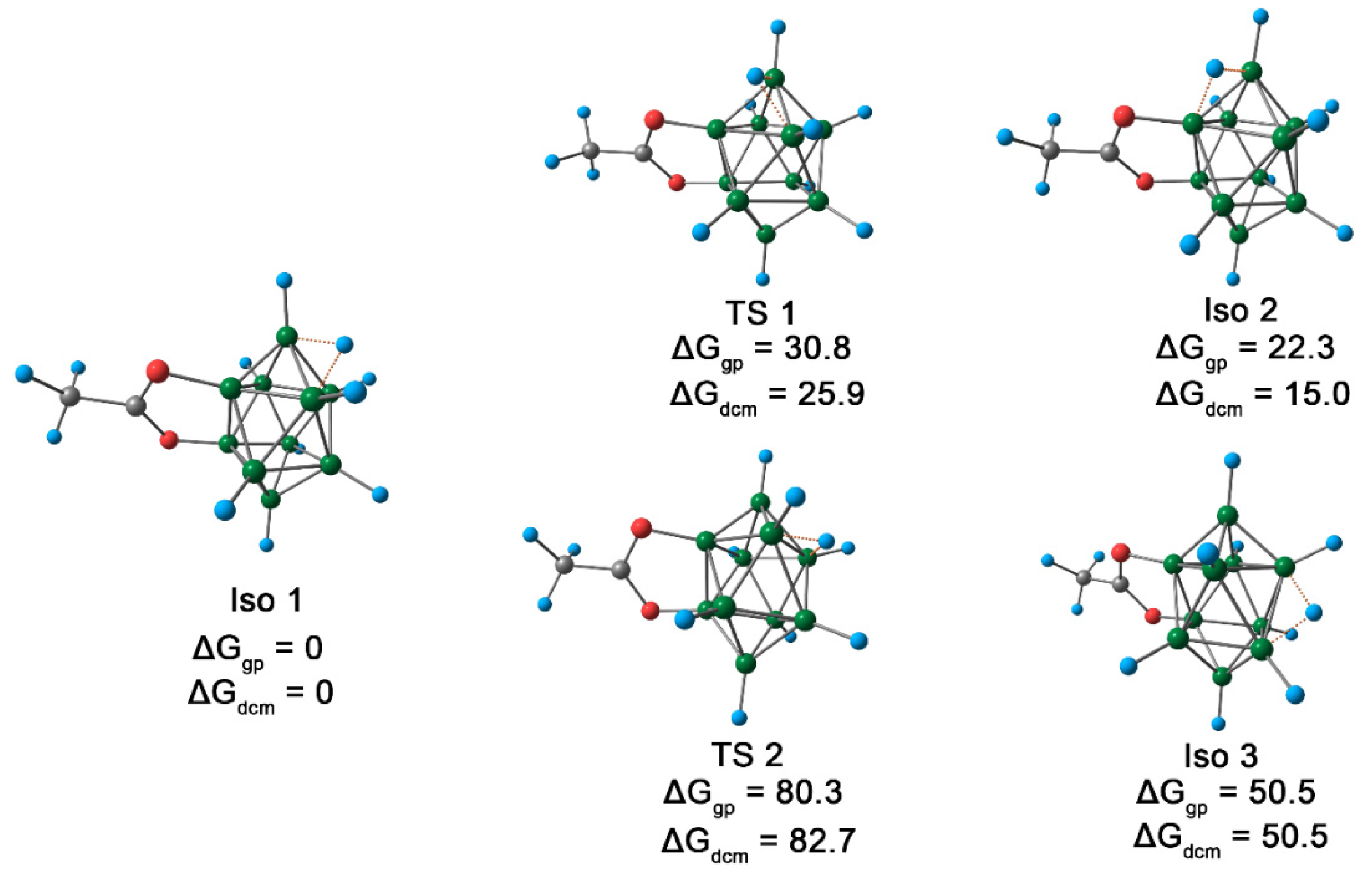
2.3. Protonation Mechanism
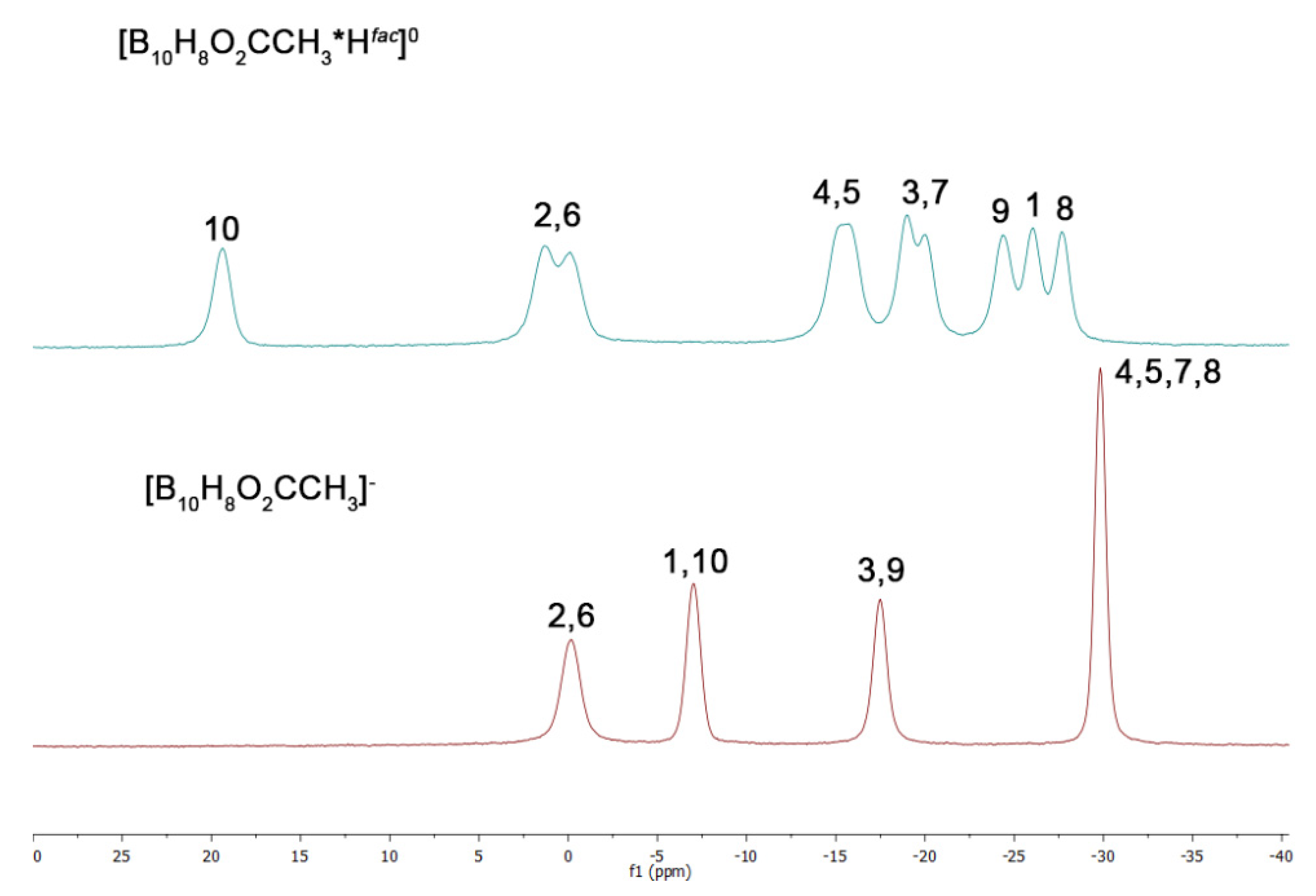
2.4. NMR Spectra Analysis
2.5. Structure Elucidation
3. Materials and Methods
3.1. Computational Details
3.2. IR Spectra
3.3. NMR Spectra
3.4. Protonation of ((C6H5)4P)[2,6-B10H8O2CCH3]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pino-Rios, R.; Inostroza, D.; Tiznado, W. Neither too Classic nor too Exotic: One-Electron Na⋅B Bond in NaBH 3− Cluster. Angew. Chem. Int. Ed. 2021, 60, 12747–12753. [Google Scholar] [CrossRef] [PubMed]
- Su, B.; Cao, Z.-C.; Shi, Z.-J. Exploration of Earth-Abundant Transition Metals (Fe, Co, and Ni) as Catalysts in Unreactive Chemical Bond Activations. Acc. Chem. Res. 2015, 48, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry. Angew. Chem. Int. Ed. 1969, 8, 781–853. [Google Scholar] [CrossRef]
- Abu-Youssef, M.A.M.; Langer, V.; Barakat, A.; Haukka, M.; Soliman, S.M. Molecular, Supramolecular Structures Combined with Hirshfeld and DFT Studies of Centrosymmetric M(II)-azido {M = Ni(II), Fe(II) or Zn(II)} Complexes of 4-Benzoylpyridine. Symmetry 2021, 13, 2026. [Google Scholar] [CrossRef]
- Gouda, M.; Ferjani, H.; El-Lateef, H.M.A.; Khalaf, M.M.; Shaaban, S.; Yousef, T.A. A Competition between Hydrogen, Stacking, and Halogen Bonding in N-(4-((3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-yl)selanyl)phenyl)acetamide: Structure, Hirshfeld Surface Analysis, 3D Energy Framework Approach, and DFT Calculation. Int. J. Mol. Sci. 2022, 23, 2716. [Google Scholar] [CrossRef]
- Kolomeychuk, F.M.; Safonova, E.A.; Polovkova, M.A.; Sinelshchikova, A.A.; Martynov, A.G.; Shokurov, A.V.; Kirakosyan, G.A.; Efimov, N.N.; Tsivadze, A.Y.; Gorbunova, Y.G. Switchable Aromaticity of Phthalocyanine via Reversible Nucleophilic Aromatic Addition to an Electron-Deficient Phosphorus(V) Complex. J. Am. Chem. Soc. 2021, 143, 14053–14058. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Discovering Chemistry with Natural Bond Orbitals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; ISBN 9781118229101. [Google Scholar]
- Lepetit, C.; Fau, P.; Fajerwerg, K.; Kahn, M.L.; Silvi, B. Topological analysis of the metal-metal bond: A tutorial review. Coord. Chem. Rev. 2017, 345, 150–181. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Guzman, F.; Bader, R. Complementarity of QTAIM and MO theory in the study of bonding in donor-acceptor complexes. Coord. Chem. Rev. 2005, 249, 633–662. [Google Scholar] [CrossRef]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Legare, D.A. Properties of atoms in molecules: Structures and reactivities of boranes and carboranes. Can. J. Chem. 1992, 70, 657–676. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Savin, P.-D.A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Angew. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Morgante, P.; Peverati, R. The devil in the details: A tutorial review on some undervalued aspects of density functional theory calculations. Int. J. Quant. Chem. 2020, 120, e26332. [Google Scholar] [CrossRef]
- Shen, Y.-F.; Xu, C.; Cheng, L.-J. Deciphering chemical bonding in BnHn2−(n = 2–17): Flexible multicenter bonding. RSC Adv. 2017, 7, 36755–36764. [Google Scholar] [CrossRef] [Green Version]
- Teixidor, C.V. The uniqueness of boron as a novel challenging element for drugs in pharmacology, medicine and for smart biomaterials. Future Med. Chem. 2013, 5, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Evamarie Hey-Hawkins, C.V.T. Boron-Based Compounds: Potential and Emerging Applications in Medicine; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2018; ISBN 978-1-119-27555-8. [Google Scholar]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Hückel’s Rule of Aromaticity Categorizes Aromatic closo Boron Hydride Clusters. Chem. A Eur. J. 2016, 22, 7437–7443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchêne, L.; Remhof, A.; Hagemann, H.; Battaglia, C. Status and prospects of hydroborate electrolytes for all-solid-state batteries. Energy Stor. Mater. 2020, 25, 782–794. [Google Scholar] [CrossRef]
- Kochnev, V.K.; Avdeeva, V.V.; Goeva, L.V.; Malinina, E.A.; Kuznetsov, N.T. The undecahydrodecaborate anion B10H11—As the starting reagent in exopolyhedral substitution and complexation: Theoretical and experimental prerequisites. Russ. J. Inorg. Chem. 2012, 57, 331–336. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, S.; Dewhurst, R.D.; Ignat’Ev, N.V.; Finze, M.; Braunschweig, H. Boron: Its Role in Energy—Related Processes and Applications. Angew. Chem. Int. Ed. 2019, 59, 8800–8816. [Google Scholar] [CrossRef]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Yang, Z.; Zhang, L.; Xie, L.; Wang, L.; Xu, H.; Josephson, L.; Liang, S.H.; Zhang, M.-R. Boron agents for neutron capture therapy. Coord. Chem. Rev. 2020, 405, 213139. [Google Scholar] [CrossRef]
- Vorobyeva, M.; Dymova, M.; Novopashina, D.; Kuligina, E.; Timoshenko, V.; Kolesnikov, I.; Taskaev, S.; Richter, V.; Venyaminova, A. Tumor Cell-Specific 2′-Fluoro RNA Aptamer Conjugated with Closo-Dodecaborate as A Potential Agent for Boron Neutron Capture Therapy. Int. J. Mol. Sci. 2021, 22, 7326. [Google Scholar] [CrossRef] [PubMed]
- Lipscomb, W.N. Framework Rearrangement in Boranes and Carboranes. Science 1966, 153, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Dobrott, R.D.; Lipscomb, W.N. Structure of Cu2B10H10. J. Chem. Phys. 1962, 37, 1779–1784. [Google Scholar] [CrossRef]
- Schwalbe, C.H.; Lipscomb, W.N. Structures of B20H182- and B20H18NO3—And conformations of the triethylammonium ion. J. Am. Chem. Soc. 1969, 91, 194–196. [Google Scholar] [CrossRef]
- Lipscomb, W.N. The Boranes and Their Relatives. Science 1977, 196, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.; Lipscomb, W.N. Theory of Polyhedral Molecules. I. Physical Factorizations of the Secular Equation. J. Chem. Phys. 1962, 36, 2179–2189. [Google Scholar] [CrossRef]
- Hoffmann, R.; Lipscomb, W.N. Theory of Polyhedral Molecules. III. Population Analyses and Reactivities for the Carboranes. J. Chem. Phys. 1962, 36, 3489–3493. [Google Scholar] [CrossRef]
- Hoffmann, R.; Lipscomb, W.N. Boron Hydrides: LCAO—MO and Resonance Studies. J. Chem. Phys. 1962, 37, 2872–2883. [Google Scholar] [CrossRef]
- Wade, K. Structural and Bonding Patterns in Cluster Chemistry. Adv. Inorg. Chem. Radiochem. 1976, 18, 1–66. [Google Scholar] [CrossRef]
- Fox, M.A.; Nervi, C.; Crivello, A.; Batsanov, A.S.; Howard, J.A.K.; Wade, K.; Low, P.J. Structural, spectroscopic, electrochemical and computational studies of C,C′-diaryl-ortho-carboranes, 1-(4-XC6H4)-2-Ph-1,2-C2B10H10 (X = H, F, OMe, NMe2, NH2, OH and O−). J. Solid State Electrochem. 2009, 13, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Nelyubin, A.V.; Selivanov, N.A.; Bykov, A.Y.; Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Karpechenko, N.Y.; Grigoriev, M.S.; Zhizhin, K.Y.; Kuznetsov, N.T. Primary Amine Nucleophilic Addition to Nitrilium Closo-Dodecaborate [B12H11NCCH3]−: A Simple and Effective Route to the New BNCT Drug Design. Int. J. Mol. Sci. 2021, 22, 13391. [Google Scholar] [CrossRef] [PubMed]
- Mingos, D.M.P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Copley, R.C.B.; Mingos, D.M.P. The novel structure of the [Au11(PMePh2)10]3+ cation: Crystal structures of [Au11(PMePh2)10][C2B9H12]3·4thf and [Au11(PMePh2)10][C2B9H12]3 (thf = tetrahydrofuran). J. Chem. Soc. Dalt. Trans. 1996, 17, 479–489. [Google Scholar] [CrossRef]
- Yan, Y.-K.; Mingos, D.M.P.; Kurmoo, M.; Li, W.-S.; Scowen, I.J.; McPartlin, M.; Coomber, A.T.; Friend, R.H. Synthesis, structure and physical properties of tetrathiafulvalenium salts of the ferracarborane complex commo-[3,3′-Fe{1-(C4H3S)-1,2-C2B9H10}2]−. J. Chem. Soc. Dalt. Trans. 1995, 17, 2851–2860. [Google Scholar] [CrossRef]
- Hertler, W.R.; Knoth, W.H.; Muetterties, E.L. Chemistry of Boranes. XXIV. Carbonylation of Derivatives of B10H102- and B12Hl22- with Oxalyl Chloride. Inorg. Chem. 1965, 4, 288–293. [Google Scholar] [CrossRef]
- Miller, H.C.; Miller, N.E.; Muetterties, E.L. Chemistry of Boranes. XX. Syntheses of Polyhedral Boranes. Inorg. Chem. 1964, 3, 1456–1463. [Google Scholar] [CrossRef]
- Chamberland, B.L.; Muetterties, E.L. Chemistry of Boranes. XVIII. Oxidation of B10H10-2 and Its Derivatives. Inorg. Chem. 1964, 231, 1450–1456. [Google Scholar] [CrossRef]
- Miller, H.C.; Hertler, W.R.; Muetterties, E.L.; Knoth, W.H.; Miller, N.E. Chemistry of Boranes. XXV. Synthesis andc Chemistry of Base Derivatives of B10H102- and B12Hl22-. Inorg. Chem. 1965, 4, 1216–1221. [Google Scholar] [CrossRef]
- Golub, I.; Filippov, O.; Belkova, N.; Epstein, L.; Shubina, E. The Reaction of Hydrogen Halides with Tetrahydroborate Anion and Hexahydro-closo-hexaborate Dianion. Molecules 2021, 26, 3754. [Google Scholar] [CrossRef]
- Černý, R.; Brighi, M.; Murgia, F. The Crystal Chemistry of Inorganic Hydroborates. Chemistry 2020, 2, 805–826. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Z.; Chen, H.; Wang, Z.; Zhou, X.; Zhang, H. Progress in three-dimensional aromatic-like closo-dodecaborate. Coord. Chem. Rev. 2021, 444, 214042. [Google Scholar] [CrossRef]
- Kamin, A.A.; Juhasz, M.A. Exhaustive Cyanation of the Dodecaborate Dianion: Synthesis, Characterization, and X-ray Crystal Structure of [B12(CN)12]2−. Inorg. Chem. 2019, 59, 189–192. [Google Scholar] [CrossRef]
- Lacroix, M.R.; Liu, Y.; Strauss, S.H. Hydrated Metal Ion Salts of the Weakly Coordinating Fluoroanions PF6−, TiF62−, B12F122−, Ga(C2F5)4−, B(3,5-C6H3(CF3)2)4−, and Al(OC(CF3)3)4−. In Search of the Weakest HOH⋯F Hydrogen Bonds. Inorg. Chem. 2019, 58, 14900–14911. [Google Scholar] [CrossRef] [PubMed]
- Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Zhizhin, K.Y.; Kuznetsov, N.T. QTAIM Analysis of Mono-Hydroxy Derivatives of closo-Borate Anions [BnHn–1OH]2 (n = 6, 10, 12). Russ. J. Inorg. Chem. 2019, 64, 1825–1828. [Google Scholar] [CrossRef]
- Kononova, E.; Klemenkova, Z. The electronic structure of nido-B10H14 and [6-Ph-nido-6-CB9H11] in terms of Bader’s theory (AIM). J. Mol. Struct. 2013, 1036, 311–317. [Google Scholar] [CrossRef]
- Mindich, A.L.; Bokach, N.A.; Dolgushin, F.M.; Haukka, M.; Lisitsyn, L.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Miltsov, S.A.; Kuznetsov, N.T.; Kukushkin, V.Y. 1,3-Dipolar Cycloaddition of Nitrones to a Nitrile Functionality in closo-Decaborate Clusters: A Novel Reactivity Mode for the Borylated C≡N Group. Organometallics 2012, 31, 1716–1724. [Google Scholar] [CrossRef]
- Burianova, V.K.; Mikherdov, A.S.; Bolotin, D.S.; Novikov, A.S.; Mokolokolo, P.P.; Roodt, A.; Boyarskiy, V.P.; Suslonov, V.V.; Zhdanov, A.P.; Zhizhin, K.Y.; et al. Electrophilicity of aliphatic nitrilium closo-decaborate clusters: Hyperconjugation provides an unexpected inverse reactivity order. J. Organomet. Chem. 2018, 870, 97–103. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Burianova, V.K.; Novikov, A.S.; Demakova, M.Y.; Pretorius, C.; Mokolokolo, P.P.; Roodt, A.; Bokach, N.A.; Suslonov, V.V.; Zhdanov, A.P.; et al. Nucleophilicity of Oximes Based upon Addition to a Nitrilium closo-Decaborate Cluster. Organometallics 2016, 35, 3612–3623. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Erokhina, S.A.; Sivaev, I.B.; Bregadze, V.I. Nitrilium derivatives of polyhedral boron compounds (boranes, carboranes, metallocarboranes): Synthesis and reactivity. Phosph. Sulf. Silicon Relat. Elem. 2019, 194, 983–988. [Google Scholar] [CrossRef]
- Golub, I.E.; Filippov, O.A.; Belkova, N.V.; Epstein, L.M.; Shubina, E.S. The Mechanism of Halogenation of Decahydro-closo-Decaborate Dianion by Hydrogen Chloride. Russ. J. Inorg. Chem. 2021, 66, 1639–1648. [Google Scholar] [CrossRef]
- Shore, S.G.; Hamilton, E.J.M.; Bridges, A.N.; Bausch, J.; Krause-Bauer, J.A.; Dou, D.; Liu, J.; Liu, S.; Bin, D.; Hall, H.; et al. The Solid State Structure of [B10H11]− and Its Dynamic NMR Spectra in Solution. Inorg. Chem. 2003, 42, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Kochnev, V.K.; Avdeeva, V.V.; Malinina, E.A.; Kuznetsov, N.T. Theoretical study of H2 elimination from [BnHn+1] monoanions (n = 6–9, 11). Russ. J. Inorg. Chem. 2014, 59, 1268–1275. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Zhang, Y.; Liu, J.; Van Der Veen, S.; Duttwyler, S. The closo-Dodecaborate Dianion Fused with Oxazoles Provides 3D Diboraheterocycles with Selective Antimicrobial Activity. Chem. A Eur. J. 2018, 24, 10364–10371. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Zhang, C.-Y.; Xu, T.-T.; Wu, J.; Wen, X.-Y.; Jiang, W.-J.; Chen, M.; Yang, J. Synthesis of Polyhedral Borane Cluster Fused Heterocycles via Transition Metal Catalyzed B-H Activation. Molecules 2020, 25, 391. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.-C.; Xing, Y.-Y.; Sun, Y.; Duttwyler, S.; Hong, X. Directed B–H functionalization of the closo-dodecaborate cluster via concerted iodination—deprotonation: Reaction mechanism and origins of regioselectivity. Org. Chem. Front. 2020, 7, 3648–3655. [Google Scholar] [CrossRef]
- Voinova, V.V.; Selivanov, N.A.; Plyushchenko, I.V.; Vokuev, M.F.; Bykov, A.Y.; Klyukin, I.N.; Novikov, A.S.; Zhdanov, A.P.; Grigoriev, M.S.; Rodin, I.A.; et al. Anion—Novel Members of Diboroheterocycle Class. Molecules 2021, 26, 248. [Google Scholar] [CrossRef]
- Klyukin, I.; Zhdanov, A.; Matveev, E.; Razgonyaeva, G.; Grigoriev, M.; Zhizhin, K.; Kuznetsov, N. Synthesis and reactivity of closo -decaborate anion derivatives with multiple carbon-oxygen bonds. Inorg. Chem. Commun. 2014, 50, 28–30. [Google Scholar] [CrossRef]
- Laskova, J.; Ananiev, I.; Kosenko, I.; Serdyukov, A.; Stogniy, M.Y.; Sivaev, I.B.; Grin, M.A.; Semioshkin, A.; Bregadze, V.I. Nucleophilic addition reactions to nitrilium derivatives [B12H11NCCH3]− and [B12H11NCCH2CH3]. Synthesis and structures of closo-dodecaborate-based iminols, amides and amidines. Dalt. Trans. 2022, 51, 3051–3059. [Google Scholar] [CrossRef]
- Avdeeva, V.V.; Polyakova, I.N.; Goeva, L.V.; Malinina, E.A.; Kuznetsov, N.T. [2,6(9)-B10H8(O)2CCH3]− and [2,7(8)- B10H8 (OC(O)CH3)2]2− derivatives in synthesis of position isomers of the [B10H8(OC(O)CH3)(OH)]2− anion with the 2,6(9)- and 2,7(8)-arrangement of functional groups. Russ. J. Inorg. Chem. 2014, 59, 1247–1258. [Google Scholar] [CrossRef]
- Klyukin, I.; Kubasov, A.; Limarev, I.; Zhdanov, A.; Matveev, E.; Polyakova, I.; Zhizhin, K.; Kuznetsov, N. The new approach to formation of exo boron-oxygen bonds from the decahydro-closo-decaborate(2-) anion. Polyhedron 2015, 101, 215–222. [Google Scholar] [CrossRef]
- Oller, J.; Pérez, P.; Ayers, P.W.; Vöhringer-Martinez, E. Global and local reactivity descriptors based on quadratic and linear energy models forα,β-unsaturated organic compounds. Int. J. Quant. Chem. 2018, 118, 1–13. [Google Scholar] [CrossRef]
- Gómez, T.; Fuentealba, P.; Robles-Navarro, A.; Cárdenas, C. Links among the Fukui potential, the alchemical hardness and the local hardness of an atom in a molecule. J. Comput. Chem. 2021, 42, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Khanum, G.; Ali, A.; Shabbir, S.; Fatima, A.; Alsaiari, N.; Fatima, Y.; Ahmad, M.; Siddiqui, N.; Javed, S.; Gupta, M. Vibrational Spectroscopy, Quantum Computational and Molecular Docking Studies on 2-[(1H-Benzimidazol-1-yl)-methyl]benzoic Acid. Crystals 2022, 12, 337. [Google Scholar] [CrossRef]
- Klyukin, I.N.; Vlasova, Y.S.; Novikov, A.S.; Zhdanov, A.P.; Hagemann, H.R.; Zhizhin, K.Y.; Kuznetsov, N.T. B-F bonding and reactivity analysis of mono- and perfluoro-substituted derivatives of closo-borate anions (6, 10, 12): A computational study. Polyhedron 2022, 211, 115559. [Google Scholar] [CrossRef]
- Klyukin, I.; Vlasova, Y.; Novikov, A.; Zhdanov, A.; Zhizhin, K.; Kuznetsov, N. Theoretical Study of closo-Borate Anions [BnHn]2− (n = 5–12): Bonding, Atomic Charges, and Reactivity Analysis. Symmetry 2021, 13, 464. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Improved algorithms for reaction path following: Higher-order implicit algorithms. J. Chem. Phys. 1991, 95, 5853–5860. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New vistas in localized and delocalized chemical bonding theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Adrienko, G.A. Chemcraft, Version 1.8 (Build 530). 2017. Available online: http://www.chemcraftprog.com (accessed on 9 April 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klyukin, I.N.; Kolbunova, A.V.; Novikov, A.S.; Nelyubin, A.V.; Selivanov, N.A.; Bykov, A.Y.; Klyukina, A.A.; Zhdanov, A.P.; Zhizhin, K.Y.; Kuznetsov, N.T. Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation. Int. J. Mol. Sci. 2022, 23, 4190. https://doi.org/10.3390/ijms23084190
Klyukin IN, Kolbunova AV, Novikov AS, Nelyubin AV, Selivanov NA, Bykov AY, Klyukina AA, Zhdanov AP, Zhizhin KY, Kuznetsov NT. Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation. International Journal of Molecular Sciences. 2022; 23(8):4190. https://doi.org/10.3390/ijms23084190
Chicago/Turabian StyleKlyukin, Ilya N., Anastasia V. Kolbunova, Alexander S. Novikov, Aleksey V. Nelyubin, Nikita A. Selivanov, Alexander Yu. Bykov, Alexandra A. Klyukina, Andrey P. Zhdanov, Konstantin Yu. Zhizhin, and Nikolay T. Kuznetsov. 2022. "Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation" International Journal of Molecular Sciences 23, no. 8: 4190. https://doi.org/10.3390/ijms23084190
APA StyleKlyukin, I. N., Kolbunova, A. V., Novikov, A. S., Nelyubin, A. V., Selivanov, N. A., Bykov, A. Y., Klyukina, A. A., Zhdanov, A. P., Zhizhin, K. Y., & Kuznetsov, N. T. (2022). Protonation of Borylated Carboxonium Derivative [2,6-B10H8O2CCH3]−: Theoretical and Experimental Investigation. International Journal of Molecular Sciences, 23(8), 4190. https://doi.org/10.3390/ijms23084190