
Mechanism of GAPDH Redox Signaling by H2O2 Activation of a Two−Cysteine Switch


Abstract
:1. Introduction
2. Results
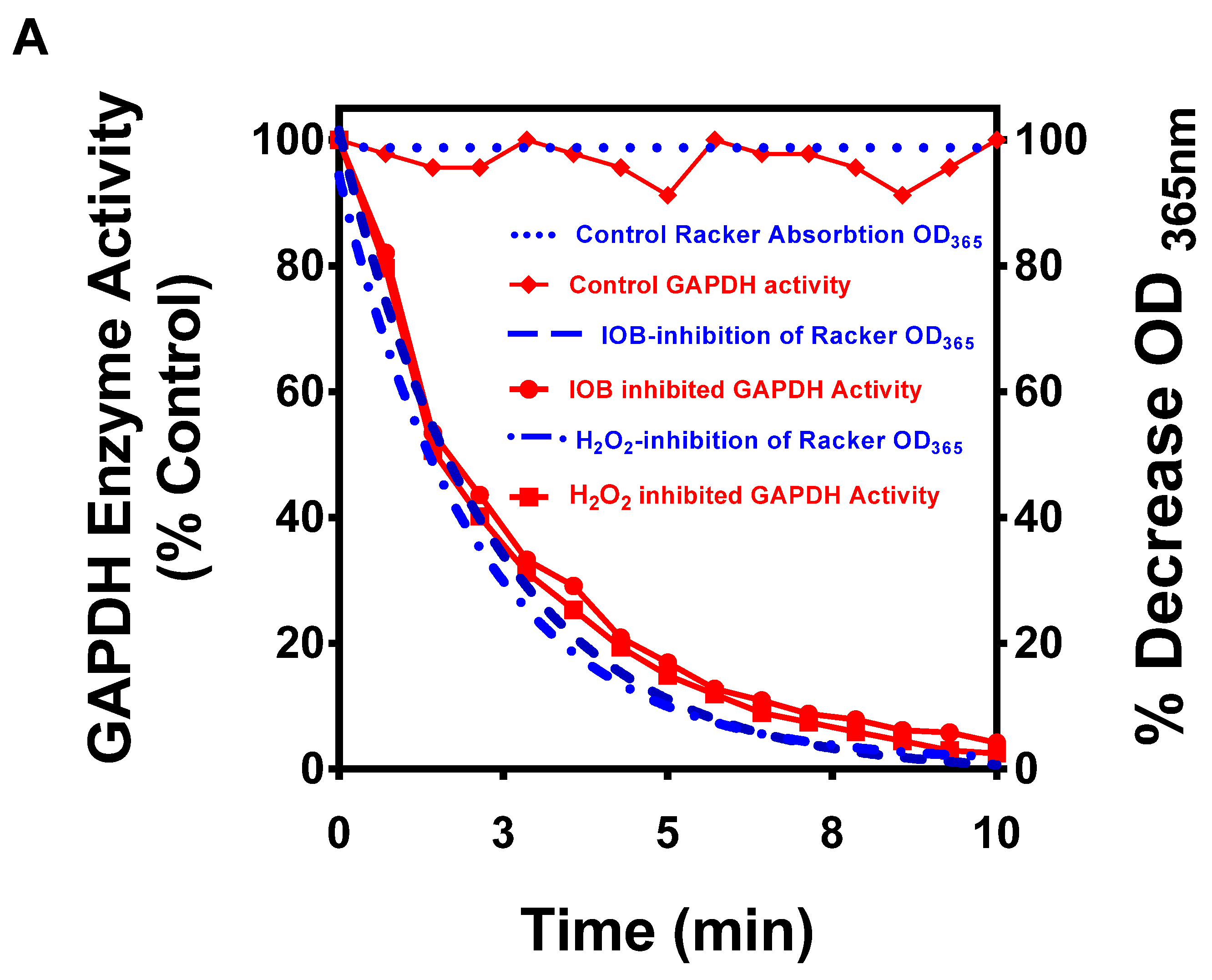
2.1. Stoichiometry and pH−Dependent Kinetics of GAPDH Oxidation
2.2. Identification of Redox−Active Cysteine Intermediates
2.3. Steps 1 and 2 Are Exclusively Consecutive
2.4. In Vitro Evidence That Cv(SH) Is a Major Factor in Mediating Irreversible GAPDH Activity by H2O2
2.5. Subunit Unfolding following H2O2 Oxidation
2.6. H2O2 Oxidation Perturbs Subunit Structure
2.7. MD Analysis of CvSH Oxidation by H2O2
2.8. MDS Analysis of the Secondary Structure of Oxidized GAPDH
2.9. Reaction Pathway for Condensation of the Sulfenic Acids
2.10. Pathway for the Formation of a Thiosulfonic Ester
2.11. Subunit Instability following Thioester Formation
2.12. The Redox−Balanced Reactions of H2O2 Oxidation of GAPDH
3. Discussion
4. Materials and Methods
4.1. GAPDH Preparation
4.2. GAPDH Activity Assay
4.3. H2O2 Standardization and Assay
4.4. Measurement of Reduced Cysteines
4.5. Oxidation of GAPDH by H2O2 and Cysteine Titration
4.6. Demonstration That both Oxidation Steps Irreversibly Inactivate Enzyme Activity
4.7. Detection of Disulfides Using NTSB
4.8. Kinetics of DTNB Reaction with Native and H2O2−Oxidized GAPDH
4.9. SDS−PAGE Analysis
4.10. Thiosulfinic Ester Determination

4.11. GAPDH Size Exclusion Gel Filtration Chromatography
4.12. CD Spectroscopy of Native and Oxidized GAPDH
4.13. Measurement of NAD+ Dissociation during H2O2 Oxidation of GAPDH
4.14. Mass Spectrometry Methods
4.15. Protein Identification by nano−LC−QTOF Peptide Sequencing and Database Search
4.16. Peptide Mapping with Protein Digestion and LC/MS
4.17. Database Searching and Data Analysis
4.18. Preparation of GAPDH Purified from L. plantarum and L. acidophilus
4.19. h−GAPDH Active Site Computational Model Methods
4.20. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hyslop, P.A.; Hinshaw, D.B.; Scraufstatter, I.U.; Cochrane, C.G.; Kunz, S.; Vosbeck, K. Hydrogen peroxide as a potent bacteriostatic antibiotic: Implications for host defense. Free Radic. Biol. Med. 1995, 19, 31–37. [Google Scholar] [CrossRef]
- Hyslop, P.A. Section Review Anti−infectives: Natural mediators of host−defence: The role of H2O2 in the regulation of bacteriostasis. Expert Opin. Investig. Drugs 1996, 5, 1013–1020. [Google Scholar] [CrossRef]
- Hyslop, P.A.; Zhang, Z.; Pearson, D.V.; Phebus, L.A. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: Correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995, 671, 181–186. [Google Scholar] [CrossRef]
- Hinshaw, D.B.; Miller, M.T.; Omann, G.M.; Beals, T.F.; Hyslop, P.A. A cellular model of oxidant−mediated neuronal injury. Brain Res. 1993, 615, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.K.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.T.; Hata, F.; Inui, T.; Takeuchi, T. The Active Site Cysteine of the Proapoptotic Protein Glyceraldehyde−3−phosphate Dehydrogenase Is Essential in Oxidative Stress−induced Aggregation and Cell Death. J. Biol. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef] [PubMed]
- Sawa, A.; Khan, A.A.; Hester, L.D.; Snyder, S.H. Glyceraldehyde−3−phosphate dehydrogenase: Nuclear translocation participates in neuronal and nonneuronal cell death. Proc. Natl. Acad. Sci. USA 1997, 94, 11669–11674. [Google Scholar] [CrossRef] [Green Version]
- Dastoor, Z.; Dreyer, J.L. Potential role of nuclear translocation of glyceraldehyde−3−phosphate dehydrogenase in apoptosis and oxidative stress. J. Cell Sci. 2001, 114 Pt 9, 1643–1653. [Google Scholar] [CrossRef]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.-I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric oxide−induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Guzhova, I.V.; Margulis, B.A. Glyceraldehyde−3−phosphate Dehydrogenase is a Multifaceted Therapeutic Target. Pharmaceutics 2020, 12, 416. [Google Scholar] [CrossRef]
- Tossounian, M.A.; Zhang, B.; Gout, I. The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants 2020, 9, 1288. [Google Scholar] [CrossRef] [PubMed]
- Butera, G.; Mullappilly, N.; Masetto, F.; Palmieri, M.; Scupoli, M.T.; Pacchiana, R.; Donadelli, M. Regulation of Autophagy by Nuclear GAPDH and Its Aggregates in Cancer and Neurodegenerative Disorders. Int. J. Mol. Sci. 2019, 20, 2062. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.; Park, J.B.; Lee, S.D.; Kim, J.H.; Ha, S.H.; Hasumi, K.; Endo, A.; Suh, P.G.; Ryu, S.H. Hydrogen peroxide induces association between glyceraldehyde 3−phosphate dehydrogenase and phospholipase D2 to facilitate phospholipase D2 activation in PC12 cells. J. Neurochem. 2003, 85, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Sirover, M.A. The role of posttranslational modification in moonlighting glyceraldehyde−3−phosphate dehydrogenase structure and function. Amino Acids 2021, 53, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde−3−phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J. Alzheimer’s Dis. 2010, 20, 369–393. [Google Scholar] [CrossRef] [Green Version]
- Sirover, M.A. Moonlighting glyceraldehyde−3−phosphate dehydrogenase: Posttranslational modification, protein and nucleic acid interactions in normal cells and in human pathology. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Gerszon, J.; Rodacka, A. Oxidatively modified glyceraldehyde−3−phosphate dehydrogenase in neurodegenerative processes and the role of low molecular weight compounds in counteracting its aggregation and nuclear translocation. Ageing Res. Rev. 2018, 48, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Semenyuk, P.; Barinova, K.; Muronetz, V. Glycation of alpha−synuclein amplifies the binding with glyceraldehyde−3−phosphate dehydrogenase. Int. J. Biol. Macromol. 2019, 127, 278–285. [Google Scholar] [CrossRef]
- Delport, A.; Kins, S.; Hewer, R. The amyloid precursor protein affects glyceraldehyde 3−phosphate dehydrogenase levels, organelle localisation and thermal stability. Mol. Biol. Rep. 2020, 47, 3019–3024. [Google Scholar] [CrossRef]
- Sekar, S.; Taghibiglou, C. Nuclear accumulation of GAPDH, GluA2 and p53 in post−mortem substantia nigral region of patients with Parkinson’s disease. Neurosci. Lett. 2020, 716, 134641. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Fan, H.; Wen, C.; Ji, Z.; Liang, S. GAPDH siRNA Regulates SH−SY5Y Cell Apoptosis Induced by Exogenous alpha−Synuclein Protein. Neuroscience 2021, 469, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.W.; Tsai, C.F.; Lin, K.H.; Chen, W.J.; Lin, M.S.; Hsieh, C.C.; Lin, C.C. An investigation of the correlation between the S−glutathionylated GAPDH levels in blood and Alzheimer’s disease progression. PLoS ONE 2020, 15, e0233289. [Google Scholar] [CrossRef] [PubMed]
- Gui, H.; Gong, Q.; Jiang, J.; Liu, M.; Li, H. Identification of the Hub Genes in Alzheimer’s Disease. Comput. Math. Methods Med. 2021, 2021, 6329041. [Google Scholar] [CrossRef]
- Hara, M.R.; Snyder, S.H. Nitric Oxide−GAPDH−Siah: A Novel Cell Death Cascade. Cell. Mol. Neurobiol. 2006, 26, 525–536. [Google Scholar] [CrossRef]
- Li, C.; Feng, J.J.; Wu, Y.P.; Zhang, G.Y. Cerebral ischemia−reperfusion induces GAPDH S−nitrosylation and nuclear translocation. Biochemistry 2012, 77, 671–678. [Google Scholar] [CrossRef]
- Nakajima, H.; Itakura, M.; Kubo, T.; Kaneshige, A.; Harada, N.; Izawa, T.; Azuma, Y.T.; Kuwamura, M.; Yamaji, R.; Takeuchi, T. Glyceraldehyde−3−phosphate Dehydrogenase (GAPDH) Aggregation Causes Mitochondrial Dysfunction during Oxidative Stress−induced Cell Death. J. Biol. Chem. 2017, 292, 4727–4742. [Google Scholar] [CrossRef] [Green Version]
- Muronetz, V.I.; Medvedeva, M.V.; Sevostyanova, I.A.; Schmalhausen, E.V. Modification of Glyceraldehyde−3−Phosphate Dehydrogenase with Nitric Oxide: Role in Signal Transduction and Development of Apoptosis. Biomolecules 2021, 11, 1656. [Google Scholar] [CrossRef]
- Parker, D.J.; Allison, W.S. The mechanism of inactivation of glyceraldehyde 3−phosphate dehydrogenase by tetrathionate, o−iodosobenzoate, and iodine monochloride. J. Biol. Chem. 1969, 244, 180–189. [Google Scholar] [CrossRef]
- You, K.S.; Benitez, L.V.; McConachie, W.A.; Allison, W.S. The conversion of glyceraldehyde−3−phosphate dehydrogenase to an acylphosphatase by trinitroglycerin and inactivation of this activity by azide and ascorbate. Biochim. Biophys. Acta 1975, 384, 317–330. [Google Scholar] [CrossRef]
- Woo, H.A.; Rhee, S.G. Immunoblot detection of proteins that contain cysteine sulfinic or sulfonic acids with antibodies specific for hyperoxidized cysteine−containing sequences. Methods Redox Signal. 2010, 4, 19–23. [Google Scholar]
- Hwang, N.R.; Yim, S.H.; Kim, Y.M.; Jeong, J.; Song, E.J.; Lee, Y.; Lee, J.H.; Choi, S.; Lee, K.J. Oxidative modifications of glyceraldehyde−3−phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 2009, 423, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Jung, Y.; Na, S.; Jeong, J.; Lee, E.; Kim, M.S.; Choi, S.; Shin, D.H.; Paek, E.; Lee, H.Y.; et al. Novel oxidative modifications in redox−active cysteine residues. Mol. Cell. Proteom. 2011, 10, M110.000513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Doka, E.; Van Laer, K.; Nagy, P.; Grater, F.; Dick, T.P. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Lia, A.; Dowle, A.; Taylor, C.; Santino, A.; Roversi, P. Partial catalytic Cys oxidation of human GAPDH to Cys−sulfonic acid. Wellcome Open Res. 2020, 5, 114. [Google Scholar] [CrossRef]
- Jenkins, J.L.; Tanner, J.J. High−resolution structure of human D−glyceraldehyde−3−phosphate dehydrogenase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62 Pt 3, 290–301. [Google Scholar] [CrossRef]
- Barinova, K.V.; Serebryakova, M.V.; Eldarov, M.A.; Kulikova, A.A.; Mitkevich, V.A.; Muronetz, V.I.; Schmalhausen, E.V. S−glutathionylation of human glyceraldehyde−3−phosphate dehydrogenase and possible role of Cys152−Cys156 disulfide bridge in the active site of the protein. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129560. [Google Scholar] [CrossRef]
- Vaidyanathan, V.V.; Sastry, P.S.; Ramasarma, T. Regulation of the activity of glyceraldehyde 3−phosphate dehydrogenase by glutathione and H2O2. Mol. Cell. Biochem. 1993, 129, 57–65. [Google Scholar] [CrossRef]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human Glyceraldehyde−3−phosphate Dehydrogenase Plays a Direct Role in Reactivating Oxidized Forms of the DNA Repair Enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef] [Green Version]
- Mountassif, D.; Baibai, T.; Fourrat, L.; Moutaouakkil, A.; Iddar, A.; El Kebbaj, M.S.; Soukri, A. Immunoaffinity purification and characterization of glyceraldehyde−3−phosphate dehydrogenase from human erythrocytes. Acta Biochim. Biophys. Sin. 2009, 41, 399–406. [Google Scholar] [CrossRef] [Green Version]
- Elkina, Y.L.; Kuravsky, M.L.; El’darov, M.A.; Stogov, S.V.; Muronetz, V.I.; Schmalhausen, E.V. Recombinant human sperm−specific glyceraldehyde−3−phosphate dehydrogenase: Structural basis for enhanced stability. Biochim. Biophys. Acta 2010, 1804, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.P.; Packer, J.E.; Sims, R.J. Kinetics of the reaction of hydrogen peroxide with cysteine and cysteamine. J. Chem. Soc. Perkin Trans. II 1973, 11, 1547–1549. [Google Scholar] [CrossRef]
- Harris, J.I.; Waters, M. Glyceraldehyde−3−phosphate dehydrogenase. In The Enzymes; Boyer, P.D., Ed.; Academic Press: Boca Raton, FL, USA, 1976; Volume Chapter 1, pp. 1–49. [Google Scholar]
- Nagy, P.; Ashby, M.T. Reactive sulfur species: Kinetics and mechanism of the hydrolysis of cysteine thiosulfinate ester. Chem. Res. Toxicol. 2007, 20, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Miron, T.; Rabinkov, A.; Mirelman, D.; Weiner, L.; Wilchek, M. A spectrophotometric assay for allicin and alliinase (Alliin lyase) activity: Reaction of 2−nitro−5−thiobenzoate with thiosulfinates. Anal. Biochem. 1998, 265, 317–325. [Google Scholar] [CrossRef]
- Rath, A.; Glibowicka, M.; Nadeau, V.G.; Chen, G.; Deber, C.M. Detergent binding explains anomalous SDS−PAGE migration of membrane proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 1760–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mampuys, P.; McElroy, C.R.; Clark, J.H.; Orru, R.V.A.; Maes, B.U.W. Thiosulfonates as Emerging Reactants: Synthesis and Applications. Adv. Synth. Catal. 2019, 362, 3–64. [Google Scholar] [CrossRef] [Green Version]
- Hamann, M.; Zhang, T.; Hendrich, S.; Thomas, J.A. Quantitation of protein sulfinic and sulfonic acid, irreversibly oxidized protein cysteine sites in cellular proteins. Methods Enzymol. 2002, 348, 146–156. [Google Scholar]
- Strus, M.; Brzychczy−Wloch, M.; Kochan, P.; Heczko, P. Hydrogen peroxide produced by Lactobacillus species as a regulatory molecule for vaginal microflora. Med. Doświadczalna Mikrobiol. 2004, 56, 67–77. [Google Scholar]
- Strus, M.; Brzychczy−Wloch, M.; Gosiewski, T.; Kochan, P.; Heczko, P.B. The in vitro effect of hydrogen peroxide on vaginal microbial communities. FEMS Immunol. Med. Microbiol. 2006, 48, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Turell, L.; Botti, H.; Carballal, S.; Ferrer−Sueta, G.; Souza, J.M.; Duran, R.; Freeman, B.A.; Radi, R.; Alvarez, B. Reactivity of sulfenic acid in human serum albumin. Biochemistry 2008, 47, 358–367. [Google Scholar] [CrossRef]
- Wassarman, P.M.; Major, J.P. The reactivity of the sulfhydryl groups of lobster muscle glyceraldehyde 3−phosphate dehydrogenase. Biochemistry 1969, 8, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Hyslop, P.A.; Hinshaw, D.B.; Halsey, W.A., Jr.; Schraufstatter, I.U.; Sauerheber, R.D.; Spragg, R.G.; Jackson, J.H.; Cochrane, C.G. Mechanisms of oxidant−mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. J. Biol. Chem. 1988, 263, 1665–1675. [Google Scholar] [CrossRef]
- Patel, S.; Balaji, P.V.; Sasidhar, Y.U. The sequence TGAAKAVALVL from glyceraldehyde−3−phosphate dehydrogenase displays structural ambivalence and interconverts between alpha−helical and beta−hairpin conformations mediated by collapsed conformational states. J. Pept. Sci. 2007, 13, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.I.; Hara, M.R.; Cascio, M.B.; Wellington, C.L.; Hayden, M.R.; Ross, C.A.; Ha, H.C.; Li, X.J.; Snyder, S.H.; Sawa, A. Mutant Huntingtin: Nuclear translocation and cytotoxicity mediated by GAPDH. Proc. Natl. Acad. Sci. USA 2006, 103, 3405–3409. [Google Scholar] [CrossRef] [Green Version]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Janin, J.; Sternberg, M.J. Protein flexibility, not disorder, is intrinsic to molecular recognition. F1000 Biol. Rep. 2013, 5, 2. [Google Scholar] [CrossRef]
- El Kadmiri, N.; Slassi, I.; El Moutawakil, B.; Nadifi, S.; Tadevosyan, A.; Hachem, A.; Soukri, A. Glyceraldehyde−3−phosphate dehydrogenase (GAPDH) and Alzheimer’s disease. Pathol. Biol. 2014, 62, 333–336. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Nikotina, A.D.; Semenyuk, P.I.; Evstafyeva, D.B.; Mikhaylova, E.R.; Muronetz, V.I.; Shevtsov, M.A.; Tolkacheva, A.V.; Dobrodumov, A.V.; Shavarda, A.L.; et al. Small molecules preventing GAPDH aggregation are therapeutically applicable in cell and rat models of oxidative stress. Free Radic. Biol. Med. 2016, 92, 29–38. [Google Scholar] [CrossRef]
- Zaffagnini, M.; Marchand, C.H.; Malferrari, M.; Murail, S.; Bonacchi, S.; Genovese, D.; Montalti, M.; Venturoli, G.; Falini, G.; Baaden, M.; et al. Glutathionylation primes soluble glyceraldehyde−3−phosphate dehydrogenase for late collapse into insoluble aggregates. Proc. Natl. Acad. Sci. USA 2019, 116, 26057–26065. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Tsolaki, M.; Mikhaylova, E.R.; Benken, K.A.; Shevtsov, M.A.; Nikotina, A.D.; Lechpammer, M.; Mitkevich, V.A.; Makarov, A.A.; Moskalev, A.A.; et al. Extracellular GAPDH Promotes Alzheimer Disease Progression by Enhancing Amyloid−beta Aggregation and Cytotoxicity. Aging Dis. 2021, 12, 1223–1237. [Google Scholar] [CrossRef]
- Cyrne, L.; Antunes, F.; Sousa−Lopes, A.; Diaz−Berrio, J.; Marinho, H.S. Glyceraldehyde−3−phosphate dehydrogenase is largely unresponsive to low regulatory levels of hydrogen peroxide in Saccharomyces cerevisiae. BMC Biochem. 2010, 11, 49. [Google Scholar] [CrossRef] [Green Version]
- Schraufstatter, I.U.; Hyslop, P.A.; Hinshaw, D.B.; Spragg, R.G.; Sklar, L.A.; Cochrane, C.G. Hydrogen peroxide−induced injury of cells and its prevention by inhibitors of poly(ADP−ribose) polymerase. Proc. Natl. Acad. Sci. USA 1986, 83, 4908–4912. [Google Scholar] [CrossRef] [Green Version]
- Barinova, K.V.; Serebryakova, M.V.; Muronetz, V.I.; Schmalhausen, E.V. S−glutathionylation of glyceraldehyde−3−phosphate dehydrogenase induces formation of C150−C154 intrasubunit disulfide bond in the active site of the enzyme. Biochim. Biophys. Acta 2017, 1861, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Spragg, R.G.; Hinshaw, D.B.; Hyslop, P.A.; Schraufstatter, I.U.; Cochrane, C.G. Alterations in adenosine triphosphate and energy charge in cultured endothelial and P388D1 cells after oxidant injury. J. Clin. Investig. 1985, 76, 1471–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velick, S.F.; Hayes, J.E., Jr. Phosphate binding and the glyceraldehyde−3−phosphate dehydrogenase reaction. J. Biol. Chem. 1953, 203, 545–562. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Atkins, P.; Atkins, P.W.; de Paula, J. The rates of chemical reactions. In Atkins’ Physical Chemistry, 8th ed.; W.H. Freeman: New York, NY, USA, 2006. [Google Scholar]
- Thannhauser, T.W.; Konishi, Y.; Scheraga, H.A. Sensitive quantitative analysis of disulfide bonds in polypeptides and proteins. Anal. Biochem. 1984, 138, 181–188. [Google Scholar] [CrossRef]
- Lawson, L.D.; Wood, S.G.; Hughes, B.G. HPLC analysis of allicin and other thiosulfinates in garlic clove homogenates. Planta Med. 1991, 57, 263–270. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Modifying Alkylating Agent: IAM | R.T. [min] | Peak Area | *Estimated Isotopic Ratio | |
| IVSNASCc[+48]TTNCv[+57]LAPK | Cc(SO3H) Cv(Carbamidomethyl) | 15.33 | 8.79 × 107 | 17% (16O3) | |
| IVSNASCc[+50]TTNCv[+57]LAPK | Cc(S16O218OH) Cv(Carbamidomethyl) | 15.52 | 2.7 × 108 | 83% (16O2,18O) | |
| IVSNASCc[+32]TTNCv[+57]LAPK | Cc(SO2H) Cv(Carbamidomethyl) | 15.18 | 8.6 × 107 | 21% (16O2) | |
| IVSNASCc[+34]TTNCv[+57]LAPK | Cc(S18O16OH) Cv(Carbamidomethyl) | 15.4 | 2.3 × 108 | 79% (16O,18O) | |
| Peptide Sequence | Charge | Modifying Alkylating Agents: NEM then IAM | R.T. [min] | Peak Area | Summed Relative Peak Area (%) |
| IVSNAS*CTTN*CLAPK | 2+ | *2 × Carbamidomethyl | 24.38 | 5.39 × 105 | 3.31 |
| IVSNAS*CTTN*CLAPK | 3+ | *2 × Carbamidomethyl | 24.38 | 5.83 × 105 | 3.31 |
| IVSNAS*CTTN*CLAPK | 2+ | *C(SO3H), *C(Carbamidomethyl) | 27.29 | 1.74 × 107 | 76.42 |
| IVSNAS*CTTN*CLAPK | 3+ | *C(SO3H), *C(Carbamidomethyl) | 27.29 | 8.42 × 106 | 76.42 |
| IVSNAS*CTTN*CLAPK | 2+ | *C(SO3H), *C(Ethyl pyrrolidinedione) | 30.56 | 5.07 × 106 | 20.26 |
| IVSNAS*CTTN*CLAPK | 3+ | *C(SO3H), *C(Ethyl pyrrolidinedione) | 30.56 | 1.79 × 105 | 20.26 |
| Lactobacillus Species | Accession No. | Active Site | H2O2 Secretor? |
|---|---|---|---|
| L. plantarum | Q88YH6 | SCTTNC | No |
| L. fermentum | B2GAL7 | SCTTSC | No |
| L. rhamnosus | C2JVV2 | SCTTNC | No |
| L. brevis | U2QJ09 | SCTTNC | No |
| L. dulbrueckii | O32755 | SCTTNS | Yes |
| L. acidophilus | Q5FL51 | SCTTNS | Yes |
| L. crispatus | Q5K118 | SCTTNS | Yes |
| L. johnsonii | C2E5E9 | SCTTNS | Yes |
| L. gasseri | DIYHE7 | SCTTNS | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyslop, P.A.; Chaney, M.O. Mechanism of GAPDH Redox Signaling by H2O2 Activation of a Two−Cysteine Switch. Int. J. Mol. Sci. 2022, 23, 4604. https://doi.org/10.3390/ijms23094604
Hyslop PA, Chaney MO. Mechanism of GAPDH Redox Signaling by H2O2 Activation of a Two−Cysteine Switch. International Journal of Molecular Sciences. 2022; 23(9):4604. https://doi.org/10.3390/ijms23094604
Chicago/Turabian StyleHyslop, Paul A., and Michael O. Chaney. 2022. "Mechanism of GAPDH Redox Signaling by H2O2 Activation of a Two−Cysteine Switch" International Journal of Molecular Sciences 23, no. 9: 4604. https://doi.org/10.3390/ijms23094604