Abstract
Vibrio cincinnatiensis is a poorly understood pathogenic Vibrio species, and the underlying mechanisms of its genetic diversity, genomic plasticity, evolutionary dynamics, and pathogenicity have not yet been comprehensively investigated. Here, a comparative genomic analysis of V. cincinnatiensis was constructed. The open pan-genome with a flexible gene repertoire exhibited genetic diversity. The genomic plasticity and stability were characterized by the determinations of diverse mobile genetic elements (MGEs) and barriers to horizontal gene transfer (HGT), respectively. Evolutionary divergences were exhibited by the difference in functional enrichment and selective pressure between the different components of the pan-genome. The evolution on the Chr I and Chr II core genomes was mainly driven by purifying selection. Predicted essential genes in V. cincinnatiensis were mainly found in the core gene families on Chr I and were subject to stronger evolutionary constraints. We identified diverse virulence-related elements, including the gene clusters involved in encoding flagella, secretion systems, several pili, and scattered virulence genes. Our results indicated the pathogenic potential of V. cincinnatiensis and highlighted that HGT events from other Vibrio species promoted pathogenicity. This pan-genome study provides comprehensive insights into this poorly understood species from the genomic perspective.
1. Introduction
Vibrio is a genus of ubiquitous bacteria found to be widely distributed in aquatic and marine ecosystems worldwide [1]. The genus Vibrio contains over 100 described species, a dozen of which cause infections in humans and aquatic animals. Non-cholera Vibrio species, such as well-known V. vulnificus, V. parahaemolyticus, and V. alginolyticus, cause vibriosis infections, including gastroenteritis, soft-tissue infections, septicemia, and meningitis [2]. These infections are usually associated with foreign travel, seawater exposure, and contaminated food ingestion. Most Vibrio species share several biological and genomic features, including two differently sized chromosomes, genetic diversity associated with mobile genetic elements (MGEs), recombination, and horizontal gene transfer (HGT) [3,4]. Comparative genomic analysis of less-studied Vibrio species allows researchers to investigate fundamental questions regarding the genetic diversity, pathogenic mechanisms, evolutionary relationships, and genomic dynamics within the Vibrio genus.
V. cincinnatiensis, represented as a species in the genus Vibrio, is a halophilic Gram-negative bacterium [5]. V. cincinnatiensis was first isolated from a 70-year-old male at the University of Cincinnati Hospital in 1986 as an infectious agent of septicemia and meningitis [6]. Then, in 1993, it was reported to be isolated from a diarrheic immunocompromised patient [7]. Furthermore, V. cincinnatiensis was found to be associated with antimicrobial resistance and harbor several resistance-related genes, including sul1, sul2, aadA2, and the class 1 integron [8]. It also appeared resistant to the complement-mediated lysis of humoral fluids of amphioxus [9]. V. cincinnatiensis as veterinary isolates with zoonotic potential have also been reported. In China, as one of the bacterial pathogens, it caused mud crab mortalities and serious economic losses [10]. Recently, Jäckel et al. collected more than 100 suspected veterinary V. cincinnatiensis isolates from the veterinary laboratory at Dresden over a period of 20 years [11]. Bai et al. determinated several virulence-related genes in V. cincinnatiensis using PCR amplification, including haemolysin, toxS, flaB, and flaC [12]. However, the underlying mechanisms of pathogenicity need to be comprehensively investigated from a whole-genome perspective.
V. cincinnatiensis has been reported to be distributed in aquatic environments worldwide [13]. It was found in water samples collected from the Choptank River in Chesapeake Bay of USA [13]. The occurrence of V. cincinnatiensis in mussels harvested from Adriatic Sea (Italy) was reported [14]. In addition, V. cincinnatiensis was also isolated from the freshwater environment of Hiroshima (Japan) and Lima (Peru) [15,16]. Global marine habitat is the ecological niche of many microorganisms, which promote genetic diversity by the acquisition of genetic material by HGT from other organisms [3]. Thus, the genetic diversity, mobile-related genetic elements, and potential role of HGT in the V. cincinnatiensis pan-genome deserve comprehensive research.
Until now, due to limited studies that focus on V. cincinnatiensis, very little molecular data have been presented. Comparative analyses by whole-genome data could offer tremendous advantages for the evaluation of genetic diversity and phylogenetic relationship and determining MGEs, virulence-related elements, and resistance genes [17,18,19,20]. Presently, there are six available genomes of V. cincinnatiensis in National Centre for Biotechnology Information (NCBI) GenBank up to 30 January 2022. In this study, we constructed a pan-genome analysis of the species V. cincinnatiensis to evaluate the genetic diversity. Then, the genomic plasticity was evaluated by the analysis of MGEs and barriers to HGT. Comparison analyses were performed to investigate the evolutionary divergence in biological function and selective pressure between each component of the pan-genome and each chromosome. Phylogenetic analysis was constructed to explore the phylogenetic position of V. cincinnatiensis. Genetic properties (e.g., fimbrial operon, secretion system operon, and virulence genes) that were observed in the pan-genome revealed the underlying pathogenic mechanisms of V. cincinnatiensis.
2. Results and Discussion
2.1. Genomic Information for V. cincinnatiensis
All available sequenced V. cincinnatiensis genomes were collected from the NCBI GenBank database (Table S1; (accessed on 30 January 2022)). The collection contained six genomes, of which four genomes were complete. These genomes were estimated to be of 98.7 ± 0.2% completeness with 1.9 ± 0.1% contamination (Table S1). Then, gene prediction and annotation were performed using a consensus approach based on RAST server. The general characteristics of the six genomes of V. cincinnatiensis are shown in Table 1. The average genome size was 3.8 ± 0.05 Mb with 43.8 ± 0.04% GC-content and 3509 ± 40 RAST-predicted protein-coding genes. The estimated size of chromosome I (Chr I) averaged 2.8 ± 0.04 Mb with 44.2 ± 0.07% GC-content and 2554 ± 24 genes, whereas, for chromosome II (Chr II), the estimated size averaged 1.007 ± 0.02 Mb with 42.6 ± 0.1% GC-content and 955 ± 35 genes.

Table 1.
General characteristics of the six genomes of V. cincinnatiensis included in this study.
2.2. Pan-Genome Analyses Revealed That a High Degree of Genetic Variability on Chr II Promoted the Genetic Diversity of V. cincinnatiensis
We characterized the pan-genome among six V. cincinnatiensis genomes to assess their genetic diversity. A total of 4126 gene families were identified (Figure 1A and Table S2). Among these, 2984 (72.3%) gene families represented the core genome and the remaining 1142 (27.7%) variable gene contents consisted of the accessory genome (648; 15.7%) and strain-specific genes (494; 12.0%). As shown in Figure 1B, most variable genes were not broadly distributed. The size of the accessory genome in V. cincinnatiensis genomes ranged from 172 to 383 gene families with an average of 334.0 ± 80.4 gene families. The size of the strain-specific gene contents ranged from 10 to 272 genes with an average of 82.3 ± 97 genes. Thus, the composition of each genome, concerning the variable gene contents, is remarkably divergent. In addition, we identified 31 gene families shared by both chromosomes, which included 17 core gene families and 14 accessory gene families.
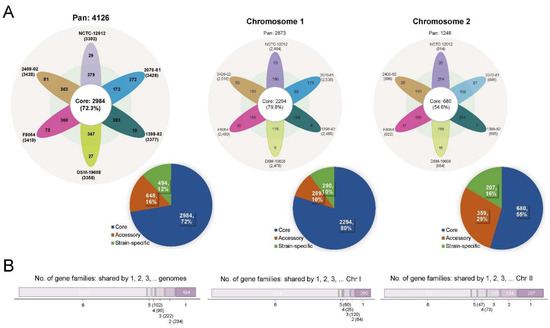
Figure 1.
The pan-genome of the V. cincinnatiensis genome, Chr I, and Chr II. (A) Flower plots represent the number of the core, accessory, and strain-specific gene families shared by each strain. Pie charts represent the percentage of the core, accessory, and strain-specific gene families participating in the pan-genome of the V. cincinnatiensis genome, Chr I, and Chr II, respectively; (B) distribution of the gene families in the pan-genome of the V. cincinnatiensis genome, Chr I, and Chr II.
In previous papers, Chibani et al., constructed the pan-genome analysis of 15 Vibrio alginolyticus genomes and found that the core genome (3876 gene families) becomes smaller than the accessory gene pool (4967 gene families) [21]. Meanwhile, Nathamuni et al., revealed that the pan-genome of seven Vibrio species (35 genomes) consisted of 17,033 genes, which included 2004 (11.8%) core, 8249 (48.4%) accessory, and 6780 (39.8%) unique genes, respectively [22]. Thus, our results might represent a smaller genetic diversity than the actual value due to the limitation of only six genomes. This is further limited by strains NCTC-12012 and DSM-19608 being the same type strain while strain 1398-82 is highly similar to both (99.99% ANI) and may be the same strain. A comprehensive dataset, which represents an even distribution of V. cincinnatiensis genomic diversity, would be required in future work.
With respect to the chromosomes, the analysis of the pan-genome resulted in 2873 gene families for Chr I and 1246 gene families for Chr II (Figure 1A). Notably, the core pan-genome for Chr I made up 79.8% (2294 gene families) of the Chr I pan-genome, whilst only 55.4% (690 gene families) was core pan-genome for the Chr II pan-genome. The accessory pan-genomes were 289 and 359 gene families for Chr I and Chr II, respectively. The strain-specific gene families for Chr I and Chr II were 290 and 207, respectively. The Chr I pan-genome is larger and more stable, whereas the Chr II pan-genome is smaller and more variable. The Chr II pan-genome accounts for more of the genetic variability of V. cincinnatiensis. The comparison of both chromosomes is shown in Figure 1.
2.3. An Open Pan-Genome of V. cincinnatiensis Exhibited the Potential for Genetic Innovation, Especially for Chr II
The V. cincinnatiensis pan-genome growth curves showed a clear linear progression fitting Heap’s law (n = ) pan-genome model [23] (Figure 2A). The pan-genome size steadily increased with the addition of each additional genome, with ~119 new genes added on average to the pan-genome as each new V. cincinnatiensis genome was added. The growth exponent values of parameter γ were 0.1 for the genome versus 0.08 for Chr I and 0.2 for Chr II, which were above the critical threshold γ = 0 [23], indicating an open pan-genome. Similarly, some aquatic bacteria, such as Aeromonas and Plesiomonas shigelloides, also have open pan-genomes and represent ecological adaptation with genetic diversity [24,25]. However, it is notable that when strain-specific genes are excluded from a pan-genome, a plateau in the pan-genome curves results, indicating that most undiscovered genes are likely not broadly distributed. Moreover, the upward trend and the growth exponent value of the Chr II pan-genome curve are greater than that of the Chr I pan-genome curve (Figure 2A), suggesting Chr II may have more frequent genetic exchange events. In contrast to the increase in the size of the pan-genome, the core genome curves decreased with additional genomes, reaching minimum values of 2984, 2294, and 880 for genome, Chr I, and Chr II, respectively.

Figure 2.
(A) The progressive curves for the core, pan-genome, and pan-genome excluding strain-specific genes of the V. cincinnatiensis genome, Chr I, and Chr II. The curves showed the downward trend of the core gene families and the upward trend of the pan-gene families with additional genomes. The deduced mathematical functions of pan-genome curves were shown below the curves; (B) distribution of mobile genetic elements; (C) distribution of multiple barriers to horizontal gene transfer.
2.4. Distribution of tRNAs, Mobile Genetic Elements, and Multiple Barriers in the V. cincinnatiensis Genomes
MGEs can mediate the transmission of genetic material and facilitate the expansion of gene pools of bacterial taxa [26]. Notably, MGEs facilitate the acquisition of genes conferring pathogenicity and antibiotic resistance during the emergence of pathogenicity [27,28]. In the V. cincinnatiensis genomes, we identified the tRNA loci and multiple types of MGEs, including genomic islands (GIs), prophages, and insertion sequences (ISs) (Table S3). On average, one genome contained 96.8 ± 16.1 tRNAs, 24.8 ± 1.9 GIs (370.3 ± 73.4 kb in size), 2.2 ± 1.0 prophages (36.9 ± 24.2 kb in size), and 18.8 ± 7.4 ISs. As shown in Figure 2B, numerous MGEs are heterogeneously distributed in the V. cincinnatiensis genomes. Most tRNA loci were located on Chr I. GIs spanned almost 10% of the genome. The number (14.5 ± 1.0) and covering regions (205.3 ± 52.0 kb) of GIs for Chr II were more than Chr I (10.3 ± 1.0; 165.0 ± 28.7 kb). The distribution of prophages and ISs presents individual differences. For example, 2409-02 genome harbored three prophages sized 54.8 kb, whereas F8054 genome had only one prophage sized 11.8 kb. Similarly, 29 ISs were located in NCTC-12012 genome, and only 11 ISs were present in F8054. Similarly, Chibani et al., demonstrated in V. alginolyticus isolates that the formation of ecotypes and speciation is mainly driven by HGT through MGEs [21]. Thus, the existence of diverse MGEs contributed to the expansion of the open pan-genome of V. cincinnatiensis.
Several genetic elements as barriers to HGT have been proposed that control genome stability in bacteria [29]. These elements defend microbes against recurrent bacteriophage and plasmid infection and prevent foreign DNA uptake [30]. In this study, we also identified multiple types of barriers in the V. cincinnatiensis genomes, including restriction-modification (RM) system, toxin/antitoxin (TA) systems, and clustered repetitively interspaced palindromic repeat (CRISPR) (Figure 2C). Interestingly, all identified TA operons were heterogeneously distributed on Chr II; 2409-02 and F84054 Chr II harbored four and five TA operons, respectively, and the remaining strains contained only one TA operon. In contrast, most identified RM operons were present on Chr I; F84054 and 2070-81 genomes have no RM operons. Furthermore, we also identified CRISPR-Cas loci and CRISPR (without Cas protein) loci in V. cincinnatiensis. The number of the spacers in these CRISPR loci is variable. The type I-F CRISPR-Cas system was located on Chr I of all genomes, and type I-C CRISPR-Cas system was only present on Chr II of the 2070-81 genome. The existence of these barriers with HGT might play an important role in the genome stability of V. cincinnatiensis.
2.5. Comparison of Functional Characteristics for Pan-Genome Revealed the Evolutionary Divergence
To gain insight into the functional characteristics of the pan-genome, we categorized the functions of the core, accessory, and strain-specific gene families using the clusters of orthologous groups (COG) assignments. The gene families not annotated with COG functional categories were defined as “hypothetical proteins”. The core gene families were significantly associated with metabolism categories (e.g., “C: energy production and conversion”, “E: amino acid transport and metabolism”, “H: coenzyme transport and metabolism”, and “P: inorganic ion transport and metabolism”) and “J: translation, ribosomal structure and biogenesis” (isher’s exact test p value < 0.05), indicating the conservation in metabolic functions (Figure 3A). Gene families involved in “L: replication, recombination and repair” and “hypothetical proteins” (Fisher’s exact test p value < 0.01) were significantly enriched in the accessory genome and strain-specific gene content (Figure 3A), indicating the presence of potential HGT events promoted the genetic diversity of the V. cincinnatiensis pan-genome.

Figure 3.
Comparison of functional enrichment for pan-genome. (A) Distribution of clusters of orthologous groups (COG) functional categories for pan-genome, core genome, accessory genome, and strain-specific genes, respectively. *: Fisher’s exact test p-value < 0.05; **: Fisher’s exact test p-value < 0.01; (B) comparison of COG functional categories for pan-genome, core genome, accessory genome, and strain-specific genes between Chr I and Chr II, respectively. *: t-test p-value < 0.05; **: t-test p-value < 0.01.
The difference of genetic variability occurring at the chromosome level was observed earlier in the pan-genome analysis. Here, to explore the potential functional difference between Chr I and Chr II, we compared the functional categories of the core, accessory, and strain-specific gene families at the chromosome level. As shown in Figure 3B, significant differences in functional categories were observed in different components of the pan-genome. In the core genome, Chr I harbored higher percentages for gene families involved in “J: translation, ribosomal structure, and biogenesis”, “U: intracellular trafficking, secretion, and vesicular transport” (t-test, p < 0.01), “N: cell motility”, and “H: coenzyme transport and metabolism” (t-test, p < 0.05), whereas the core gene families of Chr II were enriched in “G: carbohydrate transport and metabolism” and “hypothetical proteins” (t-test, p < 0.01). Interestingly, the Chr I accessory gene families were significantly associated with “L: replication, recombination, and repair” and “V: defense mechanisms” (t-test, p < 0.01) (Figure 3B), highlighting that Chr I also was closely related to HGT events and their barriers despite the lack of genetic variability. Meanwhile, gene families assigned to “T: signal transduction mechanisms” and “N: cell motility” were prominently represented in the Chr II accessory genome (t-test, p < 0.05). The Chr I strain-specific genes were significantly responsible for “K: transcription”, “M: cell wall/membrane/envelope biogenesis”, “G: carbohydrate transport and metabolism”, and “P: inorganic ion transport and metabolism” and were enriched on Chr I (t-test, p < 0.05). Significantly, the Chr II had a high proportion of strain-specific genes with “hypothetical proteins” (t-test, p < 0.01). These unknown functional genes with limited distribution contributed to the Chr II genetic diversity and their biological roles require further research.
2.6. Selective Pressure Analysis Exhibited the Dynamics of Natural Selection in Core Genome
To explore how natural selection shapes the V. cincinnatiensis genetic properties, we performed a codon-level analysis of natural selection on the 2529 mutational core gene families that represented many housekeeping functions for V. cincinnatiensis. Signatures of the evolution of these core gene families were measured by the nonsynonymous/synonymous rate ratio (dN/dS). The dN/dS rates of most of the core gene families (n = 2459, 97.2%; average dN/dS = 0.2 ± 0.3) were less than 1, exhibiting a predominant action of purifying selection in the V. cincinnatiensis core genome. These remarkable evolutionary constraints might be due to the functional importance of these core gene families, and they would maintain a stable and adapted genomic core. We further investigated the evolutionary dynamics in each functional category. Gene families assigned to “D: cell cycle control, cell division, and chromosome partitioning” (average dN/dS = 0.07 ± 0.1), “F: nucleotide transport and metabolism” (average dN/dS = 0.1 ± 0.2), and “J: translation, ribosomal structure, and biogenesis” (average dN/dS = 0.1 ± 0.2) exhibited stronger evolutionary constraints (Figure 4A). By contrast, genes related to “Q: secondary metabolites biosynthesis, transport, and catabolism” (average dN/dS = 0.4 ± 0.4), “hypothetical proteins” (average dN/dS = 0.4 ± 0.4), and “O: posttranslational modification, protein turnover, and chaperones” (average dN/dS = 0.3 ± 0.3) underwent weaker evolutionary constraints (Figure 4A). The results indicated that different functional categories have undergone different degrees of evolutionary constraints, indicating that different evolutionary strategies operated on the V. cincinnatiensis core genome. A total of 34 core gene families were identified as positively selected (dN/dS > 1) (Table S4). These gene families undergoing positive selection were involved in several functional categories, such as “S: function unknown”, “hypothetical proteins”, and “T: signal transduction mechanisms” (Figure 4B).

Figure 4.
Selective pressure on functional categories in the V. cincinnatiensis core genome. (A) Distribution of the nonsynonymous (dN)/synonymous (dS) rates of the core gene families in COG functional categories; (B) distribution of COG categories for gene families with positively selected sites; (C) comparisons of the dN/dS rates of core gene families between Chr I and Chr II in COG functional categories. ** t-test p-value < 0.01.
We also compared the evolutionary signatures of the core genome between Chr I and Chr II. Overall, the Chr I core gene families (average dN/dS = 0.2 ± 0.3) experienced a similar degree of selective pressure as the Chr II core gene families (average dN/dS = 0.2 ± 0.3) (Figure 4C). Furthermore, the similar degree of evolutionary pressure was also observed in the majority of functional categories between Chr I and Chr II, except for “K: Transcription” (Chr I: average dN/dS = 0.1 ± 0.2; Chr II: average dN/dS = 0.3 ± 0.3; (-test, p < 0.01)).
2.7. Evaluation of Potential Essential Genes in V. cincinnatiensis
Essential genes play key roles in critical biological processes and their disruption will lead to the non-viability of the organism [31]. Previous studies have reported the essential genes in several Vibrio species [32,33,34]. Here, we identified the potential essential genes in V. cincinnatiensis through a comparison of the V. cholerae essential gene set (including 343 essential genes and 91 essential domains) identified by Chao et al. using high-density transposon mutagenesis [34]. A total of 320 (73.7%) of the V. cholerae essential genes have homologs in the V. cincinnatiensis pan-genome. Among these, 316 (98.8%) represented the core gene families and the remaining only four (1.2%) represented the accessory gene families (Figure 5A). Most of the essential gene families (n = 295; 91.6%) were located on Chr I; only 23 (7.2%) essential gene families were present on Chr II; two gene families were shared by Chr I and Chr II (Figure 5A).
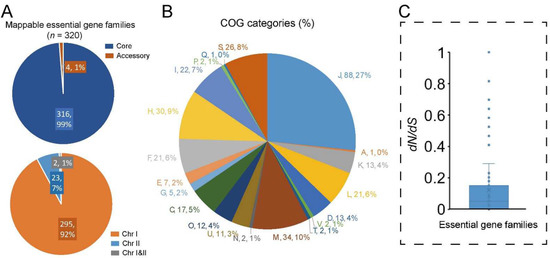
Figure 5.
Analyses of the potential essential gene families in V. cincinnatiensis: (A) distribution of the potential essential gene families in pan-genome and chromosome location; (B) distribution of COG functional categories for the potential essential gene families; (C) the dN/dS rates of the potential essential gene families.
Based on the COG annotation, essential gene families were significantly involved in conserved multicomponent pathways required for fundamental metabolic and structural functions (Figure 5B), including “J: translation, ribosomal structure, and biogenesis”, “D: cell cycle control, cell division, and chromosome partitioning”, “M: cell wall/membrane/envelope biogenesis”, “F: nucleotide transport and metabolism”, “H: coenzyme transport and metabolism”, and “I: lipid transport and metabolism” (Fisher’s exact test p value < 0.01). The dN/dS rates were estimated to investigate the conservation and evolutionary pressure for each essential gene family. Typically, the whole set of essential gene families was confirmed to have undergone significantly stronger evolutionary constraints (average dN/dS = 0.1 ± 0.2) than the whole core gene families (average dN/dS = 0.2 ± 0.3) (t-test, p = 0.02) (Figure 5C). Interestingly, the gene family that encoded DNA ligase (NAD(+)) (EC 6.5.1.2) was determinated to have undergone positive selection (dN/dS = 1.3). DNA ligase that catalyzes the formation of phosphodiester linkages using NAD as a coenzyme and as the energy source is essential for DNA replication and repair of damaged DNA [35]. Hence, the potential functional change caused by the mutation of DNA ligase sequences in V. cincinnatiensis deserves further attention.
2.8. Identification of Potential Horizontal Gene Families in V. cincinnatiensis
HGT is the major driver of bacterial genomic evolution and genetic diversity [26]. Here, we explored the potential horizontal genes in V. cincinnatiensis genomes and quantified their composition and chromosome location. We identified 240 potential horizontal gene families, which included 163 core gene families (67.9%), 49 accessory gene families (20.4%), and 28 strain-specific genes (11.7%) (Figure 6A). On average, one genome harbored 196.5 ± 6.3 horizontal genes consisting of 160.2 ± 1.8 core genes, 31.7 ± 9.6 accessory genes, and 4.7 ± 5.6 strain-specific genes (Figure 6B). This result indicated that HGT contributed to the core genome, which conferred to V. cincinnatiensis species-specific properties during process of speciation. Most of the potential horizontal gene families (n = 163; 67.9%) were located on Chr I; only 72 (30.0%) gene families were present on Chr II; five gene families (2.1%) were shared by Chr I and Chr II (Figure 6A). A genome harbored an average of 133.8 ± 6.2 horizontal genes in Chr I and 62.7 ± 7.1 horizontal genes in Chr II (Figure 6B).

Figure 6.
Analyses of the potential horizontal gene families in V. cincinnatiensis: (A) distribution of the potential horizontal gene families in pan-genome and chromosome location; (B) distribution and composition of the horizontal genes in V. cincinnatiensis genomes; (C) distribution of COG functional categories for the potential horizontal gene families; (D) the potential donor bacterial taxa providing donor genes for HGT.
Based on the COG annotation, these horizontal gene families were significantly involved in several metabolism categories, including “C: energy production and conversion”, “G: carbohydrate transport and metabolism”, “P: inorganic ion transport and metabolism”, and “Q: secondary metabolites biosynthesis, transport, and catabolism”) (Fisher’s exact test p value < 0.05) (Figure 6C). It can be inferred that the acquisition of the novel metabolism-related genetic properties driven by HGT promoted the adaptation of V. cincinnatiensis into diverse niches. Furthermore, a total of 41 potential donor bacterial families were identified (Figure 6D). Enterobacteriaceae (46.3 ± 3.2), Pseudoalteromonadaceae (15.3 ± 1.03), Oceanospirillaceae (13.2 ± 0.4), Aeromonadaceae (10.8 ± 0.8), and Alteromonadaceae (9.0 ± 0.6) appeared to be the most common donors, indicating that V. cincinnatiensis might share some properties with these families.
2.9. Phylogenetic Position of V. Cincinnatiensis in the Genus Vibrio
To further explore the evolutionary dynamics of V. cincinnatiensis, we performed a phylogenetic analysis to determinate the evolutionary position of this species. Firstly, the reference 16S rRNA sequence (FUXB01000057) of V. cincinnatiensis was subjected to similarity-based searches against the taxonomically united 16S rRNA database in EzBioCloud [36] to identify similar species. All the 16S rRNA sequences (Table S5) of closely related species were collected from the EzBioCloud database and used to construct the maximum-likelihood (ML) tree (Figure S1). Secondly, nine reference genomes of the closest-related Vibrio species on the 16S tree (Figure S1) were collected in combination with six V. cincinnatiensis genomes for subsequent analyses. A high-resolution phylogeny based on 1673 single-copy core gene families that shared all 15 genomes was generated (Figure 7A). In the core genome tree, all V. cincinnatiensis members formed a monophyletic clade with a long branch length and were deeply nested within Vibrio, suggesting evolutionary divergence occurred between V. cincinnatiensis and other members of Vibrio. The nearest species to V. cincinnatiensis is V. fujianensis (FJ201301T), and they together with V. metschnikovii (2012V-1020) and V. injenensis (M12-1144T) formed a main clade.

Figure 7.
Phylogenetic, whole-genome nucleotide, and amino acid identity analysis. (A) Phylogenetic tree based on SNPs across 1673 single-copy core gene families shared by 6 V. cincinnatiensis genomes and nine reference genome of other Vibrio species was constructed by the ML method with 100 replicates; (B) the values next to the tree indicate average nucleotide identity (ANI) and average amino acid identity (AAI) values. The heatmap presents ANI (blue matrix) and AAI (red matrix) values.
The average nucleotide identity (ANI) and average amino acid identity (AAI) values were calculated to measure the genetic relatedness between V. cincinnatiensis and other members of Vibrio (Figure 7B). The strains shared high ANI (>99.1%) and AAI (>99.1%) values between each other. The ANI and AAI values determined from comparisons between V. cincinnatiensis and other members of Vibrio were 83.3–85.9% and 72.2–89.1%, respectively, which were lower than the 95% threshold value for species [37].
2.10. HGT Promoted Genotypic Profile of Virulence in V. cincinnatiensis
Several pathogenic phenotypes of V. cincinnatiensis have been reported. To evaluate the potential pathogenicity, we identified the virulence-related elements, including fimbrial operon, secretion system operon, and virulence genes (Tables S6 and S7). A total of nine gene clusters and 13 genes, homologous to virulence-related elements identified in other pathogens, were identified in the V. cincinnatiensis genomes. These gene clusters were involved in encoding flagella, mannose-sensitive hemagglutinin (MSHA) pilus; type I (T1SS), II (T2SS), V (T5SS), and VI (T6SS) secretion systems; type IV pilus (T4P); and two types of Tad pili. The scattered virulence genes were associated with adherence (ilpA, tufA, htpB, and ompU), stress survival (clpP, katB, and sodB), quorum sensing (luxS and cqsA), exotoxin (AHA_3493), biofilm formation (algU), iron uptake (fepC), and others (icl). The distribution of these elements in all 15 Vibrio genomes is shown in Figure 8A. Most of these identified virulence-related elements are present in all six V. cincinnatiensis genomes except T1SS, which indicates that these virulence-related genotypic profiles represent a general property for V. cincinnatiensis. Parts of elements are absent in other related Vibrio genomes, such as MSHA, T1SS, T5SS, T6SS, Tad-1 and Tad-2 operon, cqsA, and fepC, suggesting that this is the result of HGT events. A tBLASTn search of the NCBI nucleotide collection (nt) databases using these elements showed that the majority of the highest homologs were identified in other Vibrio species, such as V. furnissii (harboring the highest homologs of T1SS and Tad-1), V. navarrensis (harboring the highest homolog of T6SS), and V. fluvialis (harboring the highest homolog of MSHA) (Figure 8B). Our results highlight that the potential HGT events from other Vibrio species promoted the pathogenicity of V. cincinnatiensis. Interestingly, although higher variability was exhibited by Chr II, most virulence-related elements were located on Chr I, except for T1SS operon, T6SS operon, katB, and cqsA (Figure 8A), indicating that Chr I plays an important role in the pathogenicity of V. cincinnatiensis.

Figure 8.
The genotypic profiles of virulence-related elements across six V. cincinnatiensis genomes. (A) Distribution of virulence-related elements. Color coding for virulence gene is based on the BLASTp parameters (E-value cutoff value of <1 × 10−6, identity value of >60%, and coverage value of >60%) calculated when the genomic data were screened against the reference sequences of VFDB database. Color coding for the gene clusters is based on the percentage of genes on a cluster that are present in a genome (defined as the BLASTp parameters of query gene with E-value cutoff value of <1 × 10−6, identity value of >60%, and coverage value of >60%); (B) the genetic organization of virulence-related elements. Homologous genes are shown in the same color and linked by dotted lines. The percentages of protein identities of homologous genes are shown.
The flagellum, MSHA pilus, T4P, T2SS, Tad-1 pilus, and Tad-2 pilus have been reported to contribute to the virulence of pathogenic Vibrio through motility, adhesion, or biofilm formation [38,39,40,41,42]. Furthermore, Sikora et al. reported that T2SS could confer fitness of V. cholerae in different ecological niches via the secretion of multiple proteins, including chitinases, proteases, DNase, and pilin [43]. T1SS operon was present only in one V. cincinnatiensis genome (2070-81), and T1SS could secrete multiple proteins involved in pathogenesis, nutrient acquisition, and antibacterial activity [44]. Interestingly, we did not identify any homologs with high identity (>40.0%) of the V. cincinnatiensis T5SS. Sequence analysis showed that this novel T5SS contained 549 amino acids, encoding one polypeptide-transport-associated ShlB-type domain (IPR013686) and one haemolysin activator HlyB C-terminal domain (IPR005565) (Figure 8B). The biological function of this T5SS deserves further research. T6SS in Vbrio has been reported to be associated with antimicrobial properties, antieukaryotic activities, and pathoadaptive fashion [45,46]. Finally, the identified genotypic profile of virulence highlighted the potential of V. cincinnatiensis. Future studies are required to confirm the function of these virulence-related elements in V. cincinnatiensis and their potential role in pathogenicity. In addition, V. cincinnatiensis has been reported to be associated with antimicrobial resistance and harbors several resistance-related genes (e.g., sul1, sul2, aadA2, and the class 1 integron) [8]. In this work, we did not mine any resistance genes in all six V. cincinnatiensis genomes.
3. Materials and Methods
3.1. Genome Collection and Annotation
All six V. cincinnatiensis genomes were collected and downloaded from NCBI GenBank database; nine other genomes were selected as representatives of Vibrio species related to V. cincinnatiensis. The estimates for genome completeness and contamination were performed using CheckM [47]. Gene prediction and annotation were performed using a consensus approach based on RAST server [48]. A detailed account of the genome dataset, including accession No., strain names, assembly type, genomic characteristics, completeness, and contamination is presented in Table S1. The scaffolds and contigs in draft genomes (NCTC-12012 and DSM-19608) were aligned to the chromosomes of the reference complete genome (2409-02) using Mauve Genome Alignment software 2.4.0 [49]. The scaffolds or contigs were mapped to the reference genome to determine their locations (Chr I or Chr II), with a cutoff of 60% coverage. All scaffolds in NCTC-12012 were mapped to the chromosome location. Only Contigs with 1372 (0.04%) bp length in DSM-19608 were unidentifiable.
3.2. Pan-Genome Analysis
Orthologous groups of protein-coding gene families of pan-genome were delimited using OrthoFinder 2.0 software [50,51]. The sequence files deposited in Orthogroup_Sequences folder were used to extract pan-genome families (the totality of all gene families found in V. cincinnatiensis), core genome families (genes shared among six genomes), accessory genome families (genes shared among more than one genome, but not in all), and strain-specific genes (genes found only in one genome). Curve fitting of the pan-genome was performed using a power-law regression based on Heap’s law (n = ) [23,52], where N is the number of genomes, κ is a proportionality constant, and the growth exponent γ > 0 indicates an open pan-genome. A descriptive statistical analysis was generated using OriginPro 9 software with Allometric1 model. The gene families of the pan-genome were functionally characterized by the COG functional category [53] using eggNOG-mapper 1.0 software [54].
3.3. Identification of MGEs and Barriers to HGT
The tRNA loci were collected from the RAST annotation files [48]. The prophages were predicted using the online interface of phage search tool—enhanced release (PHASTER) [55]. The online interface of IslandViewer 4 [56] (integrated three different methods: SIGI-HMM [57], IslandPath-DIMOB [58], and IslandPick [59]) was utilized to identify the genomic islands. Insertion sequences were predicted using the online interface of ISfinder [60]. The TA systems were identified by BLASTp search of the dataset from TADB 2.0 database [61]. The RM systems were identified using the online interface of Restriction-ModificationFinder 1.1 [62]. The CRISPRs were predicted using the CRISPRCasFinder 4.2.2 software with default parameters [63].
3.4. Selective Pressure Analysis
Selective pressure in core gene families was estimated by calculating the ratio of the nonsynonymous substitution rate to the dN/dS rate. ParaAT software was used to codon-based align the orthologous sequences [64]. Then, the fast unconstrained Bayesian approximation (FUBAR) pipeline [65] within HYPHY software was used to measure the dN/dS ratio at each site in each core gene family.
3.5. Identification of Potential Horizontal Genes
HGTector software was used to identify the potential horizontal genes in V. cincinnatiensis species [66]. The V. cincinnatiensis (rank: species; taxon ID: 675) and Vibrionaceae (rank: family; taxon ID: 641) were set as self-group and close-group, respectively.
3.6. Phylogenetic Analysis
The core genome phylogenetic analysis was performed based on SNPs across single-copy core gene families extracted from the sequence files deposited in Orthogroup_Sequences folder. Nucleotide sequences of the single-copy core gene families were extracted according to the protein accession numbers and then aligned using the MAFFT 1.0 software [67]. The set of single-nucleotide polymorphisms (SNPs) presented in single-copy core gene families was extracted and then integrated according to the arrangement of the genes on the 2409-02 genome (complete genome). To avoid phylogenetic confusion, we identified and removed the putative recombinational regions from the SNPs set using ClonalFrameML 1.0 software [68]. The maximum likelihood (ML) tree was constructed using MEGA 7 [69] with the general time-reversible (GTR) model and 100 bootstrap replicates.
3.7. Genomic Characteristic Analysis
The JSpecies 1.2.1 software based on MUMmer method (ANIm) [37,70] and CompareM 2.0 software (https://github.com/dparks1134/CompareM, accessed on 30 January 2022) were used to calculate the average nucleotide identity (ANI) and amino acid identity (AAI), respectively.
3.8. Identification of Virulence-Related Genetic Elements and Resistance Genes
The detection and visualization of Macromolecular systems were performed using the programs MacSyFinder [71] and TXSScan [72] within Galaxy workflow system (https://galaxy.pasteur.fr/) (accessed on 30 January 2022) on the default parameters. To identify the virulence factors, the protein sequences of all genomes were aligned using BLASTp with an E-value cutoff < ×10−6, identity > 60%, and coverage > 60% against the dataset from the Virulence Factors Database (VFDB). The detection of the homologs of virulence-related elements was performed by tBLASTn against the NCBI Nucleotide collection (nt) databases. The domains of T5SS were predicted by InterPro annotation [73]. The resistance genes were predicted using ResFinder 4.0 software [74].
4. Conclusions
V. cincinnatiensis was found to distribute in aquatic environments worldwide. This poorly understood species is regarded as a potential human pathogen. In this work, we constructed a comprehensive comparative genomic analysis with six V. cincinnatiensis genomes to investigate the genetic diversity, genomic plasticity, evolutionary dynamics, phylogenetic position, and underlying mechanisms of pathogenicity of V. cincinnatiensis. The pan-genome exhibited genetic diversity with a flexible gene repertoire. The Chr II pan-genome harbored extensive genomic variability, about 45% of the pan-genome was variable. The genomic plasticity of V. cincinnatiensis was exhibited by tRNA loci and multiple types of MGEs, including GIs, prophages, and ISs. Meanwhile, the genomic stability was determinated by the barriers to HGT, including RM systems, TA systems, and CRISPR systems. Most HGT-related elements were heterogeneously distributed in two chromosomes. Comparative analysis revealed a significant difference in the functional enrichment between different components of pan-genome and chromosomes. The Chr I and Chr II core genomes mainly underwent a similar degree of evolutionary constraints of purifying selection. The potential essential genes in V. cincinnatiensis were mostly in the core gene families on Chr I and experienced significantly stronger evolutionary constraints than the whole core gene families. The potential horizontal HGT appears to have been an important evolutionary driver of genetic diversity, shaping the V. cincinnatiensis genomic evolution. Our phylogenetic analysis revealed that V. cincinnatiensis was closely related to V. fujianensis, V. metschnikovii, and V. injenensis. Our results also identified diverse virulence-related profiles in V. cincinnatiensis, including fimbrial operon, multiple pilus operons (encoding MSHA pilus, type IV pilus, and two types of Tad pili), secretion system operons (T1SS, T2SS, T5SS, and T6SS), and several scattered virulence genes. Most of them were located on Chr I. The distribution of these virulence-related elements in other related members of Vibrio species indicated that some of them seem to be the result of HGT events. Thus, HGT might play a key role in the pathogenicity of V. cincinnatiensis. It is important to note that that our conclusion is limited by the low number of V. cincinnatiensis genomes available included in the comparative genomic analyses. Furthermore, some potential informations especially for MGEs are missed from draft genomes (NCTC-12012 and DSM-19608). Thus, in favor of a more comprehensive analysis, more complete genomes, which represent an even distribution of V. cincinnatiensis genomic diversity, would be required in the future work.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms23094520/s1.
Author Contributions
Conceptualization, Y.D. and Z.Y.; methodology, Y.D., Y.J. and Z.Y.; software, Y.J. and B.L.; validation, Y.D., Z.Y. and J.Y.; formal analysis, Y.D.; investigation, Y.D. and Y.J.; resources, Y.D. and B.L.; data curation, Y.J. and B.L.; writing—original draft preparation, Y.D. and Y.J.; writing—review and editing, Z.Y. and J.Y.; visualization, J.Y.; supervision, J.Y.; project administration, Z.Y. and J.Y.; funding acquisition, Z.Y. and J.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Shandong Provincial Natural Science Foundation (ZR2021QC208), Scientific Research Foundation of Shandong Agricultural University (010/721000), and the National Science and Technology Major Project of Infectious Diseases (2018ZX10101-003-001-008).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baker-Austin, C.; Trinanes, J.; Gonzalez-Escalona, N.; Martinez-Urtaza, J. Non-Cholera Vibrios: The Microbial Barometer of Climate Change. Trends Microbiol. 2017, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Baker-Austin, C.; Oliver, J.D.; Alam, M.; Ali, A.; Waldor, M.K.; Qadri, F.; Martinez-Urtaza, J. Vibrio spp. infections. Nat. Rev. Dis. Prim. 2018, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, F.; Blokesch, M. Eco-evolutionary Dynamics Linked to Horizontal Gene Transfer in Vibrios. Annu. Rev. Microbiol. 2018, 72, 89–110. [Google Scholar] [CrossRef] [PubMed]
- Hazen, T.H.; Pan, L.; Gu, J.-D.; Sobecky, P.A. The contribution of mobile genetic elements to the evolution and ecology of Vibrios. FEMS Microbiol. Ecol. 2010, 74, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, J.; Zhang, X.; Jiang, Y. Entry of Vibrio cincinnatiensis into viable but nonculturable state and its resuscitation. Lett. Appl. Microbiol. 2009, 48, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Bode, R.B.; Brayton, P.R.; Colwell, R.R.; Russo, F.M.; Bullock, W.E. A new Vibrio species, Vibrio cincinnatiensis, causing meningitis: Successful treatment in an adult. Ann. Intern. Med. 1986, 104, 55–56. [Google Scholar] [CrossRef] [PubMed]
- Wuthe, H.H.; Aleksić, S.; Hein, W. Contribution to some phenotypical characteristics of Vibrio cincinnatiensis. Studies in one strain of a diarrhoeic human patient and in two isolates from aborted bovine fetuses. Zentralbl. Bakteriol. 1993, 279, 458–465. [Google Scholar] [CrossRef]
- Li, L.; Heidemann Olsen, R.; Ye, L.; Yan, H.; Nie, Q.; Meng, H.; Shi, L. Antimicrobial Resistance and Resistance Genes in Aerobic Bacteria Isolated from Pork at Slaughter. J. Food Prot. 2016, 79, 589–597. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, S.; Wang, C.; Pang, Q. Complement-mediated killing of Vibrio species by the humoral fluids of amphioxus Branchiostoma belcheri: Implications for a dual role of O-antigens in the resistance to bactericidal activity. Fish Shellfish Immunol. 2008, 24, 215–222. [Google Scholar] [CrossRef]
- Liu, W.; Qian, D.; Yan, X.J. Studies on pathogenicity and prevalence of white spot syndrome virus in mud crab, Scylla serrata (Forskal), in Zhejiang Province, China. J. Fish Dis. 2011, 34, 131–138. [Google Scholar] [CrossRef]
- Jäckel, C.; Hammerl, J.A.; Arslan, H.-H.-T.; Göllner, C.; Vom Ort, N.; Taureck, K.; Strauch, E. Phenotypic and Genotypic Characterization of Veterinary Vibrio cincinnatiensis Isolates. Microorganisms 2020, 8, 739. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Pang, L.; Qi, Z.; Chen, J.; Austin, B.; Zhang, X.-H. Distribution of five vibrio virulence-related genes among Vibrio harveyi isolates. J. Gen. Appl. Microbiol. 2008, 54, 71–78. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Heidelberg, J.F.; Heidelberg, K.B.; Colwell, R.R. Seasonality of Chesapeake Bay bacterioplankton species. Appl. Environ. Microbiol. 2002, 68, 5488–5497. [Google Scholar] [CrossRef] [PubMed]
- Ripabelli, G.; Sammarco, M.L.; Grasso, G.M.; Fanelli, I.; Caprioli, A.; Luzzi, I. Occurrence of Vibrio and other pathogenic bacteria in Mytilus galloprovincialis (mussels) harvested from Adriatic Sea, Italy. Int. J. Food Microbiol. 1999, 49, 43–48. [Google Scholar] [CrossRef]
- Venkateswaran, K.; Kiiyukia, C.; Takaki, M.; Nakano, H.; Matsuda, H.; Kawakami, H.; Hashimoto, H. Characterization of toxigenic vibrios isolated from the freshwater environment of Hiroshima, Japan. Appl. Environ. Microbiol. 1989, 55, 2613–2618. [Google Scholar] [CrossRef]
- Sulca, M.A.; Orozco, R.; Alvarado, D.E. Antimicrobial resistance not related to 1,2,3 integrons and Superintegron in Vibrio spp. isolated from seawater sample of Lima (Peru). Mar. Pollut. Bull. 2018, 131, 370–377. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, J.; Du, B.; Ruan, H.H.; Huo, Y.X.; Du, Y.; Qiao, J. Whole-Genome-Based Survey for Polyphyletic Serovars of Salmonella enterica subsp. enterica Provides New Insights into Public Health Surveillance. Int. J. Mol. Sci. 2007, 21, 5226. [Google Scholar] [CrossRef]
- Yin, Z.; Yuan, C.; Du, Y.; Yang, P.; Qian, C.; Wei, Y.; Zhang, S.; Huang, D.; Liu, B. Comparative genomic analysis of the Hafnia genus reveals an explicit evolutionary relationship between the species alvei and paralvei and provides insights into pathogenicity. BMC Genom. 2019, 20, 768. [Google Scholar] [CrossRef]
- Yin, Z.; Wang, X.; Hu, Y.; Zhang, J.; Li, H.; Cui, Y.; Zhao, D.; Dong, X.; Zhang, X.; Liu, K.; et al. Metabacillus dongyingensis sp. nov. Is Represented by the Plant Growth-Promoting Bacterium BY2G20 Isolated from Saline-Alkaline Soil and Enhances the Growth of Zea mays L. under Salt Stress. mSystems 2022, 8, e0142621. [Google Scholar] [CrossRef]
- Yuan, C.; Wei, Y.; Zhang, S.; Cheng, J.; Cheng, X.; Qian, C.; Wang, Y.; Zhang, Y.; Yin, Z.; Chen, H. Comparative Genomic Analysis Reveals Genetic Mechanisms of the Variety of Pathogenicity, Antibiotic Resistance, and Environmental Adaptation of Providencia Genus. Front. Microbiol. 2020, 11, 572642. [Google Scholar] [CrossRef]
- Chibani, C.M.; Roth, O.; Liesegang, H.; Wendling, C.C. Genomic variation among closely related Vibrio alginolyticus strains is located on mobile genetic elements. BMC Genom. 2020, 21, 354. [Google Scholar] [CrossRef] [PubMed]
- Nathamuni, S.; Jangam, A.K.; Katneni, V.K.; Selvaraj, A.; Krishnan, K.; Kumar, S.; Avunje, S.; Balasubramaniam, S.; Grover, M.; Alavandi, S.V.; et al. Insights on genomic diversity of Vibrio spp. through Pan-genome analysis. Ann. Microbiol. 2019, 69, 1547–1555. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.M.D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Han, M.; Yang, P.; Chen, C.; Yu, H.; Wang, L.; Ning, K. Comprehensive Analysis Reveals the Evolution and Pathogenicity of Aeromonas, Viewed from Both Single Isolated Species and Microbial Communities. mSystems 2019, 4, e00252-19. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, S.; Wei, Y.; Wang, M.; Ma, S.; Yang, S.; Wang, J.; Yuan, C.; Jiang, L.; Du, Y. Horizontal Gene Transfer Clarifies Taxonomic Confusion and Promotes the Genetic Diversity and Pathogenicity of Plesiomonas shigelloides. mSystems 2020, 5, e00448-20. [Google Scholar] [CrossRef]
- Ochman, H.; Lawrence, J.G.; Groisman, E. A Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Wang, M.; Fan, Y.; Liu, P.; Liu, Y.; Zhang, J.; Jiang, Y.; Zhou, C.; Yang, L.; Wang, C.; Qian, C.; et al. Genomic insights into evolution of pathogenicity and resistance of multidrug-resistant Raoultella ornithinolytica WM1. Ann. N. Y. Acad. Sci. 2021, 1497, 74–90. [Google Scholar] [CrossRef]
- Yin, Z.; Liu, X.; Qian, C.; Sun, L.; Pang, S.; Liu, J.; Li, W.; Huang, W.; Cui, S.; Zhang, C.; et al. Pan-Genome Analysis of Delftia tsuruhatensis Reveals Important Traits Concerning the Genetic Diversity, Pathogenicity, and Biotechnological Properties of the Species. Microbiol. Spectr. 2022, 11, e0207221. [Google Scholar] [CrossRef]
- Argemi, X.; Matelska, D.; Ginalski, K.; Riegel, P.; Hansmann, Y.; Bloom, J.; Pestel-Caron, M.; Dahyot, S.; Lebeurre, J.; Prévost, G. Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genom. 2018, 19, 621. [Google Scholar] [CrossRef]
- Shariat, N.; Dudley, E.G. CRISPRs: Molecular Signatures Used for Pathogen Subtyping. Appl. Environ. Microbiol. 2014, 80, 430–439. [Google Scholar] [CrossRef]
- Jordan, I.K.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E. V Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002, 12, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Bekaert, M.; Goffin, N.; McMillan, S.; Desbois, A.P. Essential Genes of Vibrio anguillarum and Other Vibrio spp. Guide the Development of New Drugs and Vaccines. Front. Microbiol. 2021, 12, 755801. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, T.P.; Chao, M.C.; Abel, S.; Blondel, C.J.; Abel Zur Wiesch, P.; Zhou, X.; Davis, B.M.; Waldor, M.K. Genetic analysis of Vibrio parahaemolyticus intestinal colonization. Proc. Natl. Acad. Sci. USA 2016, 113, 6283–6288. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.C.; Pritchard, J.R.; Zhang, Y.J.; Rubin, E.J.; Livny, J.; Davis, B.M.; Waldor, M.K. High-resolution definition of the Vibrio cholerae essential gene set with hidden Markov model-based analyses of transposon-insertion sequencing data. Nucleic Acids Res. 2013, 41, 9033–9048. [Google Scholar] [CrossRef]
- Sriskanda, V.; Shuman, S. A second NAD(+)-dependent DNA ligase (LigB) in Escherichia coli. Nucleic Acids Res. 2001, 29, 4930–4934. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Watnick, P.I.; Lauriano, C.M.; Klose, K.E.; Croal, L.; Kolter, R. The absence of a flagellum leads to altered colony morphology, biofilm development and virulence in Vibrio cholerae O139. Mol. Microbiol. 2001, 39, 223–235. [Google Scholar] [CrossRef]
- Utada, A.S.; Bennett, R.R.; Fong, J.C.N.; Gibiansky, M.L.; Yildiz, F.H.; Golestanian, R.; Wong, G.C.L. Vibrio cholerae use pili and flagella synergistically to effect motility switching and conditional surface attachment. Nat. Commun. 2014, 5, 4913. [Google Scholar] [CrossRef]
- Shime-Hattori, A.; Iida, T.; Arita, M.; Park, K.-S.; Kodama, T.; Honda, T. Two type IV pili of Vibrio parahaemolyticus play different roles in biofilm formation. FEMS Microbiol. Lett. 2006, 264, 89–97. [Google Scholar] [CrossRef]
- Johnson, T.L.; Fong, J.C.; Rule, C.; Rogers, A.; Yildiz, F.H.; Sandkvist, M. The Type II secretion system delivers matrix proteins for biofilm formation by Vibrio cholerae. J. Bacteriol. 2014, 196, 4245–4252. [Google Scholar] [CrossRef] [PubMed]
- Duong-Nu, T.-M.; Jeong, K.; Hong, S.H.; Puth, S.; Kim, S.Y.; Tan, W.; Lee, K.H.; Lee, S.E.; Rhee, J.H. A stealth adhesion factor contributes to Vibrio vulnificus pathogenicity: Flp pili play roles in host invasion, survival in the blood stream and resistance to complement activation. PLoS Pathog. 2019, 15, e1007767. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.E. Proteins Secreted via the Type II Secretion System: Smart Strategies of Vibrio cholerae to Maintain Fitness in Different Ecological Niches. PLoS Pathog. 2013, 9, e1003126. [Google Scholar] [CrossRef] [PubMed]
- Kanonenberg, K.; Schwarz, C.K.W.; Schmitt, L. Type I secretion systems—A story of appendices. Res. Microbiol. 2013, 164, 596–604. [Google Scholar] [CrossRef]
- MacIntyre, D.L.; Miyata, S.T.; Kitaoka, M.; Pukatzki, S. The Vibrio cholerae type VI secretion system displays antimicrobial properties. Proc. Natl. Acad. Sci. USA 2010, 107, 19520–19524. [Google Scholar] [CrossRef]
- Ishikawa, T.; Sabharwal, D.; Bröms, J.; Milton, D.L.; Sjöstedt, A.; Uhlin, B.E.; Wai, S.N. Pathoadaptive conditional regulation of the type VI secretion system in Vibrio cholerae O1 strains. Infect. Immun. 2012, 80, 575–584. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Heaps, H.S. Information Retrieval-Computational and Theoretical Aspects; Academic Press: Orlando, FL, USA, 1978. [Google Scholar]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef]
- Hsiao, W.; Wan, I.; Jones, S.J.; Brinkman, F.S.L. IslandPath: Aiding detection of genomic islands in prokaryotes. Bioinformatics 2003, 19, 418–420. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Hsiao, W.W.L.; Brinkman, F.S.L. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 2008, 9, 329. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef]
- Xie, Y.; Wei, Y.; Shen, Y.; Li, X.; Zhou, H.; Tai, C.; Deng, Z.; Ou, H.-Y. TADB 2.0: An updated database of bacterial type II toxin-antitoxin loci. Nucleic Acids Res. 2018, 46, D749–D753. [Google Scholar] [CrossRef]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, S.; Hasman, H. Is the Evolution of Salmonella enterica subsp. enterica Linked to Restriction-Modification Systems? mSystems 2016, 1, e00009–16. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xiao, J.; Wu, J.; Zhang, H.; Liu, G.; Wang, X.; Dai, L. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012, 419, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Kosoy, M.; Dittmar, K. HGTector: An automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 2014, 15, 717. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability Article Fast Track. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient Inference of Recombination in Whole Bacterial Genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Touchon, M.; Rocha, E.P.C.; Abby, S.S.; Ne, B. MacSyFinder: A Program to Mine Genomes for Molecular Systems with an Application to CRISPR-Cas Systems. PLoS ONE 2014, 9, e110726. [Google Scholar] [CrossRef]
- Abby, S.S.; Rocha, E.P.C. Identification of protein secretion systems in bacterial genomes using MacSyFinder. Methods Mol. Biol. 2017, 1615, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; Cattoir, V.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).