Abstract
Biallelic gene defects in MFSD8 are not only a cause of the late-infantile form of neuronal ceroid lipofuscinosis, but also of rare isolated retinal degeneration. We report clinical and genetic data of seven patients compound heterozygous or homozygous for variants in MFSD8, issued from a French cohort with inherited retinal degeneration, and two additional patients retrieved from a Swiss cohort. Next-generation sequencing of large panels combined with whole-genome sequencing allowed for the identification of twelve variants from which seven were novel. Among them were one deep intronic variant c.998+1669A>G, one large deletion encompassing exon 9 and 10, and a silent change c.750A>G. Transcript analysis performed on patients’ lymphoblastoid cell lines revealed the creation of a donor splice site by c.998+1669A>G, resulting in a 140 bp pseudoexon insertion in intron 10. Variant c.750A>G produced exon 8 skipping. In silico and in cellulo studies of these variants allowed us to assign the pathogenic effect, and showed that the combination of at least one severe variant with a moderate one leads to isolated retinal dystrophy, whereas the combination in trans of two severe variants is responsible for early onset severe retinal dystrophy in the context of late-infantile neuronal ceroid lipofuscinosis.
1. Introduction
Inherited retinal degenerations (IRD) affect about two million people worldwide and share a common feature of progressive degeneration of photoreceptors and/or retinal pigment epithelium. Three sub-types of IRD—macular dystrophy (MD), cone dystrophy (COD) and cone–rod dystrophy (CORD)—manifest in primary loss of central vision, photophobia and colour vision disturbances. Inherited MD first affects the central zone of the retina. However, some MD cases evolve into a more widespread retinal disease at the late stages due to variable cone or cone–rod dysfunction [1]. The archetypal MD is Stargardt disease (STGD1, OMIM # 248200). COD and CORD constitute a heterogeneous group of disorders primarily affecting cone photoreceptors [2,3]. COD differs from CORD by the absence of night blindness, which occurs in the latter due to concomitant rod degeneration. However, most of the patients with COD develop rod degeneration later in the progression of disease [3,4].
Since the introduction of next-generation sequencing in the field of IRD, a growing number of variants have been identified in genes known to be associated with syndromic disorders, despite a clinical presentation restricted to the retina. Isolated retinal degeneration can thus be caused by variants in USH2A [5,6,7], BBS1 [8], or more recently, CLN3 [9,10] and CLN5 [11]. Lately, MFSD8 was incriminated in autosomal recessive MD, COD or CORD [12,13,14,15,16,17]. MFSD8 (major facilitator superfamily domain-containing 8) (MIM * 611124, NM_152778.3) was originally associated with neuronal ceroid lipofuscinosis (NCLs), a group of severe neurodegenerative disorders, characterized by the lysosomal accumulation of abnormal autofluorescent material (lipofuscin-like ceroid lipopigments) leading to selective damage and loss of neurons [18,19]. Pathogenic variants in MFSD8 cause a variant late-infantile form of NCL (vLINCL) called CLN7 disease (MIM # 610951) [20]. The clinical course includes psychomotor decline, seizures, visual failure and reduced lifespan [21]. MFSD8 encodes a CLN7 protein localized to the lysosomal membrane [22,23,24]. Its exact function remains unclear, but roles in lysosome trafficking [25,26] have been supported.
In this study, we report ocular phenotype of patients with MFSD8-related isolated retinal dystrophy and LINCL, describe novel MFSD8 variants and their functional repercussion, and present a tentative of genotype–phenotype correlation.
2. Results
2.1. Patients Description
MFSD8 variants have been identified in six unrelated patients with isolated retinal dystrophy and in one patient with late-infantile neuronal ceroid lipofuscinosis. Clinical data of patients on initial examination are summarized in Table 1 and follow-up data for the five oldest patients in Table S1. At first examination, two patients had a clinical picture of MD, three patients had COD, one patient presented CORD and one patient early onset severe retinal dystrophy (EOSRD).

Table 1.
Clinical findings.
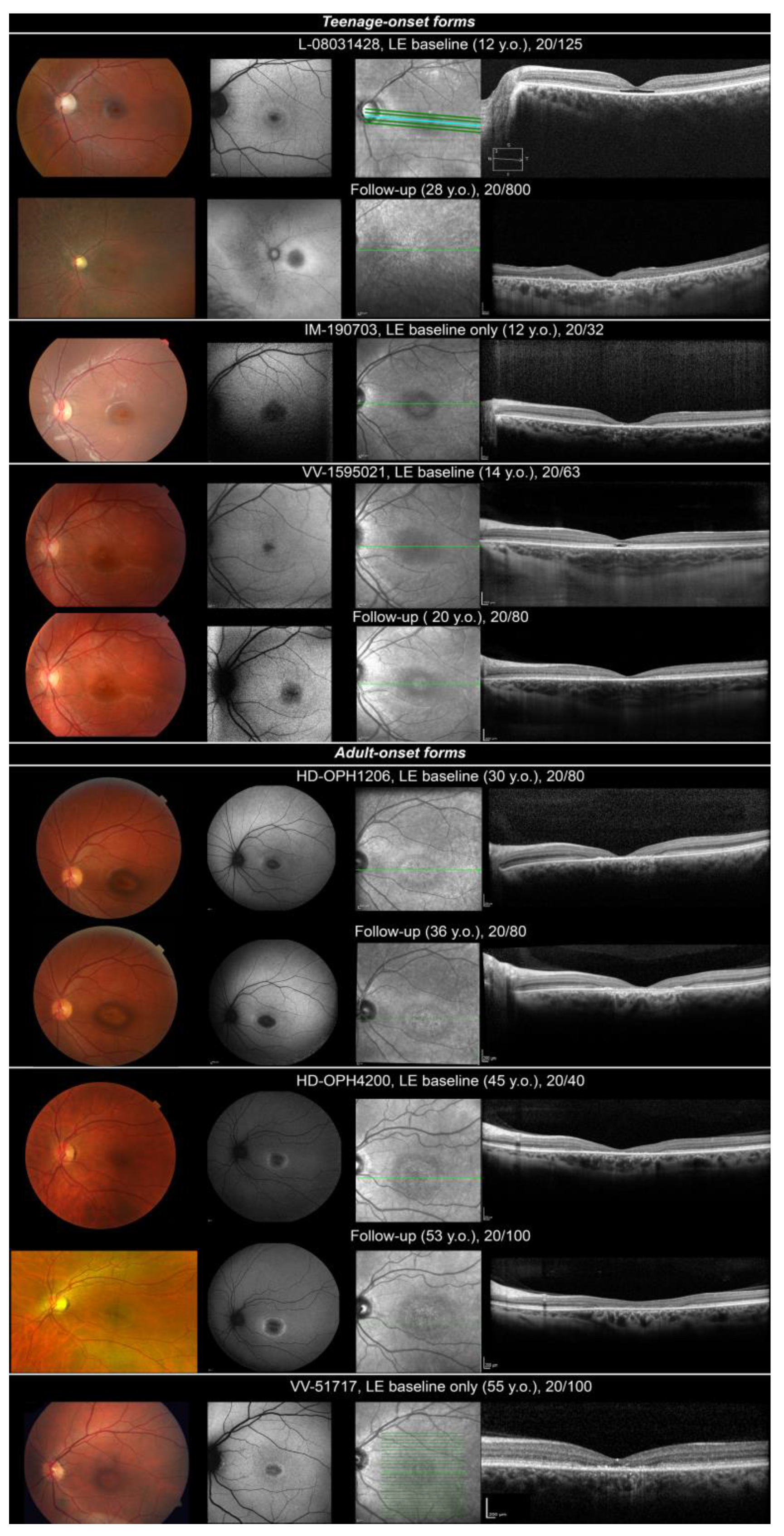
Three patients (L-08031428, VV-1595021, IM-190703) presented with teenage-onset (age 12, 12 and 14, respectively) MD, COD or CORD. First complaints were progressive visual loss, poor colour discrimination and photophobia. When realized, visual fields depicted relative central scotoma. Colour vision tests were variably affected (from normal colour perception to without-axis severe errors on ranking tests). ffERG was normal for one patient (VV-1595021: MD). Light-adapted responses only were reduced in one case (IM-190703: COD). One patient depicted a cone–rod pattern of retinal dysfunction (L-08031428: CORD, Figure S1). Fundus examination revealed an optic disc pallor and a punched-out atrophic round macular lesion with slightly hyperpigmented borders (Figure 1, teenage-onset forms). Foveal lesion appeared dark with hyperautofluorescent edges on SW-FAF. SD-OCT found either an aspect of foveal cavitation in outer reflective layers (L-08031428, VV-1595021) or a larger destruction of outer reflective layers (IM-190703) in the macula. Long-term follow-up data were available for two patients (L-08031428, VV-1595021). BCVA gradually worsened. Patient L-08031428 became legally blind at 25 years old. Light-adapted ffERG responses became undetectable at 28 years. ffERG was not repeated for VV-1595021. Initial foveal lesion progressed to a bull’s eye maculopathy in both patients and then to mid-peripheral retinal depigmentation with bone spicule-like pigment migration in L-08031428. SD-OCT aspect of foveal cavitation collapsed by 20 years in both patients and then progressed to a widespread destruction of outer reflective layers in L-08031428.
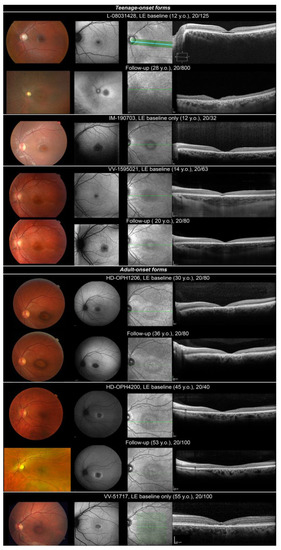
Figure 1.
Ocular findings in isolated retinal dystrophy linked to MFSD8 variants. Fundus photography, SW-FAF, IRR and HD-OCT are shown in successive rows. Top, teenage-onset maculopathy/COD/CORD. Optic disc pallor and a punched-out atrophic foveal lesion with slightly hyperpigmented borders. Foveal lesion looked dark with hyperautofluorescent edges on SW-FAF. SD-OCT found either a temporary aspect of foveal cavitation in outer reflective layers (L-08031428, VV-1595021) or a larger destruction of outer layers (IM-190703) in the macula. Slow progression of macular lesions at follow-up, progression to a more widespread retinal lesions with mid-peripheral involvement in patient L-08031428. Bottom, adult-onset maculopathy/COD. Bull’s eye maculopathy. Dark fovea with hyperautofluorescent edges on SW-FAF corresponding to a more or less large interruption of outer reflective layers on SD-OCT. Slow progression of macular lesion at follow-up.
Three patients (HD-OPH1206, HD-OPH4200, VV-51717) presented adult-onset (age 30, 37 and 55, respectively) maculopathy or COD. Retinal picture was quite similar. Patients complained of progressive BCVA loss (mostly reading difficulties) and photophobia. Visual field discovered central scotoma. ffERG was normal (VV-51717, MD, Figure S1) or depicted a reduction in light-adapted responses (HD-OPH1206 and HD-OPH4200: COD). Fundus depicted bull’s eye maculopathy (Figure 1, adult-onset forms). It was a dark fovea with hyperautofluorescent edges on SW-FAF corresponding to a more or less large interruption of outer reflective layers on SD-OCT. On follow-up, patients depicted slow progression of macular lesions.
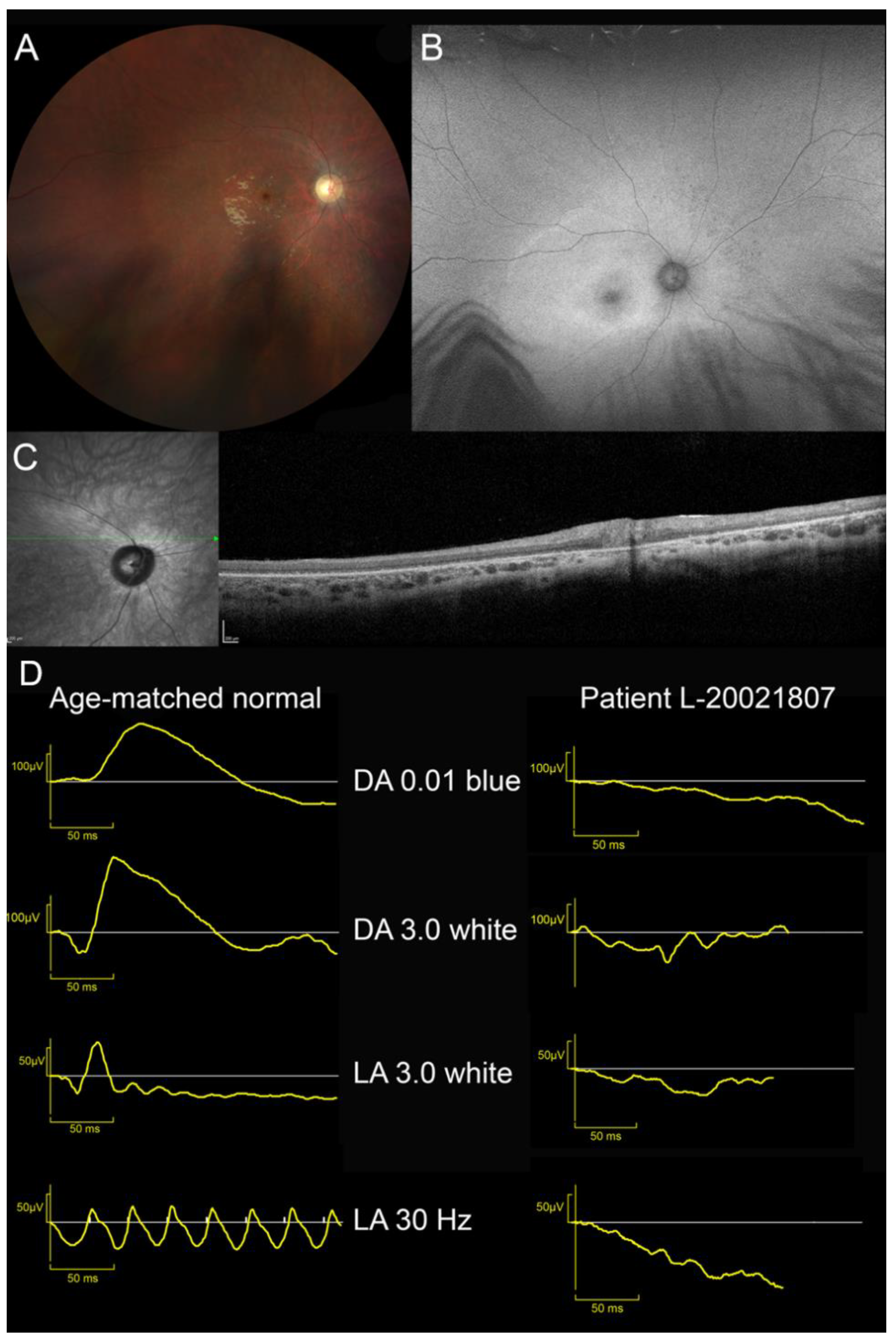
One patient presenting LINCL was also identified (L-20021807). Visual acuity loss was first reported at the age of 6. He complained of mild photophobia. Myoclonic seizures appeared at 8 and required polymedication (levetiracetam, carbamazepine). Despite this therapy, seizures were only partially controlled. Brain MRI discovered upper vermis atrophy. At the age of 9, parents noted behavioural changes: attention deficit and aggressivity. This patient was referred to our department at 10 years old. BCVA was limited to 20/400 OU; patient had eccentric fixation with “overlooking”. Kinetic perimetry disclosed a 20° relative central scotoma at III3e target. Peripheral isopter at V4e target was full. ffERG was unrecordable (Figure S1). Fundus examination revealed a waxy pallor of the optic disc, severe vascular attenuation and macular depigmentation. There was a cellophane light reflex and yellowish discoloration of the macula (Figure 2). SW-FAF showed a large area of increased autofluorescence with indistinct border in the posterior pole and the second more narrow ring of increased autofluorescence around the fovea. Both foveae were irregularly hypoautofluorescent. The peripheral retina was isoautofluorescent. On SD-OCT, the retina was thin with indistinct lamination and disappearance of outer reflective layers.
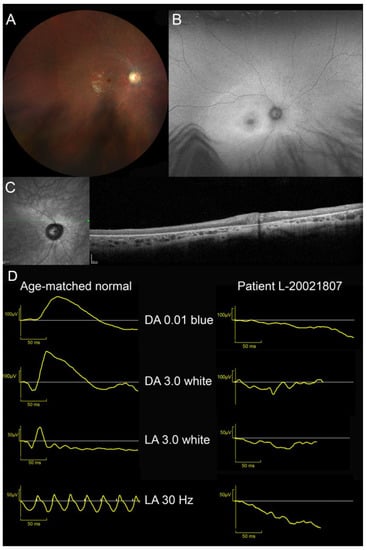
Figure 2.
Ocular findings in patient with late-infantile neuronal ceroid lipofuscinosis (MFSD8-LINCL). (A) Waxy pallor of optic disc, severe vascular attenuation and whitish, depigmented appearance of posterior pole and midperipheral retina. Cellophane light reflex from the macula. (B) SW-FAF showed a large area of increased autofluorescence with indistinct border in the posterior pole and the second more narrow annulus of increased autofluorescence around the fovea. Fovea was irregularly hypoautofluorescent. Peripheral retina was isoautofluorescent. (C) SD-OCT. Retina was thin with indistinct lamination and widespread disappearance of outer layers (ONL, EZ and RPE). (D) ffERG. Unrecordable responses under dark- and light-adapted conditions.
2.2. Genotyping and Transcript Analysis
Variants in the MFSD8 gene have been identified in five patients in a cohort of 1049 IRD cases analysed by next-generation sequencing (NGS) of large panels, among which 340 presented MD, COD or CORD (Figure S2). Two additional patients from a Swiss cohort were added to this group. Molecular analysis revealed the presence of twelve different variants in MFSD8, among which seven were novel: four missense, one nonsense, one deep intronic variant and one large deletion encompassing two exons. A silent change found twice in our cohort was reported by Reith and colleagues during the submission of this manuscript [27].
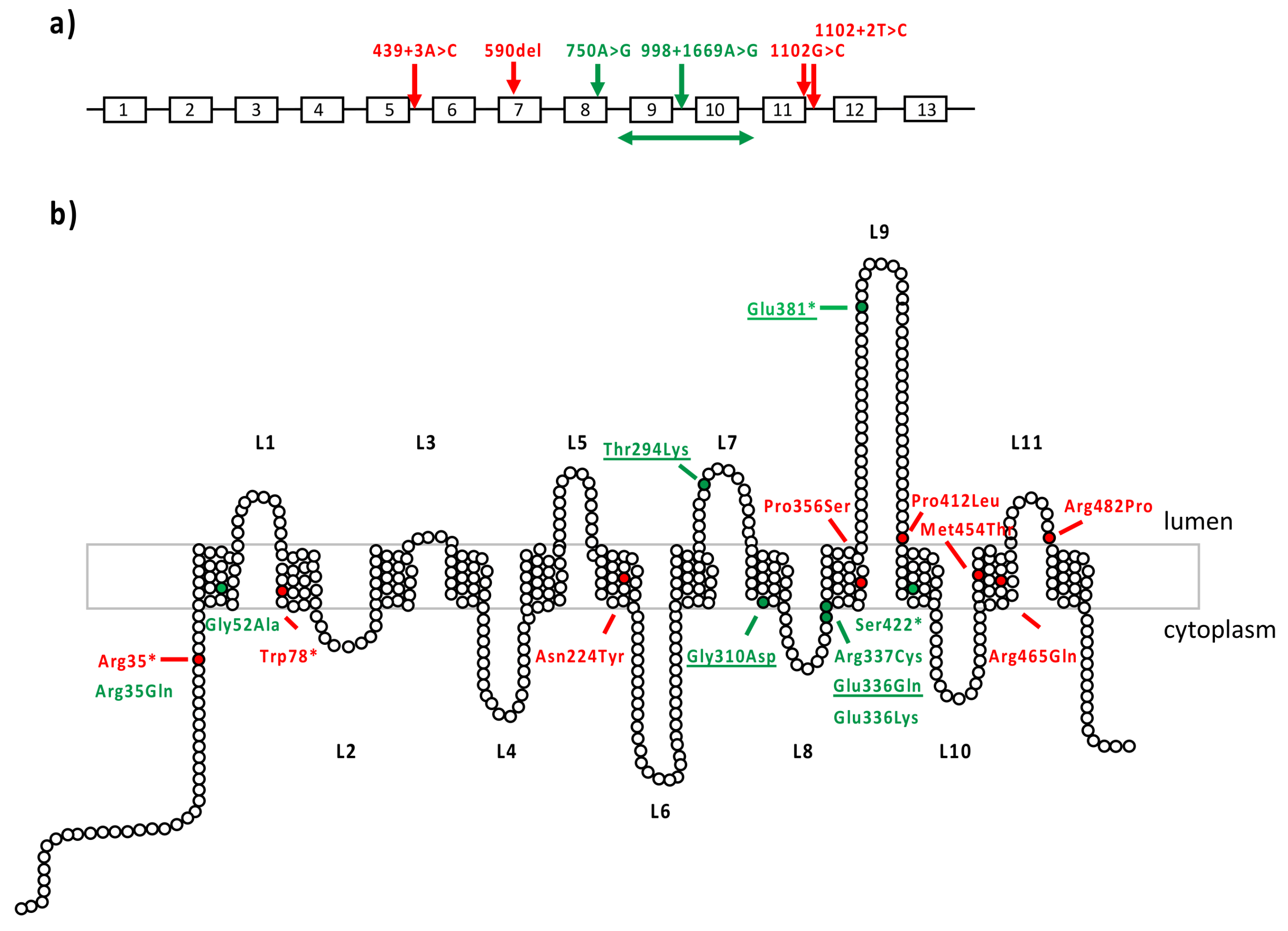
All six patients with isolated retinal dystrophy carried at least one missense variant, the second hit being either another missense or a loss-of-function variant (Table 2). Among the novel missense variants, the c.1006G>A, p.(Glu336Lys) variant has never been reported before, while the c.1006G>C p.(Glu336Gln) was previously described in MD [12,13] and identified in two patients with MD in this series. Residue Glu336 is located in the extracellular loop L9 (Figure 3). Variants c.104G>A p.(Arg35Gln) and c.155G>C p.(Gly52Ala) are associated in a complex allele. The pathogenic variant of this allele could be p.(Gly52Ala), classified as possibly pathogenic variant class 4, while p.(Arg35Gln) is predicted to be a variant of unknown significance according to ACMG 2015 criteria. The novel p.(Arg337Cys) variant is rare and presents a high CADD score.
Table 2.
MFSD8 (NM_152778.3) genotypes identified in our cohort.
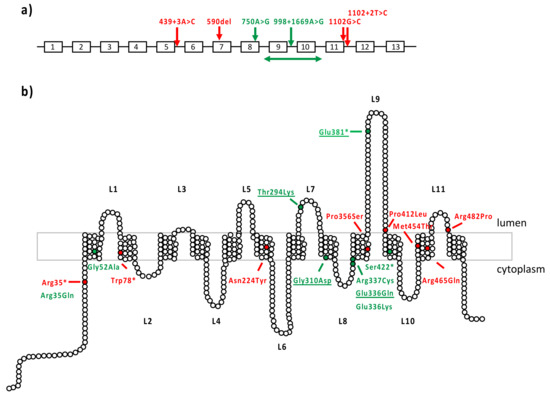
Figure 3.
MD-associated MFSD8 variants. (a) Loss-of-function variants reported in macular dystrophy. MFSD8 gene is represented with its 12 exons. Variants are annotated according to the cDNA nomenclature. In red are variants from the literature and in green are novel variants identified in this study. (b) Missense variants reported in macular dystrophy. CLN7 is composed of 12 transmembrane domains and 11 extracellular or cytoplasmic loops. The 518 amino acids of CLN7 protein are depicted as white circles. In red are variants from the literature; in green are novel variants identified in this study; underscored are the already reported variants found in our study.
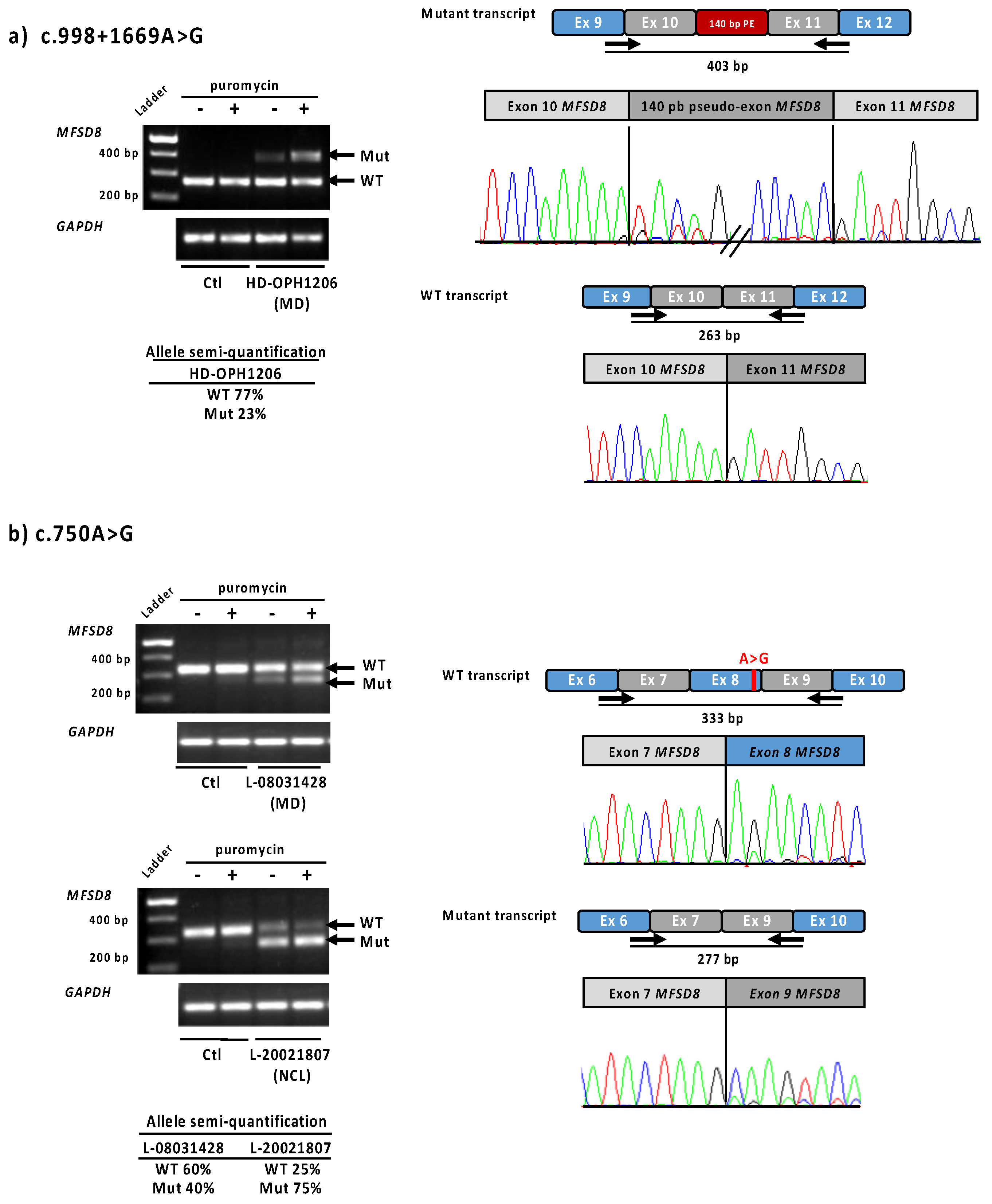
Because only one variant was found in the MFSD8 gene by NGS in patient HD-OPH1206, we completed the genetic study by whole-genome sequencing. Intron analysis enabled the identification of a second anomaly in trans: the novel deep intronic variant c.998+1669A>G (Figure S3a). This variant was predicted to create a splice donor site (SpliceAI donor gain score 0.68) (Table 2), with an acceptor site 140 bp upstream of this position (SpliceAI acceptor gain score 0.30). The transcript study in patient LCLs confirmed the insertion of a 140 bp pseudoexon between exons 10 and 11 (Figure 4a). The semi-quantification showed 77% of residual normal transcript, corresponding to the sum of the wild-type and the mutant alleles. The specific fraction due to the mutant c.998+1669A>G allele can thus be estimated at around 50%.
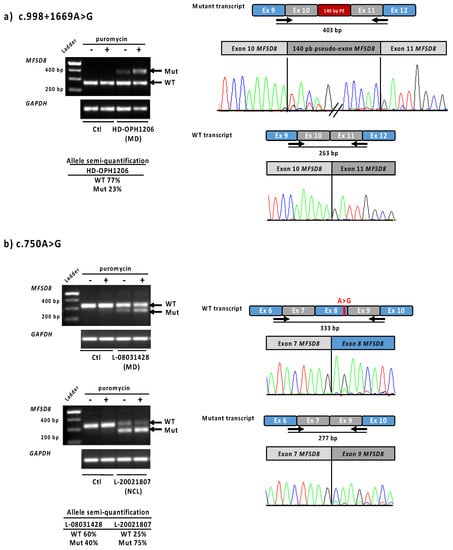
Figure 4.
Functional tests results for MFSD8 variants c.998+1669A>G and c.750A>G in LCLs Lymphoblastic cell lines from patients HD-OPH1206, L-08031428 and L-20021807 were used to analyse MFSD8 cDNA expression. (a) Overlapping primers located at the exons 9–10 and 11–12, respectively were used to show the inclusion of a 140 bp pseudoexon due to the c.998+1669A>G variant. On the right, electropherograms obtained from the upper and lower bands separated on agarose gel. (b) The exon 8 skipping due to c.750A>G variant was confirmed using primers located at exons 6–7 and 9–10 junctions. The exon 8 skipping is observed in control at a very low level, suggesting exon 8 as an alternative cassette, normally expressed, and enhanced by the presence of this variant. Semi-quantification of the bands (using primers located in exon 7 and 9) has been performed on cells not treated by puromycin. Ctl—control; MD—macular dystrophy; NCL—neuronal ceroid lipofushinose.
One patient with MD (L-08031428, 12 years old) and one with LINCL (L-20021807, 10 years old), both native from the North of France, carried the same variant c.750A>G, not reported in gnomAD. This presumably silent change variant was located five nucleotides upstream of the 3′ end of exon 8, and the SpliceAI splice prediction tool suggested a splice donor site loss (score 0.45). Transcript analysis of lymphoblastoid cell lines of both patients revealed the presence of one normal transcript and another shorter one lacking exon 8, in accordance with in silico prediction (Table 2, Figure 4b). The exon 8 loss is an out-of-frame deletion, and therefore leads to a null allele. The higher expression of the mutant in the presence of puromycin confirmed the transcript degradation by the nonsense-mediated decay (NMD) pathway. The transcript semi-quantification differed between the two families: before puromycin treatment, 25% of the normal transcript remained for patient L-20021807 (LINCL), and 60% for patient L-08031428 (MD). Of note, in LCLs, the two MFSD8 alleles cannot be separated and are both part of the semi-quantification. The remaining normal transcript amount for the LINCL case reflects the specific effect of the mutant c.750A>G allele, as the other allele carrying the large exons 9–10 deletion undergoes NMD. On the contrary, 60% of the residual normal transcript in MD patient corresponds to the sum of the wild-type and mutant MFSD8 alleles.
In patient L-20021807, the second variant is a large deletion of exons 9 and 10, c.(755-2726_998+1981)delinsGTA (Figure S3b). It was first detected by an NGS panel and then confirmed by whole-genome sequencing which defined the exact borders. This 7118 bp deletion leads also to a frameshift and therefore to a null allele. Repetitive elements were investigated at the breakpoints to decipher the causal mechanism. DNA elements and one LINE sequence have been identified at the 5′ and 3′ breakpoints, respectively.
3. Discussion
This study of seven patients carrying MFSD8 variants showed that isolated retinal diseases are associated with combination of one severe and one mild or moderately severe variant, whereas the syndromic form with an EOSRD phenotype seems to be related to severe variants. Our results confirm that MFSD8 is a rare cause of retinal degeneration as patients carrying biallelic variants in this gene represent only 5/1049 (0.47%) of the total number of cases sequenced by large NGS panels and 5/340 (1.5%) of the patients with macular dystrophy or cone/cone–rod dystrophy. This is very different from ABCA4, the most prevalent gene related to autosomal recessive MD and COD/CORD (597/1293, 46% in our cohort). However, MFSD8 should be included in NGS strategies when investigating patients with IRD. It is very likely that some of our unsolved patients analysed by a small, targeted gene panel in the past (n = 356, Figure S2) are carriers of MFSD8 variants. A complementary study would allow us to address this point.
3.1. Common or Specific Clinical Features
Patients with isolated retinal dystrophy linked to MFSD8 presented MD, COD or CORD. In two patients (L-08031428 and VV-1595021), a foveal cavitation was an early OCT feature before the collapse of inner reflective layers occurring by their twenties. Two group of age at onset were clearly distinct: early teenage onset (from 12 to 14 years in our patients) and adulthood onset (from 30 to 55 years).
The MFSD8-linked macular lesions mimic Stargardt disease and ABCA4-related COD and CORD: (i) two similar peaks of onset ages: late childhood [32,33] and late adulthood [34,35]; (ii) a typical foveal cavitation progressing to a collapse of outer retina and then to a bull’s eye macular lesion in early onset forms [36,37]; (iii) a subset of STGD1 patients had a cone or cone–rod pattern of retinal dysfunction at ffERG [38,39], as seen in patients in this series. However, macular lesions in our MFSD8 patients were grossly roundish (except for HD-OPH1206), while they are usually larger in STGD1. No patient in our series developed satellite hyperautofluorescent flecks, typical in STGD1 [40], and no peripapillary retinal sparing [41,42] was observed.
Clinical presentation of patient L-20021807 with LINCL was more precocious and severe: he was legally blind before the age of 10. Retinal degeneration with unrecordable ffERG and a widespread outer reflective layer destruction on SD-OCT was a feature at first examination. There was no similarity with STGD1, neither in retinal function (unrecordable ffERG responses), nor in fundus (waxy pallor of optic disc, vascular attenuation and posterior pole depigmentation) or in multimodal imaging appearance (ring of increased autofluorescence on SWAF and widespread outer retinal layer destruction on HD-OCT). This clinical phenotype is in keeping with EOSRD [43,44] and is reminiscent of other types of NCL [45,46,47].
3.2. Novel Splice Defects
We report here the first deep intronic variant in the MFSD8 gene, leading to the creation of a splice donor site in intron 10, and producing a 140 bp pseudoexon insertion. Introns of MFSD8 have not yet been fully investigated, and it is still unknown if this variant could be recurrent in other IRD or NCL cases. This discovery underscores the need to complete the full sequencing of MFSD8 in unsolved cases and in cases where only one pathogenic MFSD8 variant has been found. Deep intronic splice variants could be good candidates for antisense oligonucleotide therapy, as was previously shown for MFSD8 [48]. Deep intronic variants in ABCA4 are also frequently observed in Stargardt disease [49].
The second splice defect identified in our patients was c.750A>G, firstly predicted as p.(Glu250=). During the revision in this manuscript, this variant was published at the homozygous state in patients presenting a neuronal ceroid lipofuscinosis [27]. It was previously reported once in the LOVD database as likely benign, due to its supposed absence of effect on the protein. However, it has never been observed in gnomAD nor in our 3348 IRD patients’ cohort, and was found in two patients, both sharing the same geographic origin and carrying a second likely pathogenic variant in MFSD8 (Table 2). Transcript analysis in immortalized lymphocytic cell lines confirmed exon 8 skipping in the major transcript. Another splice variant, c.754+2T>A has been described by Siintola et al., in 2007 [50], who also showed exon 8 skipping with this variant. In this paper, exon 7 seems also to be alternatively spliced. According to the UCSC database, MFSD8 contains 10 transcripts differing in their first coding exon and the presence of alternative cassettes: exons 2, 6, 7 and 8 (using NM_152778.3 as a reference). We observed that exclusion of exon 8 is present at very low levels in normal conditions (Figure 4b), but that the c.750A>G variant enhanced the percentage of exclusion in patients. As shown in GTEx portal (https://gtexportal.org/home/gene/MFSD8, 6 March 2022), the alternative exon 8 is differentially expressed in tissues. The presence of the variant could modify the exonic splice regulatory sequence recognized by transcription factors, as predicted by HExoSplice [51]. The exact function of the different MFSD8 transcripts is still unknown, but a differential expression in tissues [50] might be linked to specific expression in the splice factors repertory and could lead to specific functions such as those reported for other transcripts, such as CRB1, REEP and DFNB31 [52,53,54].
3.3. Genotype–Phenotype Correlations
All MFSD8 variants associated with isolated retinal dystrophy (MD, COD/CORD) are located in the transmembrane alpha helices (Figure 3), except p.(Glu381*), located at the most extracellular side of loop 9, nearby the two proteolytic cleavage sites Asn370 and Asn376. Residue Glu336 is located in extracellular loop L9. Electrochemical charge change due to this substitution can destabilize the protein conformation in modulating interactions. However, the Glu-to-Lys substitution is considered moderately conservative as the Grantham score is 56, whereas the Glu-to-Gln change is conservative (Score: 29) [31]. A mild impact is therefore expected for both. Variants c.104G>A p.(Arg35Gln) and c.155G>C p.(Gly52Ala) are associated in a complex allele. The pathogenic variant of this allele could be p.(Gly52Ala), classified as possibly pathogenic variant class 4, while p.(Arg35Gln) is a predicted variant of unknown significance according to ACMG 2015 criteria. They are located in or close to the first transmembrane domain of the protein, p.(Arg35Gln) is on the external cytosolic side of the protein, and interacts with amino acids Ala159, and Glu164 belongs to another alpha helix domain and with Glu27 located at the junction between the first transmembrane domain and the N-terminal extremity (Supplementary Figure S4). Again, this substitution is conservative (Grantham score: 43). Variant p.(Gly52Ala) is more internal, in an helix encumbered by aromatic residues (Phe53, Tyr83, Phe180). A somewhat higher impact of this change is predicted (Grantham score: 60), but both residues are small and no dramatic destabilization is expected. The p.(Gly52Ala) variant could be the cause for the change in the complex allele, especially since another variation targeting the same residue, p.(Gly52Arg), was already reported as causal in NCL [55], but with a higher impact since it replaces a small residue by a larger and positively charged polar one. The novel p.(Arg337Cys) variant is rare and presents a high CADD score. The substitution Arg into Cys is predicted to have a high impact (Grantham score: 180) as it replaces a long, positively charged residue interacting through hydrogen bonds with Tyr503 and Glu518, located in the last transmembrane domain and in the last amino acid residue, respectively, by a short residue able to form disulphide bridges. On the other hand, the well-known variant p.(Thr294Lys) is a moderately conservative change (Grantham score: 78). Thr294 is located in loop 7, in a very buried zone, encumbered by large aromatic residues Trp300, Tyr298, Phe419 and interacting with Ile290, Thr291 Asn309 by hydrogen bonds. The substitution of a polar, uncharged amino acid residue by a positively charged amino acid residue could modify the protein conformation. Based on 3D structure changes, we observed no substantial destabilization of the protein for most of the novel missense variants reported in our series. We therefore considered them as moderately severe variants (Table 3).
Table 3.
MFSD8 variants found in both MD and NCL patients from literature analysis.
Recurrence was observed for some variants in patients presenting NCL: c.929G>A, p.(Gly310Asp) [50,55] and c.881C>A, p.(Thr294Lys) [24,55,57,58], for which a founder effect has been reported in the population originating from the former Czechoslovakia [56]. Interestingly, in NCL cases, these two variants are in trans with another severe variant (frameshift, splice site variants) or are homozygous, suggesting a severe effect (Table 3). The high pathogenicity of the p.(Thr294Lys) variant was proved by the increase in proteolytic cleavage, leading to a loss of function of the protein [24]. On the contrary, in isolated retinal dystrophy cases from our series, p.(Gly310Asp) and p.(Thr294Lys) are in combination with either a splice variant with partial effect (patient HD-OPH1206) or with p.(Glu336Gln) (patients HD-OPH4200 and VV-51717). The latter is predicted to be a mild allele by in silico analysis and was only reported in MD cases in trans with a loss of function variant [12,13] (Table 2 and Table 3). Regarding the two splice variants investigated here, both produced a partial effect with residual normal transcript, estimated at 25% for c.750A>G and at 50% for c.998+1669A>G, the mutant alleles being degraded by nonsense-mediated decay (NMD) as confirmed by puromycin treatment. The level of the remaining wild-type variant allowed us to consider c.998+1669A>G as a moderately severe variant, and c.750A>G as a severe variant. In MD cases, these two variants are in trans with a missense variant. However, in HD-OPH1206, the missense variant p.(Gly310Asp) is predicted to be the severe variant of the genotype, while in L-08031428, the severe variant should be the c.750A>G (Table 2). In the young patient with LINCL carrying the c.750A>G (L-20021807), the other allele is inactivated by the loss of two exons, leading to a very low level of protein expression. This patient, carrying two severe variants, exhibited therefore a more severe phenotype. The severe effect of this c.750A>G variant was confirmed by the recent description at the homozygous state in two patients from the same family with NCL [27]. As shown in Table 2 and Table 3, all isolated retinal dystrophy cases of our series and from literature harboured one severe and one mild or moderately severe MFSD8 variant, whereas all LINCL cases carried two severe MFSD8 variants. The combination in trans of two types of variants, and their impact on protein dysfunction, could therefore explain why a same variant can be found in both early and late phenotypes. The degree of severity is correlated with the residual protein expression in the tissue. The total expression of the CLN7 protein in patients’ cells would be necessary to further confirm this hypothesis.
4. Materials and Methods
4.1. Cohort of Patients
Patients harbouring MFSD8 variants were part of a French cohort including 3348 patients with inherited retinal degeneration (IRD) analysed in our laboratory between 2014 and 2021. Only 1049 patients analysed using a large NGS panel were considered in the following description. This cohort is divided into two groups: (i) a group of 709 patients presenting rod–cone dystrophy, Leber congenital amaurosis, congenital stationary night blindness, or Choroideremia (“Other IRD” in flowchart in Supplementary Figure S2); (ii) a group of 340 patients presenting cone or cone–rod dystrophy and macular dystrophy including Stargardt disease. Two additional patients were retrieved from an independent Swiss IRD cohort followed at the Jules-Gonin Eye Hospital and analysed in the Institute for Research in Ophthalmology. Those patients were included to strengthen the clinical and genetic description of MFSD8 variants, rarely reported in isolated macular dystrophy.
4.2. Clinical Examination
Prior to testing, written informed consent was obtained from each study participant. The study protocol adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committees (the list is provided in Institutional Review Board Statement below).
Clinical data were retrospectively collected from medical records. They included sex, age at time of first symptoms, diagnosis and examination, personal or familial history, complaints, best-corrected visual acuity (BCVA), refractive error, slit-lamp biomicroscopy, colour vision (either Ishihara album, Lanthony D-15 panel or Farnsworth 100 Hue) static and kinetic visual fields (VFs), full-field electroretinogram (ffERG according to the standards of International Society for Clinical Electrophysiology of Vision [61]), fundus photography, spectral domain optical coherence tomography (SD-OCT: Spectralis OCT, Heidelberg Engineering, Inc., Heidelberg, Germany) and short-wavelength autofluorescence (SWAF: Heidelberg Retinal Tomograph, Heidelberg Engineering, Inc., Heidelberg, Germany). Only patients presenting MD, COD or CORD were included in the study.
4.3. Genetic Assessment
Among the 3348 probands investigated in our laboratory, 1049 were genetically assessed using a large NGS panel of 156 or 230 genes [62] (the latest being an updated version of the first “156 genes” panel) (Figure S2). Both panels contained the MFSD8 gene. The second part of the cohort, comprising 2299 patients, was analysed using an NGS targeted gene panel, which did not contain the MFSD8 gene. This group was therefore excluded from the study. Among the 340 patients analysed with the large panel and presenting cone, cone–rod dystrophy and macular dystrophy, five of them harboured variants in MFSD8 gene. It should be noted that none of the 709 patients with “other IRD” carried a MFSD8 variant.
Capture oligonucleotide probes were designed using the Haloplex target enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). DNA libraries were sequenced on a NextSeq500 sequencer (Illumina Inc., San Diego, CA, USA). The data analysis was conducted using an in-house developed pipeline compiling the data obtained from Seqnext (JSI Medical System, Ettenheim, Germany) and GATK software. All single-nucleotide variations were confirmed by bidirectional Sanger sequencing of the MFSD8 exons and exon–intron boundaries (MIM * 611124, NM_152778.3). PCR and sequencing conditions are available on request (see primers list in Supplementary Table S2). Segregation analysis was performed when family members’ DNA samples were available.
A targeted MFSD8 complete gene sequencing of patient HD-OPH1206 was performed to search for the second intronic variant (BGI Genomics, Hongkong). High-quality genomic DNA samples were randomly fragmented by Covaris Technology. Fragments of 350 bp were selected. After ligation of adapters, followed by amplification by ligation-mediated PCR (LM-PCR), single-strand separation, and cyclization, DNA fragments were read through on the BGISEQ-500 platform. Sequencing-derived raw image files were processed by BGISEQ-500 base-calling Software (v1) with default parameters, and the sequence data of each individual were generated as paired-end reads. Data of each sample were mapped to the human reference genome (GRCh37/HG19) using Burrows–Wheeler Aligner software (v0.7.17). Calling was performed using Genome Analysis Toolkit. Variant calling was targeted on MFSD8 intronic variants, filtering on SpliceAI predictions (Score > 0.2).
4.4. Copy Number Variants (CNVs)
The detection of CNVs was performed by quantitative analysis of the data obtained from the bam files. For each patient and for each target (i.e., exon), we calculated the ratio of the depths of the reads for this target/depth of the reads for all targets, and divided this ratio by the full mean coverage for all control samples analysed on the same NGS run. A ratio value of 1 means that copy number is identical to the control samples, and a ratio value of 0.5 reveals only one copy of the allele and a heterozygous deletion. Borders of the CNV identified in patient L-20021807 have been obtained by whole-genome sequencing (BGI Genomics, Hongkong) according to the above protocol.
4.5. Variant Pathogenicity Assessment
Variant pathogenicity was assessed through our in-house pipeline embedding commercially available bioinformatics software and using data available from public variant databases in accordance with the Guidelines of the American College of Medical Genetics and Genomics [28].
4.6. Lymphoblastoid Cell Lines’ (LCLs) Production
To assess the pathogenicity at the RNA level, of the two splice variants, we requested novel samples for the 3 probands carrying the c.998+1669A>G and c.750G>A variants. Lymphocytes were collected from patient blood samples and EBV (Epstein–Barr Virus) immortalized using a Transfokit-EBV kit provided by the University Hospital of Reims (France). The immortalized lymphocytes were cultured in suspension in RPMI (Gibco™, LifeTechnologies, Carlsbad, CA, USA) supplemented with 15% foetal bovine serum (Gibco™, LifeTechnologies, Carlsbad, CA, USA) and 0.5% Penicillin–Streptomycin (Sigma-Aldrich, St Louis, MO, USA) at 37 °C under 5% CO2. After a few weeks, the culture was separated to prepare two cell pellets treated 5h with or without puromycin (Sigma-Aldrich, St Louis, MO, USA), and an NMD inhibitor, at a final concentration of 200 µg/mL.
4.7. Transcript Analysis
RNA extractions from the LCLs pellets were performed with the Nucleospin® RNA kit (Macherey-Nagel, Hoerdt, France) according to the supplier’s protocol. The DNased RNAs obtained were assayed with Nanodrop and the quality was checked using with the Agilent RNA 6000 Nano Chips kit (Agilent Technologies, Santa Clara, CA, USA) on Bioanalyzer 2100. cDNAs were synthesized using the AffinityScript cDNA Synthesis Kit (Agilent Technologies) according to the manufacturer’s recommendations, and regions of interest of cDNAs were amplified using the Taq DNA Polymerase Invitrogen kit (InvitrogenTM, Waltham, MA, USA). The PCR products were separated on a 1.5% agarose gel at 120V for 1 h. Quantification of bands on agarose gel were carried out with the Image Lab 6.1 software (Bio-rad). PCR products were purified and sequenced by the Sanger method with the 3730XL DNA Analyzer (Applied BiosystemsTM, Waltham, MA, USA). The sequences were read using the SeqScape™ software (Applied Biosystems™).
5. Conclusions
Isolated retinal dystrophy associated with MFSD8 gene defects manifests as MD, COD or CORD, and could mimic STGD1. Retinal degeneration in MFSD8-linked LINCL presents as EOSRD, and is clearly distinguishable from isolated phenotypes. In silico and in cellulo studies of variants identified in this study and in previously reported patients allowed us to assign a pathogenic effect to variants, and showed that the combination of at least one severe variant with a moderate one leads to isolated retinal dystrophy, whereas the combination in trans of two severe variants is responsible for an EOSRD in the context of late-infantile neuronal ceroid lipofuscinosis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23084294/s1.
Author Contributions
Conceptualization, C.-M.D., A.F.P., O.G., V.M.S.; Investigation, all co-authors; Data Curation, C.-M.D., A.F.P., O.G., V.M.S.; Writing—Original Draft Preparation, C.-M.D., A.F.P., O.G., V.M.S.; Writing—Review and Editing, all co-authors; Supervision, C.-M.D. All authors have read and agreed to the published version of the manuscript.
Funding
This work was performed within the framework of SENSGENE and ERN-EYE. C.-M.D., A.F.P., O.G, S.D.-D. and V.M.S. were supported by Retina France.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the following French research ethics committees: ID-RCB 2020-A02559-30 (for patients of V.M.S., I.D., S.D.-D., F.J., A.-G.L.), IRB-MTP-2021-11-202100959 (for patients of I.M.), EST-IV N°DC-2014-2222 (for patients of H.D.), the Swiss ethics committee DFSP 035.003-48 (for patients of V.V., M.G.T., D.F.S.). The two patients from V.M.S., I.D, S.D.-D. were indexed on https://clinicaltrials.gov/ (NCT04658251, 1 November 2020). The total Lille cohort is declared under the DC-2008-642 number.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
All data are contained within the article or Supplementary Materials.
Acknowledgments
We are grateful to C. Piriou and E. Gorecki (Lille University Hospital) for their technical assistance. The authors are thankful to the patient and family members participating in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Georgiou, M.; Kane, T.; Tanna, P.; Bouzia, Z.; Singh, N.; Kalitzeos, A.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. Am. J. Ophthalmol. 2020, 211, 159–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosing, S.; Thiadens, A.A.H.J.; Hoyng, C.B.; Klaver, C.C.W.; den Hollander, A.I.; Cremers, F.P.M. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014, 42, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboshiha, J.; Dubis, A.M.; Carroll, J.; Hardcastle, A.J.; Michaelides, M. The cone dysfunction syndromes. Br. J. Ophthalmol. 2016, 100, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ávila-Fernández, A.; Cantalapiedra, D.; Aller, E.; Vallespín, E.; Aguirre-Lambán, J.; Blanco-Kelly, F.; Corton, M.; Riveiro-Álvarez, R.; Allikmets, R.; Trujillo-Tiebas, M.J.; et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol. Vis. 2010, 16, 2550–2558. [Google Scholar] [PubMed]
- Colombo, L.; Maltese, P.E.; Castori, M.; El Shamieh, S.; Zeitz, C.; Audo, I.; Zulian, A.; Marinelli, C.; Benedetti, S.; Costantini, A.; et al. Molecular Epidemiology in 591 Italian Probands With Nonsyndromic Retinitis Pigmentosa and Usher Syndrome. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef] [PubMed]
- Goetz, K.E.; Reeves, M.J.; Gagadam, S.; Blain, D.; Bender, C.; Lwin, C.; Naik, A.; Tumminia, S.J.; Hufnagel, R.B. Genetic testing for inherited eye conditions in over 6000 individuals through the eyeGENE network. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 828–837. [Google Scholar] [CrossRef]
- Estrada-Cuzcano, A.; Koenekoop, R.K.; Senechal, A.; De Baere, E.B.W.; de Ravel, T.; Banfi, S.; Kohl, S.; Ayuso, C.; Sharon, D.; Hoyng, C.B.; et al. BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome. Arch. Ophthalmol. Chic. Ill 1960 2012, 130, 1425–1432. [Google Scholar] [CrossRef]
- Chen, F.K.; Zhang, X.; Eintracht, J.; Zhang, D.; Arunachalam, S.; Thompson, J.A.; Chelva, E.; Mallon, D.; Chen, S.-C.; McLaren, T.; et al. Clinical and molecular characterization of non-syndromic retinal dystrophy due to c.175G>A mutation in ceroid lipofuscinosis neuronal 3 (CLN3). Doc. Ophthalmol. Adv. Ophthalmol. 2019, 138, 55–70. [Google Scholar] [CrossRef]
- Smirnov, V.M.; Nassisi, M.; Solis Hernandez, C.; Méjécase, C.; El Shamieh, S.; Condroyer, C.; Antonio, A.; Meunier, I.; Andrieu, C.; Defoort-Dhellemmes, S.; et al. Retinal Phenotype of Patients with Isolated Retinal Degeneration Due to CLN3 Pathogenic Variants in a French Retinitis Pigmentosa Cohort. JAMA Ophthalmol. 2021, 139, 278–291. [Google Scholar] [CrossRef]
- Magliyah, M.S.; Geuer, S.; Alsalamah, A.K.; Lenzner, S.; Drasdo, M.; Schatz, P. Association of the Recurrent Rare Variant c.415T>C p.Phe139Leu in CLN5 with a Recessively Inherited Macular Dystrophy. JAMA Ophthalmol. 2021, 139, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Roosing, S.; van den Born, L.I.; Sangermano, R.; Banfi, S.; Koenekoop, R.K.; Zonneveld-Vrieling, M.N.; Klaver, C.C.W.; van Lith-Verhoeven, J.J.C.; Cremers, F.P.M.; den Hollander, A.I.; et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015, 122, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [Green Version]
- Zare-Abdollahi, D.; Bushehri, A.; Alavi, A.; Dehghani, A.; Mousavi-Mirkala, M.; Effati, J.; Miratashi, S.A.M.; Dehani, M.; Jamali, P.; Khorram Khorshid, H.R. MFSD8 gene mutations; evidence for phenotypic heterogeneity. Ophthalmic. Genet. 2019, 40, 141–145. [Google Scholar] [CrossRef]
- Bauwens, M.; Storch, S.; Weisschuh, N.; Ceuterick-de Groote, C.; De Rycke, R.; Guillemyn, B.; De Jaegere, S.; Coppieters, F.; Van Coster, R.; Leroy, B.P.; et al. Functional characterization of novel MFSD8 pathogenic variants anticipates neurological involvement in juvenile isolated maculopathy. Clin. Genet. 2020, 97, 426–436. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Q.; Cao, Y.; Xu, H.; Yang, Z.; Tang, L.; Xiang, J.; Li, J.; Deng, H.; Yuan, L. Novel MFSD8 Variants in a Chinese Family with Nonsyndromic Macular Dystrophy. J. Ophthalmol. 2021, 2021, 6684045. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005, 6, 107–126. [Google Scholar] [CrossRef]
- Jalanko, A.; Braulke, T. Neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta 2009, 1793, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Mole, S.E.; Cotman, S.L. Genetics of the neuronal ceroid lipofuscinoses (Batten disease). Biochim. Biophys. Acta 2015, 1852, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Mandel, H.; Cohen Katsanelson, K.; Khayat, M.; Chervinsky, I.; Vladovski, E.; Iancu, T.C.; Indelman, M.; Horovitz, Y.; Sprecher, E.; Shalev, S.A.; et al. Clinico-pathological manifestations of variant late infantile neuronal ceroid lipofuscinosis (vLINCL) caused by a novel mutation in MFSD8 gene. Eur. J. Med. Genet. 2014, 57, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Steenhuis, P.; Herder, S.; Gelis, S.; Braulke, T.; Storch, S. Lysosomal targeting of the CLN7 membrane glycoprotein and transport via the plasma membrane require a dileucine motif. Traffic 2010, 11, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, A.; Kousi, M.; Sagné, C.; Bellenchi, G.C.; Morel, L.; Darmon, M.; Hulková, H.; Ruivo, R.; Debacker, C.; El Mestikawy, S.; et al. Expression and lysosomal targeting of CLN7, a major facilitator superfamily transporter associated with variant late-infantile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2010, 19, 4497–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenhuis, P.; Froemming, J.; Reinheckel, T.; Storch, S. Proteolytic cleavage of the disease-related lysosomal membrane glycoprotein CLN7. Biochim. Biophys. Acta 2012, 1822, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Kleist, L.; Ariunbat, K.; Braren, I.; Stauber, T.; Storch, S.; Danyukova, T. A newly generated neuronal cell model of CLN7 disease reveals aberrant lysosome motility and impaired cell survival. Mol. Genet. Metab. 2019, 126, 196–205. [Google Scholar] [CrossRef]
- Huber, R.J.; Mathavarajah, S.; Yap, S.Q. Mfsd8 localizes to endocytic compartments and influences the secretion of Cln5 and cathepsin D in Dictyostelium. Cell. Signal. 2020, 70, 109572. [Google Scholar] [CrossRef]
- Reith, M.; Zeltner, L.; Schäferhoff, K.; Witt, D.; Zuleger, T.; Haack, T.B.; Bornemann, A.; Alber, M.; Ruf, S.; Schoels, L.; et al. A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. Int. J. Mol. Sci. 2022, 23, 2271. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [Green Version]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Lambertus, S.; van Huet, R.A.C.; Bax, N.M.; Hoefsloot, L.H.; Cremers, F.P.M.; Boon, C.J.F.; Klevering, B.J.; Hoyng, C.B. Early-onset stargardt disease: Phenotypic and genotypic characteristics. Ophthalmology 2015, 122, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef] [PubMed]
- Westeneng-van Haaften, S.C.; Boon, C.J.F.; Cremers, F.P.M.; Hoefsloot, L.H.; den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Nõupuu, K.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7217–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bax, N.M.; Lambertus, S.; Cremers, F.P.M.; Klevering, B.J.; Hoyng, C.B. The absence of fundus abnormalities in Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 1147–1157. [Google Scholar] [CrossRef] [Green Version]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. Chic. Ill 1960 2001, 119, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Fujinami, K.; Lois, N.; Davidson, A.E.; Mackay, D.S.; Hogg, C.R.; Stone, E.M.; Tsunoda, K.; Tsubota, K.; Bunce, C.; Robson, A.G.; et al. A longitudinal study of stargardt disease: Clinical and electrophysiologic assessment, progression, and genotype correlations. Am. J. Ophthalmol. 2013, 155, 1075–1088.e13. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Jeroen Klevering, B.; Keunen, J.E.E.; Hoyng, C.B.; Theelen, T. Fundus autofluorescence imaging of retinal dystrophies. Vis. Res. 2008, 48, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Lois, N.; Halfyard, A.S.; Bird, A.C.; Holder, G.E.; Fitzke, F.W. Fundus autofluorescence in Stargardt macular dystrophy-fundus flavimaculatus. Am. J. Ophthalmol. 2004, 138, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Swider, M.; Aleman, T.S.; Sumaroka, A.; Schwartz, S.B.; Roman, M.I.; Milam, A.H.; Bennett, J.; Stone, E.M.; Jacobson, S.G. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4739–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Weleber, R.G.; Michaelides, M.; Trzupek, K.M.; Stover, N.B.; Stone, E.M. The phenotype of Severe Early Childhood Onset Retinal Dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Preising, M.N.; Abura, M.; Jäger, M.; Wassill, K.-H.; Lorenz, B. Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life. Ophthalmic. Genet. 2017, 38, 252–259. [Google Scholar] [CrossRef]
- Wright, G.A.; Georgiou, M.; Robson, A.G.; Ali, N.; Kalhoro, A.; Holthaus, S.K.; Pontikos, N.; Oluonye, N.; de Carvalho, E.R.; Neveu, M.M.; et al. Juvenile Batten Disease (CLN3): Detailed Ocular Phenotype, Novel Observations, Delayed Diagnosis, Masquerades, and Prospects for Therapy. Ophthalmol. Retin. 2019, 4, 433–445. [Google Scholar] [CrossRef]
- Orlin, A.; Sondhi, D.; Witmer, M.T.; Wessel, M.M.; Mezey, J.G.; Kaminsky, S.M.; Hackett, N.R.; Yohay, K.; Kosofsky, B.; Souweidane, M.M.; et al. Spectrum of ocular manifestations in CLN2-associated batten (Jansky-Bielschowsky) disease correlate with advancing age and deteriorating neurological function. PLoS ONE 2013, 8, e73128. [Google Scholar] [CrossRef]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef]
- Tomkiewicz, T.Z.; Suárez-Herrera, N.; Cremers, F.P.M.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. Int. J. Mol. Sci. 2021, 22, 4621. [Google Scholar] [CrossRef]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.-Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.-K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Tubeuf, H.; Charbonnier, C.; Soukarieh, O.; Blavier, A.; Lefebvre, A.; Dauchel, H.; Frebourg, T.; Gaildrat, P.; Martins, A. Large-scale comparative evaluation of user-friendly tools for predicting variant-induced alterations of splicing regulatory elements. Hum. Mutat. 2020, 41, 1811–1829. [Google Scholar] [CrossRef]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wu, N.; Zaneveld, S.; Liu, H.; Fu, S.; Wang, K.; Bertrand, R.; Wang, J.; Li, Y.; Chen, R. Transcript isoforms of Reep6 have distinct functions in the retina. Hum. Mol. Genet. 2021, 30, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Mathur, P.D.; Zou, J.; Zheng, T.; Almishaal, A.; Wang, Y.; Chen, Q.; Wang, L.; Vashist, D.; Brown, S.; Park, A.; et al. Distinct expression and function of whirlin isoforms in the inner ear and retina: An insight into pathogenesis of USH2D and DFNB31. Hum. Mol. Genet. 2015, 24, 6213–6228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, C.; Terracciano, A.; Simonati, A.; Discepoli, G.; Cannelli, N.; Claps, D.; Crow, Y.J.; Bianchi, M.; Kitzmuller, C.; Longo, D.; et al. Mutations in MFSD8/CLN7 are a frequent cause of variant-late infantile neuronal ceroid lipofuscinosis. Hum. Mutat. 2009, 30, E530–E540. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain J. Neurol. 2009, 132, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheldof, A.; Seneca, S.; Stouffs, K.; Lissens, W.; Jansen, A.; Laeremans, H.; Verloo, P.; Schoonjans, A.-S.; Meuwissen, M.; Barca, D.; et al. Clinical implementation of gene panel testing for lysosomal storage diseases. Mol. Genet. Genomic. Med. 2019, 7, e00527. [Google Scholar] [CrossRef]
- Craiu, D.; Dragostin, O.; Dica, A.; Hoffman-Zacharska, D.; Gos, M.; Bastian, A.E.; Gherghiceanu, M.; Rolfs, A.; Nahavandi, N.; Craiu, M.; et al. Rett-like onset in late-infantile neuronal ceroid lipofuscinosis (CLN7) caused by compound heterozygous mutation in the MFSD8 gene and review of the literature data on clinical onset signs. Eur. J. Paediatr. Neurol. 2015, 19, 78–86. [Google Scholar] [CrossRef]
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal Ceroid Lipofuscinosis Caused by MFSD8 Mutations: A Common Theme Emerging. Neurogenetics 2009, 10, 307–311. [Google Scholar] [CrossRef]
- Patiño, L.C.; Battu, R.; Ortega-Recalde, O.; Nallathambi, J.; Anandula, V.R.; Renukaradhya, U.; Laissue, P. Exome Sequencing Is an Efficient Tool for Variant Late-Infantile Neuronal Ceroid Lipofuscinosis Molecular Diagnosis. PLoS ONE 2014, 9, e109576. [Google Scholar] [CrossRef]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, V.; Grunewald, O.; Muller, J.; Zeitz, C.; Obermaier, C.D.; Devos, A.; Pelletier, V.; Bocquet, B.; Andrieu, C.; Bacquet, J.-L.; et al. Novel TTLL5 Variants Associated with Cone-Rod Dystrophy and Early-Onset Severe Retinal Dystrophy. Int. J. Mol. Sci. 2021, 22, 6410. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).