
Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing

 ,
,  , and
, and
Abstract
:1. Introduction
2. Myotonic Dystrophy Type 1: Clinical Features and Pathogenetic Mechanisms
3. DM1 Cell and Animal Models
4. Molecular Therapies Acting on DMPK Gene Expression
4.1. Induction of Repeat Contractions and Transcription Inhibition at DMPK Gene
4.2. Targeting Expanded CUG RNA
4.2.1. ASOs Blocking MBNL1/2 Binding
4.2.2. ASOs Silencing Expanded CUG Transcript
4.2.3. Other Strategies
4.3. Targeting MBNL1/2
4.4. Targeting MBNL:CUG-Repeat Interactions
4.5. Targeting CELF1 and Signaling Molecules Downstream of CUG-Repeat RNA
4.6. Advantages and Limitations of DMPK-Expression Targeting Therapies
5. DMPK Gene Editing
5.1. Zinc Finger and TALEN Nucleases
5.2. CRISPR/Cas9 Gene-Editing Approaches
5.3. Specificity and Safety of Gene-Editing Approaches
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, N.E.; Butterfield, R.J.; Mayne, K.; Newcomb, T.; Imburgia, C.; Dunn, D.; Duval, B.; Feldkamp, M.L.; Weiss, R.B. Population-Based Prevalence of Myotonic Dystrophy Type 1 Using Genetic Analysis of Statewide Blood Screening Program. Neurology 2021, 96, e1045–e1053. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Myotonic Dystrophy Type 1. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1165/ (accessed on 21 March 2022).
- Harper, P.; van Engelen, B.; Eymard, B.; Wilcox, D. Myotonic Dystrophy. Present management, future therapy; Oxford University Press: Oxford, UK, 2004; ISBN 9780198527824. [Google Scholar]
- Meola, G.; Cardani, R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim. Et Biophys. Acta 2015, 1852, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef]
- Ashizawa, T.; Dunne, C.J.; Dubel, J.R.; Perryman, M.B.; Epstein, H.F.; Boerwinkle, E.; Hejtmancik, J.F. Anticipation in myotonic dystrophy. I. Statistical verification based on clinical and haplotype findings. Neurology 1992, 42, 1871–1877. [Google Scholar] [CrossRef]
- Overend, G.; Legare, C.; Mathieu, J.; Bouchard, L.; Gagnon, C.; Monckton, D.G. Allele length of the DMPK CTG repeat is a predictor of progressive myotonic dystrophy type 1 phenotypes. Hum. Mol. Genet. 2019, 28, 2245–2254. [Google Scholar] [CrossRef]
- Miller, J.N.; van der Plas, E.; Hamilton, M.; Koscik, T.R.; Gutmann, L.; Cumming, S.A.; Monckton, D.G.; Nopoulos, P.C. Variant repeats within the DMPK CTG expansion protect function in myotonic dystrophy type 1. Neurol. Genet. 2020, 6, e504. [Google Scholar] [CrossRef]
- Andre, L.M.; Ausems, C.R.M.; Wansink, D.G.; Wieringa, B. Abnormalities in Skeletal Muscle Myogenesis, Growth, and Regeneration in Myotonic Dystrophy. Front. Neurol. 2018, 9, 368. [Google Scholar] [CrossRef]
- Visconti, V.V.; Centofanti, F.; Fittipaldi, S.; Macri, E.; Novelli, G.; Botta, A. Epigenetics of Myotonic Dystrophies: A Minireview. Int. J. Mol. Sci. 2021, 22, 12594. [Google Scholar] [CrossRef]
- Alwazzan, M.; Newman, E.; Hamshere, M.G.; Brook, J.D. Myotonic dystrophy is associated with a reduced level of RNA from the DMWD allele adjacent to the expanded repeat. Hum. Mol. Genet. 1999, 8, 1491–1497. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.A.; Wymer, J.P.; Simmons, Z.; McClain, C.; Moxley, R.T., 3rd. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat. Genet. 1997, 16, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, J.R.; Huguet, A.; Nicole, A.; Munnich, A.; Gourdon, G. Transcriptionally Repressive Chromatin Remodelling and CpG Methylation in the Presence of Expanded CTG-Repeats at the DM1 Locus. J. Nucleic Acids 2013, 2013, 567435. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Cody, N.A.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutter, C.A.; Bubenik, J.L.; Oliveira, R.; Ivankovic, F.; Sznajder, L.J.; Kidd, B.M.; Pinto, B.S.; Otero, B.A.; Carter, H.A.; Vitriol, E.A.; et al. Cell-type-specific dysregulation of RNA alternative splicing in short tandem repeat mouse knockin models of myotonic dystrophy. Genes Dev. 2019, 33, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Kalsotra, A.; Xiao, X.; Ward, A.J.; Castle, J.C.; Johnson, J.M.; Burge, C.B.; Cooper, T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl. Acad. Sci. USA 2008, 105, 20333–20338. [Google Scholar] [CrossRef] [Green Version]
- Imbriano, C.; Molinari, S. Alternative Splicing of Transcription Factors Genes in Muscle Physiology and Pathology. Genes 2018, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Martinez, A.; Soblechero-Martin, P.; de-la-Puente-Ovejero, L.; Nogales-Gadea, G.; Arechavala-Gomeza, V. An Overview of Alternative Splicing Defects Implicated in Myotonic Dystrophy Type I. Genes 2020, 11, 1109. [Google Scholar] [CrossRef]
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007, 28, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.F.; Loureiro, J.R.; Bessa, J.; Silveira, I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes 2020, 11, 1418. [Google Scholar] [CrossRef]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell. 2005, 20, 483–489. [Google Scholar] [CrossRef]
- Huguet, A.; Medja, F.; Nicole, A.; Vignaud, A.; Guiraud-Dogan, C.; Ferry, A.; Decostre, V.; Hogrel, J.Y.; Metzger, F.; Hoeflich, A.; et al. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1000 CTG repeats from the human DM1 locus. PLoS Genet. 2012, 8, e1003043. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehorst, E.; Nunez-Manchon, J.; Ballester-Lopez, A.; Almendrote, M.; Lucente, G.; Arbex, A.; Chojnacki, J.; Vazquez-Manrique, R.P.; Gomez-Escribano, A.P.; Pintos-Morell, G.; et al. Characterization of RAN Translation and Antisense Transcription in Primary Cell Cultures of Patients with Myotonic Dystrophy Type 1. J. Clin. Med. 2021, 10, 5520. [Google Scholar] [CrossRef] [PubMed]
- Falcone, G.; Perfetti, A.; Cardinali, B.; Martelli, F. Noncoding RNAs: Emerging players in muscular dystrophies. BioMed Res. Int. 2014, 2014, 503634. [Google Scholar] [CrossRef] [Green Version]
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. NMD 2011, 21, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Perfetti, A.; Greco, S.; Cardani, R.; Fossati, B.; Cuomo, G.; Valaperta, R.; Ambrogi, F.; Cortese, A.; Botta, A.; Mignarri, A.; et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci. Rep. 2016, 6, 38174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 2011, 18, 840–845. [Google Scholar] [CrossRef]
- Czubak, K.; Taylor, K.; Piasecka, A.; Sobczak, K.; Kozlowska, K.; Philips, A.; Sedehizadeh, S.; Brook, J.D.; Wojciechowska, M.; Kozlowski, P. Global Increase in Circular RNA Levels in Myotonic Dystrophy. Front. Genet. 2019, 10, 649. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Cardinali, B.; Falcone, G.; Martelli, F. Circular RNAs in Muscle Function and Disease. Int. J. Mol. Sci. 2018, 19, 3454. [Google Scholar] [CrossRef] [Green Version]
- Voellenkle, C.; Perfetti, A.; Carrara, M.; Fuschi, P.; Renna, L.V.; Longo, M.; Sain, S.B.; Cardani, R.; Valaperta, R.; Silvestri, G.; et al. Dysregulation of Circular RNAs in Myotonic Dystrophy Type 1. Int. J. Mol. Sci. 2019, 20, 1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozimski, L.L.; Sabater-Arcis, M.; Bargiela, A.; Artero, R. The hallmarks of myotonic dystrophy type 1 muscle dysfunction. Biol. Rev. Camb. Philos. Soc. 2021, 96, 716–730. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Espinosa, J.; Gonzalez-Barriga, A.; Lopez-Castel, A.; Artero, R. Deciphering the Complex Molecular Pathogenesis of Myotonic Dystrophy Type 1 through Omics Studies. Int. J. Mol. Sci. 2022, 23, 1441. [Google Scholar] [CrossRef] [PubMed]
- Philips, A.V.; Timchenko, L.T.; Cooper, T.A. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 1998, 280, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Warf, M.B.; Berglund, J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 2007, 13, 2238–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arandel, L.; Polay Espinoza, M.; Matloka, M.; Bazinet, A.; De Dea Diniz, D.; Naouar, N.; Rau, F.; Jollet, A.; Edom-Vovard, F.; Mamchaoui, K.; et al. Immortalized human myotonic dystrophy muscle cell lines to assess therapeutic compounds. Dis. Models Mech. 2017, 10, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Pantic, B.; Borgia, D.; Giunco, S.; Malena, A.; Kiyono, T.; Salvatori, S.; De Rossi, A.; Giardina, E.; Sangiuolo, F.; Pegoraro, E.; et al. Reliable and versatile immortal muscle cell models from healthy and myotonic dystrophy type 1 primary human myoblasts. Exp. Cell Res. 2016, 342, 39–51. [Google Scholar] [CrossRef]
- Provenzano, C.; Cappella, M.; Valaperta, R.; Cardani, R.; Meola, G.; Martelli, F.; Cardinali, B.; Falcone, G. CRISPR/Cas9-Mediated Deletion of CTG Expansions Recovers Normal Phenotype in Myogenic Cells Derived from Myotonic Dystrophy 1 Patients. Mol. Ther. Nucleic Acids 2017, 9, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Renna, L.V.; Bose, F.; Iachettini, S.; Fossati, B.; Saraceno, L.; Milani, V.; Colombo, R.; Meola, G.; Cardani, R. Receptor and post-receptor abnormalities contribute to insulin resistance in myotonic dystrophy type 1 and type 2 skeletal muscle. PLoS ONE 2017, 12, e0184987. [Google Scholar] [CrossRef] [Green Version]
- Franck, S.; Couvreu De Deckersberg, E.; Bubenik, J.L.; Markouli, C.; Barbe, L.; Allemeersch, J.; Hilven, P.; Duque, G.; Swanson, M.S.; Gheldof, A.; et al. Myotonic dystrophy type 1 embryonic stem cells show decreased myogenic potential, increased CpG methylation at the DMPK locus and RNA mis-splicing. Biol. Open 2022, 11. [Google Scholar] [CrossRef]
- Gao, Y.; Guo, X.; Santostefano, K.; Wang, Y.; Reid, T.; Zeng, D.; Terada, N.; Ashizawa, T.; Xia, G. Genome Therapy of Myotonic Dystrophy Type 1 iPS Cells for Development of Autologous Stem Cell Therapy. Mol. Ther. 2016, 24, 1378–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueki, J.; Nakamori, M.; Nakamura, M.; Nishikawa, M.; Yoshida, Y.; Tanaka, A.; Morizane, A.; Kamon, M.; Araki, T.; Takahashi, M.P.; et al. Myotonic dystrophy type 1 patient-derived iPSCs for the investigation of CTG repeat instability. Sci. Rep. 2017, 7, 42522. [Google Scholar] [CrossRef] [PubMed]
- Martineau, L.; Racine, V.; Benichou, S.A.; Puymirat, J. Lymphoblastoids cell lines—Derived iPSC line from a 26-year-old myotonic dystrophy type 1 patient carrying (CTG)200 expansion in the DMPK gene: CHUQi001-A. Stem Cell Res. 2018, 26, 103–106. [Google Scholar] [CrossRef]
- Spitalieri, P.; Talarico, R.V.; Caioli, S.; Murdocca, M.; Serafino, A.; Girasole, M.; Dinarelli, S.; Longo, G.; Pucci, S.; Botta, A.; et al. Modelling the pathogenesis of Myotonic Dystrophy type 1 cardiac phenotype through human iPSC-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2018, 118, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garibay, X.; Ortega, M.A.; Cerro-Herreros, E.; Comelles, J.; Martinez, E.; Artero, R.; Fernandez-Costa, J.M.; Ramon-Azcon, J. Bioengineeredin vitro3D model of myotonic dystrophy type 1 human skeletal muscle. Biofabrication 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- de Haro, M.; Al-Ramahi, I.; De Gouyon, B.; Ukani, L.; Rosa, A.; Faustino, N.A.; Ashizawa, T.; Cooper, T.A.; Botas, J. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 2006, 15, 2138–2145. [Google Scholar] [CrossRef]
- Houseley, J.M.; Wang, Z.; Brock, G.J.; Soloway, J.; Artero, R.; Perez-Alonso, M.; O’Dell, K.M.; Monckton, D.G. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum. Mol. Genet. 2005, 14, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Picchio, L.; Plantie, E.; Renaud, Y.; Poovthumkadavil, P.; Jagla, K. Novel Drosophila model of myotonic dystrophy type 1: Phenotypic characterization and genome-wide view of altered gene expression. Hum. Mol. Genet. 2013, 22, 2795–2810. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335. [Google Scholar] [CrossRef]
- Jansen, G.; Groenen, P.J.; Bachner, D.; Jap, P.H.; Coerwinkel, M.; Oerlemans, F.; van den Broek, W.; Gohlsch, B.; Pette, D.; Plomp, J.J.; et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat. Genet. 1996, 13, 316–324. [Google Scholar] [CrossRef]
- Carrell, S.T.; Carrell, E.M.; Auerbach, D.; Pandey, S.K.; Bennett, C.F.; Dirksen, R.T.; Thornton, C.A. Dmpk gene deletion or antisense knockdown does not compromise cardiac or skeletal muscle function in mice. Hum. Mol. Genet. 2016, 25, 4328–4338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, S.K.; Wheeler, T.M.; Justice, S.L.; Kim, A.; Younis, H.S.; Gattis, D.; Jauvin, D.; Puymirat, J.; Swayze, E.E.; Freier, S.M.; et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J. Pharmacol. Exp. Ther. 2015, 355, 329–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charizanis, K.; Lee, K.Y.; Batra, R.; Goodwin, M.; Zhang, C.; Yuan, Y.; Shiue, L.; Cline, M.; Scotti, M.M.; Xia, G.; et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 2012, 75, 437–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanadia, R.N.; Johnstone, K.A.; Mankodi, A.; Lungu, C.; Thornton, C.A.; Esson, D.; Timmers, A.M.; Hauswirth, W.W.; Swanson, M.S. A muscleblind knockout model for myotonic dystrophy. Science 2003, 302, 1978–1980. [Google Scholar] [CrossRef] [Green Version]
- Matynia, A.; Ng, C.H.; Dansithong, W.; Chiang, A.; Silva, A.J.; Reddy, S. Muscleblind1, but not Dmpk or Six5, contributes to a complex phenotype of muscular and motivational deficits in mouse models of myotonic dystrophy. PLoS ONE 2010, 5, e9857. [Google Scholar] [CrossRef]
- Poulos, M.G.; Batra, R.; Li, M.; Yuan, Y.; Zhang, C.; Darnell, R.B.; Swanson, M.S. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet. 2013, 22, 3547–3558. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Personius, K.E.; DiFranco, M.; Dansithong, W.; Yu, C.; Srivastava, S.; Dixon, D.M.; Bhatt, D.B.; Comai, L.; Vergara, J.L.; et al. Muscleblind-Like 1 and Muscleblind-Like 3 Depletion Synergistically Enhances Myotonia by Altering Clc-1 RNA Translation. EBioMedicine 2015, 2, 1034–1047. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef]
- Thomas, J.D.; Sznajder, L.J.; Bardhi, O.; Aslam, F.N.; Anastasiadis, Z.P.; Scotti, M.M.; Nishino, I.; Nakamori, M.; Wang, E.T.; Swanson, M.S. Disrupted prenatal RNA processing and myogenesis in congenital myotonic dystrophy. Genes Dev. 2017, 31, 1122–1133. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.H.; Bundman, D.; Armstrong, D.L.; Cooper, T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005, 14, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Koshelev, M.; Sarma, S.; Price, R.E.; Wehrens, X.H.; Cooper, T.A. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, A.J.; Rimer, M.; Killian, J.M.; Dowling, J.J.; Cooper, T.A. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010, 19, 3614–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benders, A.A.; Groenen, P.J.; Oerlemans, F.T.; Veerkamp, J.H.; Wieringa, B. Myotonic dystrophy protein kinase is involved in the modulation of the Ca2+ homeostasis in skeletal muscle cells. J. Clin. Investig. 1997, 100, 1440–1447. [Google Scholar] [CrossRef] [PubMed]
- Berul, C.I.; Maguire, C.T.; Aronovitz, M.J.; Greenwood, J.; Miller, C.; Gehrmann, J.; Housman, D.; Mendelsohn, M.E.; Reddy, S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J. Clin. Investig. 1999, 103, R1–R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006, 38, 1066–1070. [Google Scholar] [CrossRef] [Green Version]
- Yadava, R.S.; Mandal, M.; Giese, J.M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Modeling muscle regeneration in RNA toxicity mice. Hum. Mol. Genet. 2021, 30, 1111–1130. [Google Scholar] [CrossRef]
- Yadava, R.S.; Yu, Q.; Mandal, M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3′UTR RNA. Hum. Mol. Genet. 2020, 29, 1440–1453. [Google Scholar] [CrossRef]
- Seznec, H.; Agbulut, O.; Sergeant, N.; Savouret, C.; Ghestem, A.; Tabti, N.; Willer, J.C.; Ourth, L.; Duros, C.; Brisson, E.; et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 2001, 10, 2717–2726. [Google Scholar] [CrossRef] [Green Version]
- Seznec, H.; Lia-Baldini, A.S.; Duros, C.; Fouquet, C.; Lacroix, C.; Hofmann-Radvanyi, H.; Junien, C.; Gourdon, G. Transgenic mice carrying large human genomic sequences with expanded CTG repeat mimic closely the DM CTG repeat intergenerational and somatic instability. Hum. Mol. Genet. 2000, 9, 1185–1194. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Hernandez, O.; Guiraud-Dogan, C.; Sicot, G.; Huguet, A.; Luilier, S.; Steidl, E.; Saenger, S.; Marciniak, E.; Obriot, H.; Chevarin, C.; et al. Myotonic dystrophy CTG expansion affects synaptic vesicle proteins, neurotransmission and mouse behaviour. Brain J. Neurol. 2013, 136, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sicot, G.; Servais, L.; Dinca, D.M.; Leroy, A.; Prigogine, C.; Medja, F.; Braz, S.O.; Huguet-Lachon, A.; Chhuon, C.; Nicole, A.; et al. Downregulation of the Glial GLT1 Glutamate Transporter and Purkinje Cell Dysfunction in a Mouse Model of Myotonic Dystrophy. Cell Rep. 2017, 19, 2718–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Nakamori, M.; Lueck, J.D.; Pouliquin, P.; Aoike, F.; Fujimura, H.; Dirksen, R.T.; Takahashi, M.P.; Dulhunty, A.F.; Sakoda, S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum. Mol. Genet. 2005, 14, 2189–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tylock, K.M.; Auerbach, D.S.; Tang, Z.Z.; Thornton, C.A.; Dirksen, R.T. Biophysical mechanisms for QRS- and QTc-interval prolongation in mice with cardiac expression of expanded CUG-repeat RNA. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef]
- Wang, G.S.; Kearney, D.L.; De Biasi, M.; Taffet, G.; Cooper, T.A. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J. Clin. Investig. 2007, 117, 2802–2811. [Google Scholar] [CrossRef] [Green Version]
- Orengo, J.P.; Chambon, P.; Metzger, D.; Mosier, D.R.; Snipes, G.J.; Cooper, T.A. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2008, 105, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.Y.; Lin, Y.M.; Wang, L.H.; Kuo, T.Y.; Cheng, S.J.; Wang, G.S. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 2017, 26, 2247–2257. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.N.; Campbell, H.M.; Guan, X.; Word, T.A.; Wehrens, X.H.; Xia, Z.; Cooper, T.A. Reversible cardiac disease features in an inducible CUG repeat RNA-expressing mouse model of myotonic dystrophy. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Valencik, M.L.; McDonald, J.A. Codon optimization markedly improves doxycycline regulated gene expression in the mouse heart. Transgenic Res. 2001, 10, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.R.; Rajapakshe, K.; Huang, S.; Coarfa, C.; Cooper, T.A. Mechanisms of skeletal muscle wasting in a mouse model for myotonic dystrophy type 1. Hum. Mol. Genet. 2018, 27, 2789–2804. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Gilabert, M.; Lopez-Castel, A.; Artero, R. Myotonic dystrophy type 1 drug development: A pipeline toward the market. Drug Discov. Today 2021, 26, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.A.; Wang, E.; Carrell, E.M. Myotonic dystrophy: Approach to therapy. Curr. Opin. Genet. Dev. 2017, 44, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Pereira, M.; Monckton, D.G. Chemically induced increases and decreases in the rate of expansion of a CAG*CTG triplet repeat. Nucleic Acids Res. 2004, 32, 2865–2872. [Google Scholar] [CrossRef] [Green Version]
- Hashem, V.I.; Sinden, R.R. Chemotherapeutically induced deletion of expanded triplet repeats. Mutat. Res. 2002, 508, 107–119. [Google Scholar] [CrossRef]
- Pineiro, E.; Fernandez-Lopez, L.; Gamez, J.; Marcos, R.; Surralles, J.; Velazquez, A. Mutagenic stress modulates the dynamics of CTG repeat instability associated with myotonic dystrophy type 1. Nucleic Acids Res. 2003, 31, 6733–6740. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Lau, R.; Marcadier, J.L.; Chitayat, D.; Pearson, C.E. Replication inhibitors modulate instability of an expanded trinucleotide repeat at the myotonic dystrophy type 1 disease locus in human cells. Am. J. Hum. Genet. 2003, 73, 1092–1105. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, V.C.; Dion, V. Modifiers of CAG/CTG Repeat Instability: Insights from Mammalian Models. J. Huntingtons Dis. 2021, 10, 123–148. [Google Scholar] [CrossRef]
- Nakamori, M.; Panigrahi, G.B.; Lanni, S.; Gall-Duncan, T.; Hayakawa, H.; Tanaka, H.; Luo, J.; Otabe, T.; Li, J.; Sakata, A.; et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet. 2020, 52, 146–159. [Google Scholar] [CrossRef]
- Nakamori, M.; Gourdon, G.; Thornton, C.A. Stabilization of expanded (CTG)*(CAG) repeats by antisense oligonucleotides. Mol. Ther. 2011, 19, 2222–2227. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.R.; Chang, C.R.; Chern, Y.; Wang, T.H.; Hsieh, W.C.; Shen, W.C.; Chang, C.Y.; Chu, I.C.; Deng, N.; Cohen, S.N.; et al. Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell 2012, 148, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, N.J.; Carlomagno, Y.; Zhang, Y.J.; Almeida, S.; Cook, C.N.; Gendron, T.F.; Prudencio, M.; Van Blitterswijk, M.; Belzil, V.; Couthouis, J.; et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 2016, 353, 708–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.M.; Chern, Y.; Chen, I.H.; Liu, C.R.; Li, S.H.; Chun, S.J.; Rigo, F.; Bennett, C.F.; Deng, N.; Feng, Y.; et al. Effects on murine behavior and lifespan of selectively decreasing expression of mutant huntingtin allele by supt4h knockdown. PLoS Genet. 2015, 11, e1005043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coonrod, L.A.; Nakamori, M.; Wang, W.; Carrell, S.; Hilton, C.L.; Bodner, M.J.; Siboni, R.B.; Docter, A.G.; Haley, M.M.; Thornton, C.A.; et al. Reducing levels of toxic RNA with small molecules. ACS Chem. Biol. 2013, 8, 2528–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siboni, R.B.; Nakamori, M.; Wagner, S.D.; Struck, A.J.; Coonrod, L.A.; Harriott, S.A.; Cass, D.M.; Tanner, M.K.; Berglund, J.A. Actinomycin D Specifically Reduces Expanded CUG Repeat RNA in Myotonic Dystrophy Models. Cell Rep. 2015, 13, 2386–2394. [Google Scholar] [CrossRef] [Green Version]
- Reddy, K.; Jenquin, J.R.; McConnell, O.L.; Cleary, J.D.; Richardson, J.I.; Pinto, B.S.; Haerle, M.C.; Delgado, E.; Planco, L.; Nakamori, M.; et al. A CTG repeat-selective chemical screen identifies microtubule inhibitors as selective modulators of toxic CUG RNA levels. Proc. Natl. Acad. Sci. USA 2019, 116, 20991–21000. [Google Scholar] [CrossRef] [Green Version]
- Overby, S.J.; Cerro-Herreros, E.; Llamusi, B.; Artero, R. RNA-mediated therapies in myotonic dystrophy. Drug Discov. Today 2018, 23, 2013–2022. [Google Scholar] [CrossRef]
- Goodchild, J.; Kim, B.; Zamecnik, P.C. The clearance and degradation of oligodeoxynucleotides following intravenous injection into rabbits. Antisense Res. Dev. 1991, 1, 153–160. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Mulders, S.A.; van den Broek, W.J.; Wheeler, T.M.; Croes, H.J.; van Kuik-Romeijn, P.; de Kimpe, S.J.; Furling, D.; Platenburg, G.J.; Gourdon, G.; Thornton, C.A.; et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 13915–13920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, T.M.; Sobczak, K.; Lueck, J.D.; Osborne, R.J.; Lin, X.; Dirksen, R.T.; Thornton, C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science 2009, 325, 336–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtkowiak-Szlachcic, A.; Taylor, K.; Stepniak-Konieczna, E.; Sznajder, L.J.; Mykowska, A.; Sroka, J.; Thornton, C.A.; Sobczak, K. Short antisense-locked nucleic acids (all-LNAs) correct alternative splicing abnormalities in myotonic dystrophy. Nucleic Acids Res. 2015, 43, 3318–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.F.; Varela, M.A.; Arandel, L.; Holland, A.; Naouar, N.; Arzumanov, A.; Seoane, D.; Revillod, L.; Bassez, G.; Ferry, A.; et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J. Clin. Investig. 2019, 129, 4739–4744. [Google Scholar] [CrossRef] [Green Version]
- Leger, A.J.; Mosquea, L.M.; Clayton, N.P.; Wu, I.H.; Weeden, T.; Nelson, C.A.; Phillips, L.; Roberts, E.; Piepenhagen, P.A.; Cheng, S.H.; et al. Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther. 2013, 23, 109–117. [Google Scholar] [CrossRef]
- NeuBase Therapeutics, Inc. NT-0200: Myotonic Dystrophy Type 1 (DM1). Available online: https://www.neubasetherapeutics.com/pipeline/ (accessed on 21 March 2022).
- Hsieh, W.C.; Bahal, R.; Thadke, S.A.; Bhatt, K.; Sobczak, K.; Thornton, C.; Ly, D.H. Design of a "Mini" Nucleic Acid Probe for Cooperative Binding of an RNA-Repeated Transcript Associated with Myotonic Dystrophy Type 1. Biochemistry 2018, 57, 907–911. [Google Scholar] [CrossRef]
- Ait Benichou, S.; Jauvin, D.; De Serres-Berard, T.; Pierre, M.; Ling, K.K.; Bennett, C.F.; Rigo, F.; Gourdon, G.; Chahine, M.; Puymirat, J. Antisense oligonucleotides as a potential treatment for brain deficits observed in myotonic dystrophy type 1. Gene Ther. 2022, 1–12. [Google Scholar] [CrossRef]
- Jauvin, D.; Chretien, J.; Pandey, S.K.; Martineau, L.; Revillod, L.; Bassez, G.; Lachon, A.; MacLeod, A.R.; Gourdon, G.; Wheeler, T.M.; et al. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol. Therapy. Nucleic Acids 2017, 7, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, T.M.; Leger, A.J.; Pandey, S.K.; MacLeod, A.R.; Nakamori, M.; Cheng, S.H.; Wentworth, B.M.; Bennett, C.F.; Thornton, C.A. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 2012, 488, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Bennett, C.F.; Cooper, T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2012, 109, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- A Safety and Tolerability Study of Multiple Doses of ISIS-DMPKRx in Adults With Myotonic Dystrophy Type 1. Available online: https://clinicaltrials.gov/ct2/show/NCT02312011 (accessed on 21 March 2022).
- Cerro-Herreros, E.; Gonzalez-Martinez, I.; Moreno, N.; Espinosa-Espinosa, J.; Fernandez-Costa, J.M.; Colom-Rodrigo, A.; Overby, S.J.; Seoane-Miraz, D.; Poyatos-Garcia, J.; Vilchez, J.J.; et al. Preclinical characterization of antagomiR-218 as a potential treatment for myotonic dystrophy. Mol. Ther. Nucleic Acids 2021, 26, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Cerro-Herreros, E.; Gonzalez-Martinez, I.; Moreno-Cervera, N.; Overby, S.; Perez-Alonso, M.; Llamusi, B.; Artero, R. Therapeutic Potential of AntagomiR-23b for Treating Myotonic Dystrophy. Mol. Ther. Nucleic Acids 2020, 21, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Overby, S.J.; Cerro-Herreros, E.; Gonzalez-Martinez, I.; Varela, M.A.; Seoane-Miraz, D.; Jad, Y.; Raz, R.; Moller, T.; Perez-Alonso, M.; Wood, M.J.; et al. Proof of concept of peptide-linked blockmiR-induced MBNL functional rescue in myotonic dystrophy type 1 mouse model. Mol. Therapy Nucleic Acids 2022, 27, 1146–1155. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, K.; Wheeler, T.M.; Wang, W.; Thornton, C.A. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 2013, 21, 380–387. [Google Scholar] [CrossRef] [Green Version]
- Study of AOC 1001 in Adult Myotonic Dystrophy Type 1 (DM1) Patients (MARINA). Available online: https://clinicaltrials.gov/ct2/show/NCT05027269?term=NCT05027269&draw=2&rank=1 (accessed on 21 March 2022).
- Kanadia, R.N.; Shin, J.; Yuan, Y.; Beattie, S.G.; Wheeler, T.M.; Thornton, C.A.; Swanson, M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. USA 2006, 103, 11748–11753. [Google Scholar] [CrossRef] [Green Version]
- Arandel, L.; Matloka, M.; Klein, A.F.; Rau, F.; Sureau, A.; Ney, M.; Cordier, A.; Kondili, M.; Polay-Espinoza, M.; Naouar, N.; et al. Reversal of RNA toxicity in myotonic dystrophy via a decoy RNA-binding protein with high affinity for expanded CUG repeats. Nat. Biomed. Eng. 2022, 6, 207–220. [Google Scholar] [CrossRef]
- Cardinali, B.; Provenzano, C.; Izzo, M.; Voellenkle, C.; Battistini, J.; Strimpakos, G.; Golini, E.; Mandillo, S.; Scavizzi, F.; Raspa, M.; et al. Time-controlled and muscle-specific CRISPR/Cas9-mediated deletion of CTG-repeat expansion in the DMPK gene. Mol. Ther. Nucleic Acids 2022, 27, 184–199. [Google Scholar] [CrossRef]
- Lo Scrudato, M.; Poulard, K.; Sourd, C.; Tome, S.; Klein, A.F.; Corre, G.; Huguet, A.; Furling, D.; Gourdon, G.; Buj-Bello, A. Genome Editing of Expanded CTG Repeats within the Human DMPK Gene Reduces Nuclear RNA Foci in the Muscle of DM1 Mice. Mol. Ther. 2019, 27, 1372–1388. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Mendez-Gomez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell. 2017, 68, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Roth, D.M.; Krach, F.; Nutter, C.A.; Tadokoro, T.; Thomas, J.D.; Sznajder, L.J.; Blue, S.M.; Gutierrez, H.L.; et al. The sustained expression of Cas9 targeting toxic RNAs reverses disease phenotypes in mouse models of myotonic dystrophy type 1. Nat. Biomed. Eng. 2021, 5, 157–168. [Google Scholar] [CrossRef]
- Gonzalez-Barriga, A.; Mulders, S.A.; van de Giessen, J.; Hooijer, J.D.; Bijl, S.; van Kessel, I.D.; van Beers, J.; van Deutekom, J.C.; Fransen, J.A.; Wieringa, B.; et al. Design and analysis of effects of triplet repeat oligonucleotides in cell models for myotonic dystrophy. Mol. Ther. Nucleic Acids 2013, 2, e81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, T.M.; Lueck, J.D.; Swanson, M.S.; Dirksen, R.T.; Thornton, C.A. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J. Clin. Investig. 2007, 117, 3952–3957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koebis, M.; Kiyatake, T.; Yamaura, H.; Nagano, K.; Higashihara, M.; Sonoo, M.; Hayashi, Y.; Negishi, Y.; Endo-Takahashi, Y.; Yanagihara, D.; et al. Ultrasound-enhanced delivery of morpholino with Bubble liposomes ameliorates the myotonia of myotonic dystrophy model mice. Sci. Rep. 2013, 3, 2242. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Barriga, A.; Kranzen, J.; Croes, H.J.; Bijl, S.; van den Broek, W.J.; van Kessel, I.D.; van Engelen, B.G.; van Deutekom, J.C.; Wieringa, B.; Mulders, S.A.; et al. Cell membrane integrity in myotonic dystrophy type 1: Implications for therapy. PLoS ONE 2015, 10, e0121556. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Kim, E.; Antoury, L.; Li, J.; Gonzalez-Perez, P.; Rutkove, S.B.; Wheeler, T.M. Antisense oligonucleotide and adjuvant exercise therapy reverse fatigue in old mice with myotonic dystrophy. Mol. Ther. Nucleic Acids 2021, 23, 393–405. [Google Scholar] [CrossRef] [PubMed]
- MDA. Ionis Reports Setback on DMPKRx Program for Myotonic Dystrophy. Available online: https://strongly.mda.org/ionis-reports-setback-dmpkrx-program-myotonic-dystrophy/ (accessed on 10 March 2022).
- Ionis Pharmaceuticals Reports on DMPKRx Phase 1/2 Clinical Trial. Available online: https://us8.campaign-archive.com/?e=[UNIQID]&u=8f5969cac3271759ce78c8354&id=8cc67ae9b8 (accessed on 22 March 2022).
- Ostergaard, M.E.; Jackson, M.; Low, A.; Alfred, E.C.; Richard, G.L.; Peralta, R.Q.; Yu, J.; Kinberger, G.A.; Dan, A.; Carty, R.; et al. Conjugation of hydrophobic moieties enhances potency of antisense oligonucleotides in the muscle of rodents and non-human primates. Nucleic Acids Res. 2019, 47, 6045–6058. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Antoury, L.; Baran, T.M.; Mitra, S.; Bennett, C.F.; Rigo, F.; Foster, T.H.; Wheeler, T.M. Non-invasive monitoring of alternative splicing outcomes to identify candidate therapies for myotonic dystrophy type 1. Nat. Commun. 2018, 9, 5227. [Google Scholar] [CrossRef]
- Tanner, M.K.; Tang, Z.; Thornton, C.A. Targeted splice sequencing reveals RNA toxicity and therapeutic response in myotonic dystrophy. Nucleic Acids Res. 2021, 49, 2240–2254. [Google Scholar] [CrossRef]
- Francois, V.; Klein, A.F.; Beley, C.; Jollet, A.; Lemercier, C.; Garcia, L.; Furling, D. Selective silencing of mutated mRNAs in DM1 by using modified hU7-snRNAs. Nat Struct. Mol. Biol. 2011, 18, 85–87. [Google Scholar] [CrossRef]
- Bisset, D.R.; Stepniak-Konieczna, E.A.; Zavaljevski, M.; Wei, J.; Carter, G.T.; Weiss, M.D.; Chamberlain, J.R. Therapeutic impact of systemic AAV-mediated RNA interference in a mouse model of myotonic dystrophy. Hum. Mol. Genet. 2015, 24, 4971–4983. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Masuda, A.; Konishi, H.; Ohkawara, B.; Ito, M.; Kinoshita, M.; Kiyama, H.; Matsuura, T.; Ohno, K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci. Rep. 2016, 6, 25317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargiela, A.; Sabater-Arcis, M.; Espinosa-Espinosa, J.; Zulaica, M.; Lopez de Munain, A.; Artero, R. Increased Muscleblind levels by chloroquine treatment improve myotonic dystrophy type 1 phenotypes in in vitro and in vivo models. Proc. Natl. Acad. Sci. USA 2019, 116, 25203–25213. [Google Scholar] [CrossRef] [PubMed]
- Cerro-Herreros, E.; Fernandez-Costa, J.M.; Sabater-Arcis, M.; Llamusi, B.; Artero, R. Derepressing muscleblind expression by miRNA sponges ameliorates myotonic dystrophy-like phenotypes in Drosophila. Sci. Rep. 2016, 6, 36230. [Google Scholar] [CrossRef] [Green Version]
- Cerro-Herreros, E.; Sabater-Arcis, M.; Fernandez-Costa, J.M.; Moreno, N.; Perez-Alonso, M.; Llamusi, B.; Artero, R. miR-23b and miR-218 silencing increase Muscleblind-like expression and alleviate myotonic dystrophy phenotypes in mammalian models. Nat. Commun. 2018, 9, 2482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Bodycombe, N.E.; Haskell, K.M.; Sun, Y.L.; Wang, E.T.; Morris, C.A.; Jones, L.H.; Wood, L.D.; Pletcher, M.T. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum. Mol. Genet. 2017, 26, 3056–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernat, V.; Disney, M.D. RNA Structures as Mediators of Neurological Diseases and as Drug Targets. Neuron 2015, 87, 28–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelbello, A.J.; Gonzalez, A.L.; Rzuczek, S.G.; Disney, M.D. Development of pharmacophore models for small molecules targeting RNA: Application to the RNA repeat expansion in myotonic dystrophy type 1. Bioorg. Med Chem. Lett. 2016, 26, 5792–5796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.H.; Richardson, S.L.; Ho, Y.J.; Lucas, A.M.; Tuccinardi, T.; Baranger, A.M.; Zimmerman, S.C. Investigating the binding mode of an inhibitor of the MBNL1.RNA complex in myotonic dystrophy type 1 (DM1) leads to the unexpected discovery of a DNA-selective binder. Chembiochem 2012, 13, 2505–2509. [Google Scholar] [CrossRef] [Green Version]
- Childs-Disney, J.L.; Parkesh, R.; Nakamori, M.; Thornton, C.A.; Disney, M.D. Rational design of bioactive, modularly assembled aminoglycosides targeting the RNA that causes myotonic dystrophy type 1. ACS Chem. Biol. 2012, 7, 1984–1993. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Lopez, A.; Llamusi, B.; Orzaez, M.; Perez-Paya, E.; Artero, R.D. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. USA 2011, 108, 11866–11871. [Google Scholar] [CrossRef] [Green Version]
- Ketley, A.; Chen, C.Z.; Li, X.; Arya, S.; Robinson, T.E.; Granados-Riveron, J.; Udosen, I.; Morris, G.E.; Holt, I.; Furling, D.; et al. High-content screening identifies small molecules that remove nuclear foci, affect MBNL distribution and CELF1 protein levels via a PKC-independent pathway in myotonic dystrophy cell lines. Hum. Mol. Genet. 2014, 23, 1551–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, L.M.; Nguyen, L.; Peng, S.; Lee, J.; Lee, H.Y.; Wong, C.H.; Hergenrother, P.J.; Chan, H.Y.; Zimmerman, S.C. A Potent Inhibitor of Protein Sequestration by Expanded Triplet (CUG) Repeats that Shows Phenotypic Improvements in a Drosophila Model of Myotonic Dystrophy. ChemMedChem 2016, 11, 1428–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzuczek, S.G.; Colgan, L.A.; Nakai, Y.; Cameron, M.D.; Furling, D.; Yasuda, R.; Disney, M.D. Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 2017, 13, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Bai, Y.; Chembazhi, U.V.; Peng, S.; Yum, K.; Luu, L.M.; Hagler, L.D.; Serrano, J.F.; Chan, H.Y.E.; Kalsotra, A.; et al. Intrinsically cell-penetrating multivalent and multitargeting ligands for myotonic dystrophy type 1. Proc. Natl. Acad. Sci. USA 2019, 116, 8709–8714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondono, R.; Lirio, A.; Elvira, C.; Alvarez-Marimon, E.; Provenzano, C.; Cardinali, B.; Perez-Alonso, M.; Peralvarez-Marin, A.; Borrell, J.I.; Falcone, G.; et al. Design of novel small molecule base-pair recognizers of toxic CUG RNA transcripts characteristics of DM1. Comput. Struct. Biotechnol. J. 2021, 19, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Siboni, R.B.; Bodner, M.J.; Khalifa, M.M.; Docter, A.G.; Choi, J.Y.; Nakamori, M.; Haley, M.M.; Berglund, J.A. Biological Efficacy and Toxicity of Diamidines in Myotonic Dystrophy Type 1 Models. J. Med. Chem. 2015, 58, 5770–5780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelbello, A.J.; DeFeo, M.E.; Glinkerman, C.M.; Boger, D.L.; Disney, M.D. Precise Targeted Cleavage of a r(CUG) Repeat Expansion in Cells by Using a Small-Molecule-Deglycobleomycin Conjugate. ACS Chem. Biol. 2020, 15, 849–855. [Google Scholar] [CrossRef]
- Angelbello, A.J.; Rzuczek, S.G.; McKee, K.K.; Chen, J.L.; Olafson, H.; Cameron, M.D.; Moss, W.N.; Wang, E.T.; Disney, M.D. Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA 2019, 116, 7799–7804. [Google Scholar] [CrossRef] [Green Version]
- Childs-Disney, J.L.; Stepniak-Konieczna, E.; Tran, T.; Yildirim, I.; Park, H.; Chen, C.Z.; Hoskins, J.; Southall, N.; Marugan, J.J.; Patnaik, S.; et al. Induction and reversal of myotonic dystrophy type 1 pre-mRNA splicing defects by small molecules. Nat. Commun. 2013, 4, 2044. [Google Scholar] [CrossRef] [Green Version]
- Konieczny, P.; Selma-Soriano, E.; Rapisarda, A.S.; Fernandez-Costa, J.M.; Perez-Alonso, M.; Artero, R. Myotonic dystrophy: Candidate small molecule therapeutics. Drug Discov. Today 2017, 22, 1740–1748. [Google Scholar] [CrossRef]
- Nakamori, M.; Taylor, K.; Mochizuki, H.; Sobczak, K.; Takahashi, M.P. Oral administration of erythromycin decreases RNA toxicity in myotonic dystrophy. Ann. Clin. Transl. Neurol. 2016, 3, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Safety and Efficacy Trial of MYD-0124 for Myotonic Dystrophy Type 1. Available online: https://trialsearch.who.int/?TrialID=JPRN-jRCT2051190069 (accessed on 22 March 2022).
- Wojciechowska, M.; Taylor, K.; Sobczak, K.; Napierala, M.; Krzyzosiak, W.J. Small molecule kinase inhibitors alleviate different molecular features of myotonic dystrophy type 1. RNA Biol. 2014, 11, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.S.; Kuyumcu-Martinez, M.N.; Sarma, S.; Mathur, N.; Wehrens, X.H.; Cooper, T.A. PKC inhibition ameliorates the cardiac phenotype in a mouse model of myotonic dystrophy type 1. J. Clin. Investig. 2009, 119, 3797–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.; Wei, C.; Iakova, P.; Bugiardini, E.; Schneider-Gold, C.; Meola, G.; Woodgett, J.; Killian, J.; Timchenko, N.A.; Timchenko, L.T. GSK3beta mediates muscle pathology in myotonic dystrophy. J. Clin. Investig. 2012, 122, 4461–4472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Weng, W.C.; Stock, L.; Lindquist, D.; Martinez, A.; Gourdon, G.; Timchenko, N.; Snape, M.; Timchenko, L. Correction of Glycogen Synthase Kinase 3beta in Myotonic Dystrophy 1 Reduces the Mutant RNA and Improves Postnatal Survival of DMSXL Mice. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horrigan, J.; Gomes, T.B.; Snape, M.; Nikolenko, N.; McMorn, A.; Evans, S.; Yaroshinsky, A.; Della Pasqua, O.; Oosterholt, S.; Lochmuller, H. A Phase 2 Study of AMO-02 (Tideglusib) in Congenital and Childhood-Onset Myotonic Dystrophy Type 1 (DM1). Pediatr Neurol. 2020, 112, 84–93. [Google Scholar] [CrossRef]
- Brockhoff, M.; Rion, N.; Chojnowska, K.; Wiktorowicz, T.; Eickhorst, C.; Erne, B.; Frank, S.; Angelini, C.; Furling, D.; Ruegg, M.A.; et al. Targeting deregulated AMPK/mTORC1 pathways improves muscle function in myotonic dystrophy type I. J. Clin. Investig. 2017, 127, 549–563. [Google Scholar] [CrossRef] [Green Version]
- Ravel-Chapuis, A.; Al-Rewashdy, A.; Belanger, G.; Jasmin, B.J. Pharmacological and physiological activation of AMPK improves the spliceopathy in DM1 mouse muscles. Hum. Mol. Genet. 2018, 27, 3361–3376. [Google Scholar] [CrossRef] [Green Version]
- Loro, E.; Rinaldi, F.; Malena, A.; Masiero, E.; Novelli, G.; Angelini, C.; Romeo, V.; Sandri, M.; Botta, A.; Vergani, L. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Differ. 2010, 17, 1315–1324. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Costa, J.M.; Garcia-Lopez, A.; Zuniga, S.; Fernandez-Pedrosa, V.; Felipo-Benavent, A.; Mata, M.; Jaka, O.; Aiastui, A.; Hernandez-Torres, F.; Aguado, B.; et al. Expanded CTG repeats trigger miRNA alterations in Drosophila that are conserved in myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2013, 22, 704–716. [Google Scholar] [CrossRef] [Green Version]
- Sabater-Arcis, M.; Bargiela, A.; Furling, D.; Artero, R. miR-7 Restores Phenotypes in Myotonic Dystrophy Muscle Cells by Repressing Hyperactivated Autophagy. Mol. Ther. Nucleic Acids 2020, 19, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Arcis, M.; Bargiela, A.; Moreno, N.; Poyatos-Garcia, J.; Vilchez, J.J.; Artero, R. Musashi-2 contributes to myotonic dystrophy muscle dysfunction by promoting excessive autophagy through miR-7 biogenesis repression. Mol. Ther. Nucleic Acids 2021, 25, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Richard, G.F.; Dujon, B.; Haber, J.E. Double-strand break repair can lead to high frequencies of deletions within short CAG/CTG trinucleotide repeats. Mol. Gen. Genet. 1999, 261, 871–882. [Google Scholar] [CrossRef]
- Mittelman, D.; Moye, C.; Morton, J.; Sykoudis, K.; Lin, Y.; Carroll, D.; Wilson, J.H. Zinc-finger directed double-strand breaks within CAG repeat tracts promote repeat instability in human cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9607–9612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosbach, V.; Poggi, L.; Viterbo, D.; Charpentier, M.; Richard, G.F. TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep. 2018, 22, 2146–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, G.F.; Viterbo, D.; Khanna, V.; Mosbach, V.; Castelain, L.; Dujon, B. Highly specific contractions of a single CAG/CTG trinucleotide repeat by TALEN in yeast. PLoS ONE 2014, 9, e95611. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Gao, Y.; Jin, S.; Subramony, S.H.; Terada, N.; Ranum, L.P.; Swanson, M.S.; Ashizawa, T. Genome modification leads to phenotype reversal in human myotonic dystrophy type 1 induced pluripotent stem cell-derived neural stem cells. Stem Cells 2015, 33, 1829–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinesi, C.; Aeschbach, L.; Yang, B.; Dion, V. Contracting CAG/CTG repeats using the CRISPR-Cas9 nickase. Nat. Commun. 2016, 7, 13272. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912. [Google Scholar] [CrossRef]
- Wang, Y.; Hao, L.; Wang, H.; Santostefano, K.; Thapa, A.; Cleary, J.; Li, H.; Guo, X.; Terada, N.; Ashizawa, T.; et al. Therapeutic Genome Editing for Myotonic Dystrophy Type 1 Using CRISPR/Cas9. Mol. Ther. 2018, 26, 2617–2630. [Google Scholar] [CrossRef] [Green Version]
- van Agtmaal, E.L.; Andre, L.M.; Willemse, M.; Cumming, S.A.; van Kessel, I.D.G.; van den Broek, W.; Gourdon, G.; Furling, D.; Mouly, V.; Monckton, D.G.; et al. CRISPR/Cas9-Induced (CTGCAG)n Repeat Instability in the Myotonic Dystrophy Type 1 Locus: Implications for Therapeutic Genome Editing. Mol. Ther. 2017, 25, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastidar, S.; Ardui, S.; Singh, K.; Majumdar, D.; Nair, N.; Fu, Y.; Reyon, D.; Samara, E.; Gerli, M.F.M.; Klein, A.F.; et al. Efficient CRISPR/Cas9-mediated editing of trinucleotide repeat expansion in myotonic dystrophy patient-derived iPS and myogenic cells. Nucleic Acids Res. 2018, 46, 8275–8298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell. Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Jasin, M.; Haber, J.E. The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair 2016, 44, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Dastidar, S.; Majumdar, D.; Tipanee, J.; Singh, K.; Klein, A.F.; Furling, D.; Chuah, M.K.; VandenDriessche, T. Comprehensive transcriptome-wide analysis of spliceopathy correction of myotonic dystrophy using CRISPR-Cas9 in iPSCs-derived cardiomyocytes. Mol. Ther. 2022, 30, 75–91. [Google Scholar] [CrossRef]
- Yanovsky-Dagan, S.; Bnaya, E.; Diab, M.A.; Handal, T.; Zahdeh, F.; van den Broek, W.J.A.A.; Epsztejn-Litman, S.; Wansink, D.G.; Eiges, R. Deletion of the CTG Expansion in Myotonic Dystrophy Type 1 Reverses DMPK Aberrant Methylation in Human Embryonic Stem Cells but not Affected Myoblasts. bioRxiv 2019, 631457. [Google Scholar] [CrossRef] [Green Version]
- Andre, L.M.; van Cruchten, R.T.P.; Willemse, M.; Bezstarosti, K.; Demmers, J.A.A.; van Agtmaal, E.L.; Wansink, D.G.; Wieringa, B. Recovery in the Myogenic Program of Congenital Myotonic Dystrophy Myoblasts after Excision of the Expanded (CTG)n Repeat. Int. J. Mol. Sci. 2019, 20, 5685. [Google Scholar] [CrossRef] [Green Version]
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, J.; Weis, S.; Sippel, J.; Tulalamba, W.; Remes, A.; El Andari, J.; Herrmann, A.K.; Pham, Q.H.; Borowski, C.; Hille, S.; et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020, 11, 5432. [Google Scholar] [CrossRef]
- Kondratov, O.; Kondratova, L.; Mandel, R.J.; Coleman, K.; Savage, M.A.; Gray-Edwards, H.L.; Ness, T.J.; Rodriguez-Lebron, E.; Bell, R.D.; Rabinowitz, J.; et al. A comprehensive study of a 29-capsid AAV library in a non-human primate central nervous system. Mol. Ther. 2021, 29, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Taniguchi-Ikeda, M.; Kato, T.; Shinkai, Y.; Tanaka, S.; Hagiwara, H.; Sasaki, N.; Masaki, T.; Matsumura, K.; Sonoo, M.; et al. Unexpected Mutations by CRISPR-Cas9 CTG Repeat Excision in Myotonic Dystrophy and Use of CRISPR Interference as an Alternative Approach. Mol. Ther. Methods Clin. Dev. 2020, 18, 131–144. [Google Scholar] [CrossRef]
- Thomas, M.; Burgio, G.; Adams, D.J.; Iyer, V. Collateral damage and CRISPR genome editing. PLoS Genet. 2019, 15, e1007994. [Google Scholar] [CrossRef]
- Bennett, E.P.; Petersen, B.L.; Johansen, I.E.; Niu, Y.; Yang, Z.; Chamberlain, C.A.; Met, O.; Wandall, H.H.; Frodin, M. INDEL detection, the ‘Achilles heel’ of precise genome editing: A survey of methods for accurate profiling of gene editing induced indels. Nucleic Acids Res. 2020, 48, 11958–11981. [Google Scholar] [CrossRef]
- Allen, F.; Crepaldi, L.; Alsinet, C.; Strong, A.J.; Kleshchevnikov, V.; De Angeli, P.; Palenikova, P.; Khodak, A.; Kiselev, V.; Kosicki, M.; et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2019, 37, 64–72. [Google Scholar] [CrossRef]
- Allen, D.; Weiss, L.E.; Saguy, A.; Rosenberg, M.; Iancu, O.; Matalon, O.; Lee, C.; Beider, K.; Nagler, A.; Shechtman, Y.; et al. High-Throughput Imaging of CRISPR- and Recombinant Adeno-Associated Virus-Induced DNA Damage Response in Human Hematopoietic Stem and Progenitor Cells. CRISPR J. 2022, 5, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Tanner, M.R.; Lee, C.M.; Hurley, A.E.; De Giorgi, M.; Jarrett, K.E.; Davis, T.H.; Doerfler, A.M.; Bao, G.; Beeton, C.; et al. AAV-CRISPR Gene Editing Is Negated by Pre-existing Immunity to Cas9. Mol. Ther. 2020, 28, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
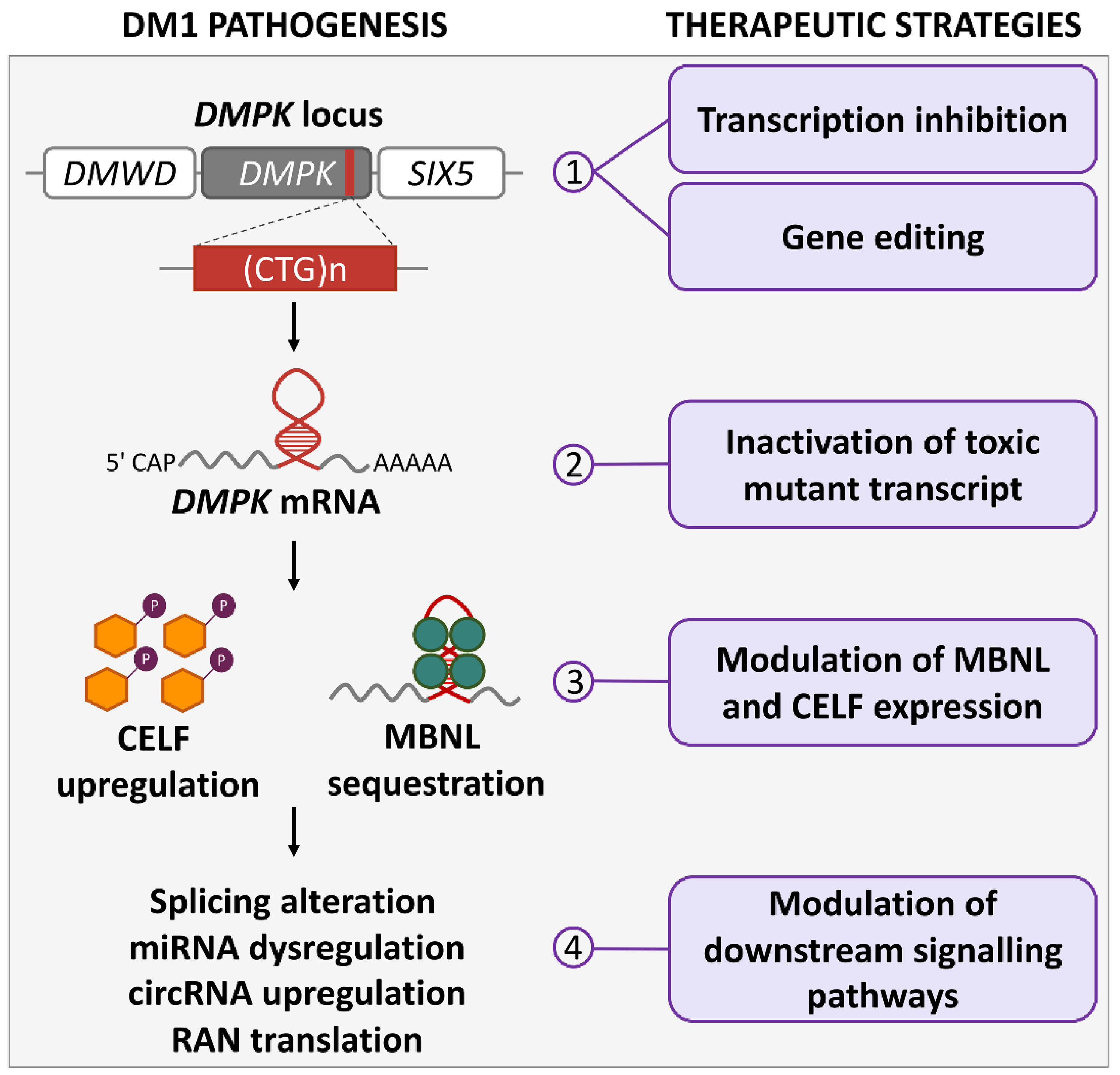

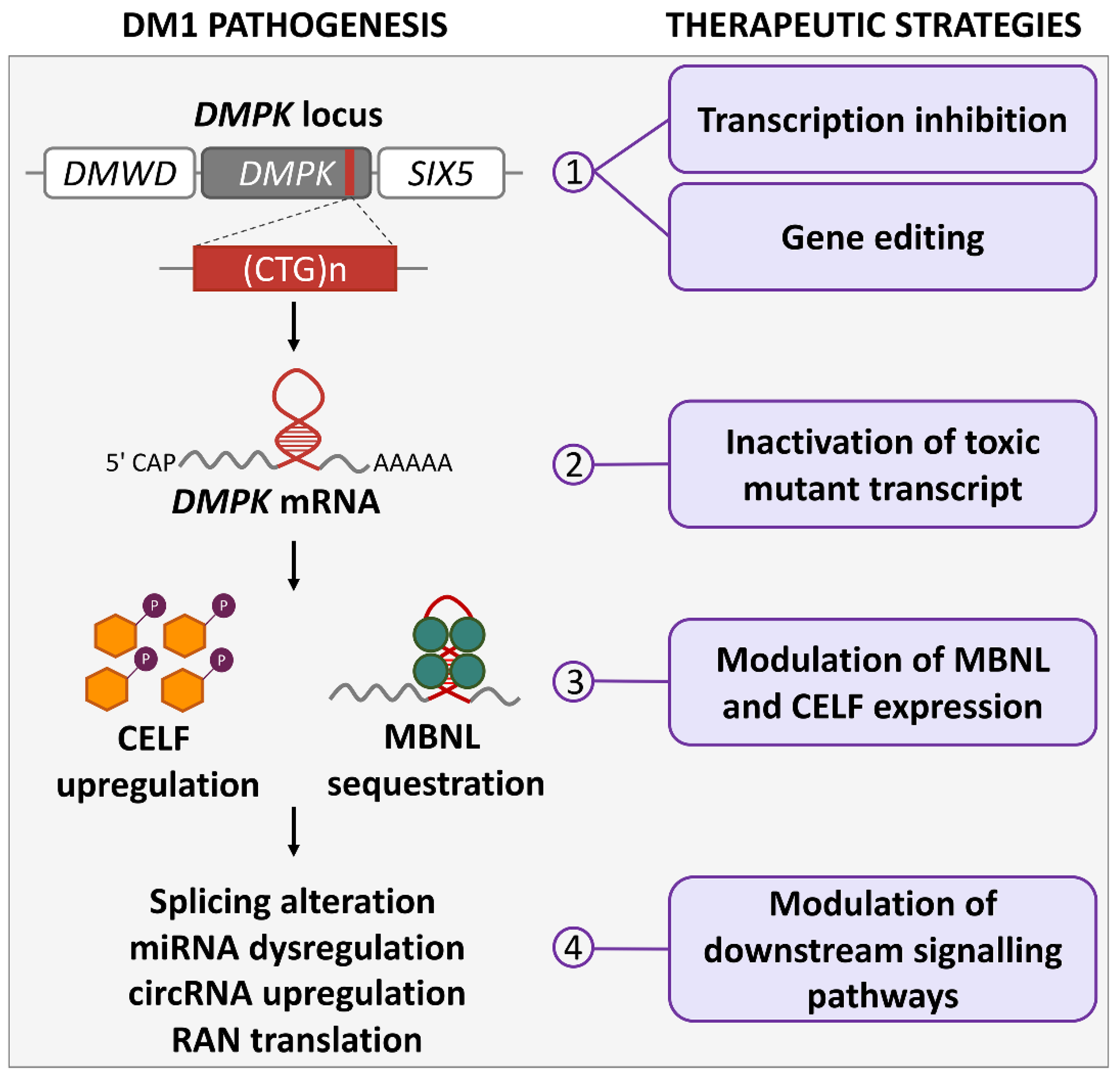{kind=link}
| (A) Knockout and Overexpressing Models | ||||||||||||
| Mouse Model | Generation Strategy | DM1-Like Features | Limitations | Research Application | Ref | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with hygromycin cassette | Late-onset mild myopathy and altered Ca++ homeostasis | Mild phenotype; possible confounding insertional effects on flanking genes; mixed genetic background | Relevance of absence of DMPK protein to DM1 phenotype | [52,66] | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with neomycin cassette | Late-onset mild myopathy; decreased force generation; altered Na+ currents in skeletal muscles; cardiac conduction defects | Mild phenotype; possible confounding insertional effects on flanking genes; mixed genetic background | Relevance of absence of DMPK protein to DM1 phenotype | [51,67] | |||||||
| DMPK-/- | Dmpk KO via replacement of 5′-UTR and exons 1-7 with hygromycin cassette | No phenotype | Failure to replicate the DM1 phenotype | Relevance of absence of DMPK protein to DM1 phenotype | [53] | |||||||
| Mbnl1ΔE3/ΔE3 | Mbnl1 KO via targeted deletion of Mbnl1 exon 3 | Mild myotonia and myopathy (centralized nuclei, split fibers); heart conduction defects; progressive cataracts; AS alterations | Mild muscle phenotype; mild brain alterations; limited spliceopathy | Evaluation of MBNL1 splicing regulation to DM1 phenotype | [56,57] | |||||||
| Mbnl2ΔE2/ΔE2 | Mbnl2 KO via targeted deletion of Mbnl2 exon 2 | Development of several CNS alterations (REM sleep propensity, deficit in spatial memory, decreased synaptic plasticity), AS alterations | Failure to replicate the DM1 muscular phenotype | Evaluation of MBNL2 splicing regulation to DM1 phenotype | [55] | |||||||
| Mbnl3ΔE2 | Mbnl3 KO via targeted deletion of Mbnl3 exon 2 (X-linked) | Progressive delay in muscle regeneration; abnormalities in embryonic muscle differentiation leading to neonatal hypotonia | Possible compensation by MBNL3 truncated isoform or other MBNl family members | Evaluation of MBNL3 contribution to DM1 phenotype | [58] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl2C/C; Myo-Cre+/- | Mbnl1 KO; skeletal-muscle specific Cre-mediated Mbnl2 KO | Small size at birth and skeletal abnormalities; myopathy and severe motor deficits; AS alterations also in brain tissues | High neonatal mortality and reduced lifespan | Evaluation of MBNL1 and MBNL2 contribution to DM1 muscular phenotype | [60] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl3ΔE2 | Mbnl1 and Mbnl3 KO via targeted deletion of Mbnl1 exon 3 and Mbnl3 exon 2 | Myotonia and myopathy; reduction in muscle strength; chloride currents alteration; AS alterations; translation defects | AS alterations similar to Mbnl1 single knock out; lack of brain alterations | Evaluation of MBNL1 and MBNL3 contribution to DM1 phenotype | [59] | |||||||
| Mbnl1ΔE3/ΔE3; Mbnl2C/C; Mbnl3C; Myo-Cre+/- | Mbnl1 KO; muscle-specific Cre-mediated Mbnl2 and Mbnl3 KO | Severe congenital myopathy and spliceopathy, severe respiratory difficulties and muscle wasting in adults; gene expression changes | High neonatal mortality and reduced lifespan | Evaluation of all MBNL proteins loss contribution to DM1 muscular phenotype | [61] | |||||||
| MCKCUGBP1 | Insertion of human CELF1 transgene under striated-muscle-specific MCK mouse promoter | Chains of central nuclei in myofibers, increased NADH reactivity, degenerating fibers and AS alterations | Neonatal lethality in mice expressing high levels of CELF1 | Contribution of CELF1 overexpression to DM1 muscular phenotype | [62] | |||||||
| TRECUGBP1 | Insertion of Tet-responsive human CELF1 transgene; heart-specific rtTA expression | Left ventricular systolic dysfunction and dilatation, AS alterations | DM1-like phenotype limited to heart defects | Contribution of CELF1 overexpression to DM1 heart phenotype | [63] | |||||||
| TRECUGBP1 | Insertion of Tet-responsive human CELF1 transgene; skeletal-muscle-specific rtTA expression | Myofibers containing central nuclei, decreased muscle weight, impaired muscle function, AS alterations | DM1-like phenotype limited to skeletal-muscle defects | Contribution of CELF1 overexpression to DM1 skeletal-muscle phenotype | [65] | |||||||
| TRECUGBP2 | Insertion of Tet-responsive human CELF2 transgene; heart-specific rtTA expression | No observed heart pathology; AS alterations similar to those observed in TRECUBP1 mice | Mild heart phenotype | Contribution of CELF2 overexpression to DM1 heart phenotype | [64] | |||||||
| (B) Transgenic Models with Repeat Expansion | ||||||||||||
| Mouse Model | Generation Strategy | (CTG)n | DM1-Like Features | Limitations | Research Application | Ref | ||||||
| DM200 | Insertion of a Tet-responsive expanded DMPK transgene where DMPK coding region is replaced by GFP | 200 | Ribonuclear foci; MBNL1 sequestration; AS alterations; myotonia, progressive cardiac conduction abnormalities | Splicing alterations in the heart have not been described | Study of DM1 phenotype associated with toxic CUG repeats; modeling muscle regeneration; test of therapeutic strategies | [68,69,70] | ||||||
| DM300 | Insertion of a 45Kb human genomic fragment containing DMWD, DMPK and SIX5 genes from a DM1 patient | ~300 | Ribonuclear foci (skeletal muscle, heart and brain); myotonia; muscle atrophy; morphological abnormalities; changes in the distribution of MAPT/Tau protein isoform; defect in glucose metabolism | High mortality; mild splicing alterations; intergenerational instability of CTG-repeat numbers | Evaluation of DMPK transcript toxicity in different tissues | [71,72] | ||||||
| DMSXL | Insertion of a 45Kb human genomic fragment containing DMWD, DMPK and SIX5 genes from a DM1 patient | >1000 | Ribonuclear foci; MBNL1 sequestration; AS alterations; deficits in motor performance; behavioral abnormalities; synaptic dysfunction; inhibition of exploratory activity and cerebellar glial dysfunction | High mortality; severe body-weight reduction; interindividual variability; decreased transgene expression with aging; mild muscular phenotype | Evaluation of DMPK transcript toxicity in different tissues and in multiple brain cell types; test of therapeutic strategies | [23,73,74] | ||||||
| HSALR | Insertion of the human skeletal actin (HSA) gene including CTG repeats in the 3’ UTR | ~250 | Ribonuclear foci; AS alterations; myotonia and muscle histopathology abnormalities (increase in central nuclei and variability in fiber size) after six months of age | Limited to skeletal muscle; does not contain DMPK gene sequence; absence of muscle weakness | Investigation of expanded-CUG-repeat toxicity in muscle fibers; test of therapeutic strategies | [75,76] | ||||||
| LC15 | Insertion of CTG expanded DMPK 3’ UTR downstream Luciferase gene driven by CMV-βA promoter | 250–400 | Ribonuclear foci, AS alteration and MBNL2 upregulation in the heart; reduced Na+ and K+ channel activity; ventricular arrhythmias | DM1-like phenotype limited to heart defects | Evaluation of biophysical mechanisms reproducing DM1-like electrocardiograph abnormalities | [77] | ||||||
| EpA960/ 𝛼-MHC-Cre | Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequences; heart-specific myosin Cre expression | 960 (CTCGA-interrupted) | Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations; cardiomyopathy, arrhythmias; systolic and diastolic dysfunction | Does not reproduce CTG-repeat continuity; mouse model no longer available | Evaluation of DMPK transcript toxicity and CELF1 overexpression in heart tissue | [78] | ||||||
| EpA960/ HSA-Cre | Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequence; skeletal-muscle-specific Cre expression | 960 (CTCGA-interrupted) | Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS defects; myotonia and progressive muscle wasting, deficits in muscle performance and histopathological abnormalities | Does not reproduce CTG-repeat continuity; mouse model no longer available | Evaluation of DMPK transcript toxicity and CELF1 overexpression in skeletal tissue | [79] | ||||||
| EpA960/ CamKII-Cre | Insertion of CTG expanded DMPK exon 15 transgene containing Cre-responsive loxP sequence; brain-specific Cre expression | 960 (CTCGA-interrupted) | Ribonuclear foci; MBNL1 sequestration; AS alterations; learning disability; neurotransmission dysfunction; brain atrophy and aging | Does not reproduce CTG-repeat continuity; mouse model no longer available | Identify mechanisms involved in CTG-dependent neuronal degeneration | [80] | ||||||
| TREDT960I/𝛼-MHC-rtTA | Insertion of Tet-responsive expanded DMPK exons 11–15 transgene; heart-specific rtTA expression | 960 (CTCGA-interrupted) | Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations ; arrhythmias | Does not reproduce CTG-repeat continuity | Study of alteration of ion transport and action potential in cardiomyocytes expressing toxic CUG | [81,82] | ||||||
| TREDT960I/ MDAF-rtTA | Insertion of Tet-responsive expanded DMPK exons 11–15 transgene; skeletal-muscle-specific rtTA expression | 960 (CTCGA-interrupted) | Ribonuclear foci; MBNL1 sequestration; CELF1 protein upregulation; AS alterations; muscle wasting and myopathy | Does not reproduce CTG-repeat continuity | Study the mechanisms of CUG-repeat-induced muscle tissue loss | [83] | ||||||
| Molecule Class | Target | Therapeutic Molecule | Mechanism | DDS | Admin. Route | Study Phase | Ref |
|---|---|---|---|---|---|---|---|
| ASOs | DMPK CUGexp | PMO-CAG25, 2′-OMe-CAG, LNA-CAG mixmers, all-LNA-CAG | MBNL1 binding block | Naked | IM | Preclinical | [92,103,104,105] |
| DMPK CUGexp | PPMO-B, PPMO-K; Pip6a-PMO | MBNL1 binding block | CPP-conj | IM, IV | Preclinical | [106,107] | |
| DMPK CUGexp | miniPEG-γ PNA | MBNL1 binding block | Polymer-conj | SC | Preclinical | [108,109] | |
| DMPK 3′UTR | MOE gapmers, c-Et gapmers, LNA gapmers | DMPK mRNA degradation | Naked | IM, SC, ICV | Preclinical | [54,70,110,111,112] | |
| DMPK CUGexp | LNA gapmers, MOE gapmers | Mutated DMPK mRNA degradation | Naked | IM | Preclinical | [113] | |
| DMPK 3′UTR | IONIS-DMPKRx | DMPK mRNA degradation | Naked | SC | Clinical(completed) | [114] | |
| DMPK 3′UTR | palmitoyl-c-Et gapmers | DMPK mRNA degradation | Lipid-conj | SC | Preclinical | [69] | |
| miRNAs targeting Mbnl1 mRNA | cholesterol-2′OMe-ASOs | AntagomiR | Lipid-conj | SC, IV | Preclinical | [115,116] | |
| Mbnl1 3′UTR | Pip9b2-PMO | BlockmiR | CPP-conj | IV | Preclinical | [117] | |
| siRNA | DMPK CUGexp | siRNA-CAG | Mutated DMPK mRNA degradation | Nacked | IM | Preclinical | [118] |
| DMPK mRNA | AOC 1001 | DMPK mRNA degradation | Ab-conj | IV | Clinical (recruiting) | [119] | |
| rAAV | DMPK downstream pathway | MBNL1 | MBNL1 overexpression | rAAV1 | IM | Preclinical | [120] |
| DMPK downstream pathway | MBNL1 | Competition for CUGexp interaction | rAAV9 | IM | Preclinical | [121] | |
| DMPK CTG spanning region | Sa/eSpCas9-sgRNAs | CTGexp removal | rAAV9 | IM | Preclinical | [122,123] | |
| DMPK CTGexp | dSaCas9-sgRNA | Transcription inhibition | rAAV6, rAAV9 | IV | Preclinical | [124] | |
| DMPK CUGexp | RCas9-sgRNA | DMPK mRNA degradation | rAAV9 | IV, TA | Preclinical | [125] |
| Nuclease | Mechanism | Effect | DM1 Model | Advantages | Limitations | Ref |
|---|---|---|---|---|---|---|
| ZNF | Induction of DNA double strand breaks at CAG/CTG repeats | Repeat contractions | Yeast cells carrying CTG repeats | Permanent reduction in CTG repeats; good cleavage efficiency | Repeat rearrangements | [171] |
| ZNF | Induction of DNA double strand breaks at CAG/CTG repeats | Repeat contractions and duplications | Mammalian cells carrying CTG repeats | Permanent reduction in CTG repeats; good cleavage efficiency | Repeat duplications | [172] |
| TALEN | Induction of DNA double strand breaks at CAG/CTG repeats | Repeat contractions | Yeast cells carrying CTG repeats | Permanent reduction in CTG repeats; good cleavage efficiency, no mutations | Application limited to yeast cells | [173,174] |
| TALEN | Insertion of a polyA signal upstream CTG repeats | Production of shorter DMPK transcripts (no CUG) | DM1-patient-derived iPSCs | Elimination of toxic CUG repeats from DMPK transcript | Production of truncated DMPK protein; retention of CTG at DMPK locus | [43,175] |
| SpCas9 D10A | Induction of DNA single strand breaks at CAG/CTG repeats | Repeat contractions | Human cells carrying CAG/CTG repeats | Permanent reduction in CTG repeats | Cell-type dependent efficiency | [176] |
| dCas9 | Block DNA transcription at CTG repeats | DMPK transcription inhibition | DM1-patient-derived cells; HSALR mice | Suppression of CUG-repeat-transcript production | Decreased DMPK protein production; retention of CTG at DMPK locus; need of repeated treatment | [124] |
| RCas9 | Cleaving single-strand RNA at CUG repeats | CUG-repeated transcript degradation | DM1-patient-derived cells; HSALR mice | Elimination of CUG-repeat transcript | Decreased DMPK protein production; retention of CTG at DMPK locus; need of repeated treatment | [125,177] |
| SpCas9 D10A | Insertion of a polyA signal upstream CTG repeats | Production of shorter DMPK transcripts (no CUG) | DM1-patient-derived iPSCs | Elimination of toxic CUG repeats from DMPK transcript | Production of truncated DMPK protein; retention of CTG at DMPK locus | [178] |
| SpCas9 or SaCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Deletion of CTG expanded region | DM1-patient-derived iPSCs | Permanent elimination of toxic CTG repeats; no off-targets | Low efficiency using SpCas9; higher efficiency but frequent inversions using SaCas9 | [178] |
| SpCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Deletion of CTG expanded region | DM1-mouse-derived myoblasts; DM1-patient derived myoblasts | Permanent elimination of CTG repeats; no off-targets | On-target indels, inversions, large deletions | [179] |
| SpCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Deletion of CTG expanded region | DM1-patient-derived MYOD1-converted fibroblasts | Permanent elimination of CTG repeats; no off-targets | On-target indels, inversions | [40] |
| SpCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Deletion of CTG expanded region | DM1-patient-derived primary myoblasts; DM1-patient-derived iPSCs | Permanent elimination of CTG repeats; good editing efficiency in iPSCs; no off-targets | On-target indels, partial deletions | [180] |
| SaCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Deletion of CTG expanded region | DM1-patient-derived myoblasts; DMSXL mice | Permanent elimination of CTG repeats; good editing efficiency in DM1 cells; no off-targets | On-target indels; low editing efficiency in mice skeletal muscle | [123] |
| eSpCas9 | Induction of two DNA double strand breaks at CTG-repeats flanking regions | Inducible deletion of CTG expanded region | DM1-patient-derived MYOD1-converted fibroblasts; DMSXL mice | Permanent elimination of CTG repeats; well-regulated editing induction; no off-targets | On-target indels, inversions, large deletions | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izzo, M.; Battistini, J.; Provenzano, C.; Martelli, F.; Cardinali, B.; Falcone, G. Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing. Int. J. Mol. Sci. 2022, 23, 4622. https://doi.org/10.3390/ijms23094622
Izzo M, Battistini J, Provenzano C, Martelli F, Cardinali B, Falcone G. Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing. International Journal of Molecular Sciences. 2022; 23(9):4622. https://doi.org/10.3390/ijms23094622
Chicago/Turabian StyleIzzo, Mariapaola, Jonathan Battistini, Claudia Provenzano, Fabio Martelli, Beatrice Cardinali, and Germana Falcone. 2022. "Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing" International Journal of Molecular Sciences 23, no. 9: 4622. https://doi.org/10.3390/ijms23094622
APA StyleIzzo, M., Battistini, J., Provenzano, C., Martelli, F., Cardinali, B., & Falcone, G. (2022). Molecular Therapies for Myotonic Dystrophy Type 1: From Small Drugs to Gene Editing. International Journal of Molecular Sciences, 23(9), 4622. https://doi.org/10.3390/ijms23094622