1. Introduction
Actinide contamination may result from an incident in the nuclear industry, a malicious act targeting a nuclear power plant, the explosion of a dirty bomb, or a degraded situation in a nuclear-powered naval vessel such as an icebreaker or submarine. Although rare in the history of nuclear facilities, radioelement contamination is particularly harmful because these elements, all alpha emitters, present both chemical and radiological toxicity. Among these, plutonium (Pu) is particularly involved in both civil and military nuclear activity. It is highly toxic whatever its isotopy and remains an emblematic radioelement linked to the nuclear industry for the general public.
Despite the panic it may inspire, one should still consider that Pu is only/mainly toxic once it has entered the organism. Indeed, alpha emitters exhibit a limited power of penetration (e.g., about 5 cm in air and only about 30 µm, representing a few cell diameters, in living tissues); however, the deposited energy is enormous (alpha particles are on the order of MeV energy). Unfortunately, human exposure to Pu can be efficiently widespread through the inhalation of small particles kicked up by wind and dust after the accident has occurred.
Inhalation is, thus, the most likely entry route into the organism. The retention and the fate of the inhaled particles depend on their size and physicochemical form [
1]. Bigger particles are either filtered by the upper respiratory region and swallowed or transferred into the throat by the lung clearance process (elimination of the mucus layer and particles through the natural motion of the bronchial cilia). In both cases, these particles are directed into the gastrointestinal tract and mainly excreted. On the contrary, the smaller particles (10 nm to 1 µm) are capable of reaching the lung alveoli [
2], where they are sequestered by alveolar macrophages [
3] and eventually transferred to lymph nodes or into lung tissues, representing long-term storage (for years), which contributes to lung cancers. The soluble forms (nitrates, citrates, and certain oxides) of inhaled Pu are absorbed more easily, pass into the bloodstream [
4], and are redistributed throughout the body. About 90% of this absorbed Pu is then equally deposited in the liver and bones, where it contributes to the distribution of the dose over very long periods causing, in particular, bone cancer. In case of ingestion of Pu, subsequent entry into the bloodstream from the digestive tract is very low (<1%) [
5]. Most of the ingested Pu is then eliminated in the feces [
6].
As for the absorption of Pu through the skin, this represents a risk only for workers in highly contaminated areas and/or in the case of wounding (cut or blast injury). In that case, Pu follows the same paths as described earlier in the case of its transfer into the blood, whereby about 90% of the Pu absorbed is retained, mostly in the liver and bones.
Therefore, these three major compartments, lung, liver, and bones, constitute a real sanctuary for sequestered Pu.
The current recommended and approved treatment for contamination with transuranic radionuclides (e.g., plutonium, americium, and curium) is chelate calcium- and zinc-diethylenetriaminepentaacetic acid (Ca and Zn-DTPA), administered intravenously (i.v.) or by nebulizer. However, DTPA exhibits a narrow biodistribution [
7,
8] and, thus, is eliminated very quickly through urine [
9]. Moreover, DTPA is only active on the soluble forms of Pu [
10]. Of course, new chelating agents, some of which are promising, such as hydroxypyridonates (e.g., 3,4,3-LI(1,2-HOPO)) are still being developed [
11]. However, as with DTPA chelation therapy, their action is mainly directed toward the soluble forms of Pu such as nitrates.
To address the limitations of DTPA, special formulations such as aerosolized Ca-DTPA for pulmonary administration have been tested [
12,
13]. Despite its effectiveness on soluble forms of Pu deposited in the lungs, it remained ineffective on insoluble oxides (PuO
2) at the primary site of contamination. However, independently of the primary site of contamination or the DTPA treatment regimen (i.v. or by aerosolized Ca-DTPA), it reduced the systemic retention (skeleton and liver). Thus, there is still a need for a more effective treatment for the remaining Pu at the primary site of contamination.
Stealth liposomes encapsulating DTPA have also been tested [
14] for their enhanced half-life in the blood and, hence, higher biodistribution. However, again, they showed improved efficiency when soluble forms such as
238Pu-phytate were injected. As for the bones, to the best of our knowledge, only one team [
15] reported a real in vivo decontamination, of uranium only, using a 3,2-hydroxypyridinone-based compound. Indeed, the lower observed actinide level in the liver and/or bones highlighted in most studies should be rather attributed to the chelation effect (subsequent to the i.v. administration) of the decontaminating agent rather than to actinide extravasation (e.g., real decontamination) from the liver and/or bones. In other words, once the Pu has been incorporated into a retention compartment, it is virtually inextricable.
In spite of its limitations (effectiveness mainly confined to circulating soluble forms of Pu), i.v. DTPA remains the most efficient treatment and constitutes a solid basis for comparison. It should be noticed that DTPA decontamination therapy is also quite well tolerated since, according to the National Council on Radiation Protection and Measurements, NCRP (Report No. 166), Bethesda, MD, 2011, the recommended dose (i.v. or nebulized inhalation) represents 1 g (in one shot) of chelate/day, while, in case of multiple treatments, total amounts as high as 500 g of DTPA could be administrated within several years. However, at the present time, no treatment fully meets all the required specifications. More specifically, no agent is currently capable, in addition to its action in the bloodstream, of (i) specifically targeting the three major biological Pu retention compartments, and (ii) extracting it from there.
Our challenge was then to propose a simple, effective, and affordable complementary method to DTPA therapy based on a polymeric platform. We postulated that a chelating polymer would indirectly target the main organs (liver and lungs) or the bones (provided an affinity for bone fixation sites would be implemented in that case). This strategy could, therefore, be entirely complementary to Ca-/Zn-DTPA therapy which, as previously seen, is the only one currently in use despite being mainly effective for the circulating (e.g., soluble) forms of Pu.
During the past years, our group has been developing a macromolecular approach based on a polyethyleneimine backbone for the decontamination of actinides [
16,
17,
18,
19]. This can provide, after adequate functionalization, polymeric analogs of DTPA, PEI-MC (polyethyleneimine methylcarboxylate, true polymeric analog) and PEI-MP (polyethyleneimine methylphosphonate, phosphonate analog) from a commercially available 25 kDa branched PEI (polyethyleneimine, see
Figure 1). One fully functionalized polymer is most likely represented by a wide polydisperse population with a mass fraction ranging from ~5–150 kDa.
The syntheses of these chelating PEIs can be carried out in a single step, with very high yields, and are very cheap. Purification via ultrafiltration is straightforward. Too many chelating agents developed so far require a greater number of tedious synthesis and purification steps. Furthermore, as these compounds are polyelectrolytes salts, they readily dissolve in water, making them very easy to use.
We have clearly demonstrated the ability of PEI-based polymers to sequester both U(VI) (uranyl) and actinides(IV) (Pu and Th as a chemical surrogate of Pu). Indeed, under (pseudo)physiological conditions, the two polymers show an EC
50 (50% effective concentration) comparable to DTPA taken as a reference. Complexation to Pu(IV) was also demonstrated by EXAFS spectroscopy with both polymers, PEI-MC and PEI-MP [
18,
19].
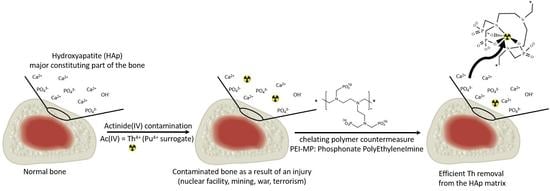
We present now, in this report, a first study on the decontamination of Th(IV) from a hydroxyapatite (HAp) matrix in which Th(IV) has been used as a chemical surrogate of Pu(IV) (see
Section 2.4). Dose–response and kinetic profiles of decontamination associated with this model are provided. Furthermore, viability experiments realized in bones constitutive cells (osteoblasts and osteoclasts) are also reported for PEI-MC and PEI-MP compared to DTPA, taken as the gold standard.
3. Materials and Methods
3.1. Reagents
Bromoacetic acid, sodium carbonate, acid chloride (37%), phosphorous acid H3PO4, branched polyethyleneimine (bPEI, 25 kDa MW), formaldehyde (37% solution), and hydroxyapatite nanopowder (<200 nm particle size (BET), ≥97%, synthetic) were purchased from Sigma-Aldrich, Saint-Louis, Mo, USA. This hydroxyapatite (HAp in the text) was provided with the following elemental analysis: Ca, 39.89%; H, 0.20%; O, 41.41%; P, 18.50%, corresponding to a formula of Ca5(OH)(PO4)3, and it was kept under inert atmosphere. Nitric acid (67–70%, Plasmapure plus degree) and ICP-MS standard solution were purchased from SCP Science, Villebon sur Yvette, France and Honeywell Fluka, Guyancourt, France. A Th(IV) stock solution was prepared from thorium nitrate solution (5.8 mg Th(NO3)4.5H2O in 1 mL of HNO3 0.1 M, (Th(IV)) = 0.01 M). Ultrafiltration was carried out with a stirred ultrafiltration cell (Millipore, 76 mm) equipped with ultrafiltration membrane disc filters Omega™ membrane, OM005076, 5 kDa MWCO (Pall corporation, port Washington, NY, USA) for the polymer purification. Ionic chromatography was performed with a Metrohm, Villebon sur Yvette, France 761 compact apparatus, equipped with a Metrosep Anion Dual 1 (3 × 150 mm) column, using 2.4 mmol/L NaHCO3/2.5 mmol/L Na2CO3 + 2% acetone (conductivity after chemical suppression approximately 16 µS/cm) as eluent for the determination of chloride content. Na+ counterions and phosphorus content were determined from ICP-AAS optima 8000 (Perkin Elmer, Villebon sur Yvette, France) with, respectively, Na standard solution and ICP P standard solution (Honeywell Fluka, Guyancourt, France) (see specific conditions below). Quantification of the thorium content was carried out by resorting to external calibration with standards of Th in HNO3 1% prepared from ICP-MS Th standard solutions plasmaCAL. (SCP Science, Villebon sur Yvette, France). All ICP-MS experiments were performed with ELAN 9000 ((Perkin Elmer, Villebon sur Yvette, France).
3.2. Synthesis of PEI Chelates
3.2.1. Synthesis of PEI-MC
Branched PEI 25 kDa (10 g, 77.52 mmol of monomeric units, C
6H
15N
3) was dissolved in 500 mL of sodium carbonate solution (0.1 M). Then, bromoacetic acid (53.9 g, 387.6 mmol, 5 eq) diluted in water was added dropwise under constant stirring. The reaction mixture was stirred at room temperature overnight. The resulting solution was acidified with HCl to pH ≈ 7, and then purified by ultrafiltration (5 kDa MWCO) with a three-step sequence procedure. First, the neutral reaction mixture coming from the functionalization step was passed through the 5 kDa MWCO membrane (under adequate pressure); the resulting residue was then rinsed thoroughly with a saturated solution of NaCl (250 mL) and then with ultrapure water (250 mL × 2). Finally, the product was freeze-dried and conserved under inert (argon) to prevent moisture addition. This yielded 26 g of the water soluble polyethyleneimine methylcarboxylate sodium salt. Microanalysis revealed a C/N mass ratio of 3.43 (C/N molar ratio of 4) indicating that a full level of methylene carboxylation was achieved. Furthermore, the counterion amount, sodium for carboxylate and eventually chloride for tertiary amine, was determined with ICP-AAS and ionic chromatography, respectively (see
Supplementary Materials). The following molecular formula per monomer was derived: C
12H
19N
3O
6Na
2 (MW 347.28 g/mol), suggesting that the dried polymer, as a sodium salt, dissociated into polyampholyte.
3.2.2. Synthesis of PEI-MP
PEI-MP was synthesized as described elsewhere [
29]. Basically, phosphorous acid H
3PO
4 (19.1 g) was dissolved in concentrated HCl solution (50 mL) and heated at 80 °C. Then, formaldehyde 37% (37.8 mL) was added dropwise. Branched 25 kDa PEI (10.0 g) was dissolved in water (48 mL), and this solution was added dropwise to the reaction mixture. The reaction mixture was stirred at 90 °C for 2 h and then cooled slowly overnight. The product was separated as a viscous oil. After decanting, this viscous oil was washed with water to form a doughy substance. This decanting/washing procedure was repeated twice. The resulting oily residue was basified with Na
2CO
3 to pH ≈ 5, concentrated under vacuum, and then purified by ultrafiltration (5 kDa MWCO) as above, before being freeze-dried and conserved under inert (argon) to prevent moisture addition. This yielded 7.8 g of the water-soluble polyethyleneimine methylphosphonate sodium salt). Microanalysis revealed a C/N mass ratio of 2.57 (C/N molar ratio of 3) indicating that a full level of methylene phosphonylation was achieved. As for the PEI-MC, counterions were determined with ICP-AAS and ionic chromatography (see
Supplementary Materials). The following molecular formula per monomer was derived: C
9H
21.5N
3P
3O
9Na
3.5 (MW 488.16 g/mol), suggesting that the dried polymer, as a sodium salt, dissociated into polyampholyte.
3.3. Toxicity of PEI Chelates toward Bone Constitutive Cells
The SAOS-2 cell line was purchased from the American Type Culture Collection. Briefly, SAOS-2 cells were maintained in McCoy’s 5A medium without phenol red (HyClone, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 15% heat-inactivated fetal bovine serum (Biowest, Nuaille, France) and antibiotics (100 IU/mL penicillin, 100 µg/mL streptomycin, Sigma-Aldrich, Saint-Louis, MO, USA). Prior to assessment of toxicity, SAOS-2 cells were plated in 96-well plates (12,000 cells/well).
The MLO-A5 (murine-like osteocytes) cell line obtained from Linda Bonewald lab [
30] was maintained at 37 °C, 5% CO
2 using rat tail collagen I-coated (Sigma-Aldrich, Saint-Louis, MO, USA) wells, in α-MEM with nucleotides and Ultraglutamine (BE02-002F, Lonza, Basel, Switzerland) supplemented with 1% penicillin/streptomycin (P/S, Sigma-Aldrich, Saint-Louis, MO, USA), 5% heat-inactivated fetal bovine serum (FBS: Hyclone SH30071.03 GE Healthcare, Chicago, IL, USA) and 5% heat-inactivated calf serum (CS: Hyclone SH30072.03, GE-Healthcare, Chicago, IL, USA).
For the assessment of toxicity, the medium was replaced by complete culture medium supplemented with DTPA, PEI-MP, or PEI-MC (0.1 µM, 1 µM, 10 µM, 100 µM, 1 mM, 10 mM, 50 mM, and 100 mM). Cells were further incubated for 1 h and washed three times with PBS buffer to remove the noninternalized polymers or DTPA. Next, 200 µL of culture medium was added, and cells were incubated for 24 or 48 h. At the end of the incubation period, cytotoxicity was assessed using the MTT assay. Briefly, culture medium was removed and replaced by 100 µL of EMEM containing MTT. After 1 h at 37 °C, EMEM containing MTT was removed and replaced by 150 µL of DMSO. After 15 min, absorbance was measured at 570 nm. The mean absorbance of nonexposed cells was taken as the reference value. The percentage of cell viability was calculated on the basis of the ratio between the absorbance of each sample compared to the average absorbance of the untreated cells. Results were expressed as the percentage mean (±SD) from two independent experiments performed in triplicate.
3.4. Determination of the Affinity of the Chelating Agents toward HAp by Thermogravimetric Analysis
HAp powder (10 mg) was dispersed in a 1.5 mL solution of DTPA, PEI-MC or PEI-MC (10 mM, based on monomer concentration), 1.5 µmol monomer/mg, and stirred overnight. Samples were then centrifuged at 15,000 rpm during 15 min and washed (three times), to remove unbound DTPA, PEI-MC, or PEI-MP. The resulting HAp–chelate powder was freeze-dried to eliminate the water excess and kept under inert atmosphere. Thermogravimetric analyses (TGA) were performed on a Mettler Toledo, Columbus, OH, USA, TGA 851e using STAR© software (version 13.00) for data analysis. Freeze-dried samples (~10 mg) were placed in 70 µL alumina pans and heated at 10 °C·min−1 from 25 to 800 °C under N2 flow (50 mL·min−1). Calculation of the mass loss percentage, in the 200–400 °C range, allowed directly determining the affinity of each chelate toward HAp their comparison.
3.5. Contamination of HAp
To avoid hydrolysis at physiological pH, Th(NO
3)
4, was converted into Th(CO
3)
4. Briefly, a Th(CO
3)
4 (1.33 mM) solution was prepared by adding 400 µL of Th(NO
3)
4 (0.1M) in 29.6 mL of Na
2CO
3 (0,1M). The resulting Th(IV) solution, 30 mL (1.33 mM), was then incorporated into the hydroxyapatite (HAp) (500 mg). This suspension was stirred for 48 h. The contaminated HAp–Th powder was submitted to three cycles of centrifugation/washing steps with ultrapure water until no Th could be detected (ICP-MS) into the last filtrate (see
Supplementary Materials). HAp–Th powder was then freeze-dried to yield 480 mg of a white powder. The incorporation level of Th(IV) into the HAp was ensured by quantifying the elementary Th via ICP-MS from different aliquots of the HAp–Th powder. It should be noted that the incorporation level of Th(IV) into the HAp matrix could be very precisely controlled using this procedure. This procedure was independently repeated onto different HAp samples, and contamination rates were found to be highly repeatable. Overall, the total Th content into the contaminated HAp samples was 0.7%.
3.6. Efficiency of Th Extraction: Dose–Response of HAp–Th Subjected to the Chelates
First, 5 mg of HAp–Th powder (0.7% Th content as determined by ICP-MS) were added to 1.5 mL of chelates (PEI-MC, PEI-MP, or DTPA), at different concentrations, in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.4) and mixed (orbitally) for 7 days. The corresponding chelate monomer molar concentrations were equal to 0 (blank), 0.010, 0.015, 0.039, 0.050, 0.077, 0.15, 0.23, 0.39, 0.77, 1.16, 1.93, 3.14, 6.29, and 10.0. Each of these samples was prepared in triplicate. After 8 days, a purification step (15,000 rpm, 15 min) was carried out. Then, 500 µL of supernatant was recovered from two centrifugations (250 µL 2×) for each sample and digested in 5 mL of 67–70% HNO3 (Plasmapure plus degree, SCP Science, Villebon sur Yvette, France) at 120 °C during 2 h. The digested samples were then evaporated to dryness at 90 °C using a heating block. Finally, 5 mL of 1.5% HNO3 was added to the tubes. Each sample was analyzed by ICP-MS (Perkin Elmer ELAN 9000). Operation conditions were daily optimized using a tuning solution. Determination of the Th concentrations was carried out by resorting to external calibration with standards of Th prepared from single-element ICP-MS standard solutions (SPEX CertiPrep, Inc., Vernon Hills, IL, USA) for thorium. An analytical blank consisting of HAp–Th without polymers or DTPA was prepared in the same conditions. Bismuth was added to each sample at a concentration of 10 ppb to correct for sample matrix effects. Dose–response curves represent the percentage of Th recovered from the filtrate versus the chelate concentration (monomer). Results were expressed as the mean (±SD) from triplicates.
3.7. Kinetics of Th Extraction from the HAp–Th Subjected to the Chelates
Firstly, 5 mg of HAp–Th powder (9.3 ppm Th content as determined by ICP-MS) was added to a 1.5 mL solution of PEI-MC, PEI-MP, or DTPA at a concentration of 6.3 mM (monomer) in TBS buffer (50 mM Tris, 150 mM NaCl, pH 7.4). Each sample was prepared in triplicate. A centrifugation step (15,000 rpm, 15 min) was carried out at different times (2, 18, 42, 66, 90, 114, 162, 186, 210, 290, 354, and 504 h). Blank samples (absence of chelates) were also evaluated in the same conditions. Next, 500 µL of supernatant was recovered from two centrifugations (250 µL 2×) for each sample and digested in 5 mL of 67–70% HNO3 (Plasmapure plus degree, SCP Science) at 120 °C for 2 h. The digested samples were then evaporated to dryness at 90 °C using a heating block. Finally, 5 mL of 1.5% HNO3 was added to each tube, and samples were analyzed by ICP-MS (Perkin Elmer ELAN 9000) as previously described. Kinetic curves represented the percentage of Th recovered from the filtrate with a 6.3 mM chelate concentration (monomer) at the specified time. Results were expressed as the mean (±SD) from triplicates.
3.8. EXAFS of HAp–Th, PEI-MP–Th, and PEI-MP–Th–HAp
3.8.1. Sample Preparation
Solid pellets were prepared by mixing HAp–Th (5mg) with polyethylene to obtain homogeneous solid pellets.
PEI-MP–Th was also prepared by using the same stock solution of Th(IV) as described above (Th(NO3)4 (0.1 M)). Then, 50 µL of Th(NO3)4), [Th] = 2.5 × 10−3 M pH = 1, was mixed with 250 µL of PEI-MP solution (5 mM of monomeric units) in Tris/NaCl buffer (50 mM, 150 mM). The pH was increased slowly to pH 7.0 by adding NaOH.
PEI-MP–Th–HAp was prepared by directly using the sample from the dose–response experiments of HAp–Th subjected to the chelates. Then, 1.5 mL of supernatant was recovered for each experiment. A centrifugation step (12,000× g, 5 min, 20 °C) was carried out on 10 kDa microcon® centrifugal filter, allowing us to concentrate the sample for suitable EXAFS measurements.
3.8.2. Data Recording and Processing
XAS data were recorded at the Th LIII edge (16,300 eV) on the MARS beamline at the SOLEIL synchrotron facility, which is dedicated to the study of radioactive materials. The optics of the beamline consisted of a water-cooled double-crystal monochromator for incident energy selection and horizontal focalization and two large water-cooled reflecting mirrors for high-energy rejection (harmonic part), vertical collimation, and focalization. All measurements were recorded in double-layered solution cells (200 µL) specifically designed for radioactive samples at room temperature. A 13-element Ge detector was used for data collection in the fluorescence mode.
Data treatment was carried out using ATHENA code of Demeter 0.9.26 package [
31]. The E
o energy was identified at the maximum of the absorption edge. Background removal was performed using a pre-edge linear function. Atomic absorption was simulated with a cubic spline function.
3.8.3. Data Fitting
The extracted EXAFS signal was fitted in R space without any additional filtering after Fourier transformation with a Hanning window in k
2 and k
3 using the ARTEMIS code of Demeter 0.9.26 package [
31]. Phases and amplitudes were calculated with Feff7 code embedded in ARTEMIS code. Only one global amplitude factor S
02 fixed to 1 and one energy threshold correction factor Δe
0 were used for all paths. The agreement factor R (in %) and quality factor-reduced χ
2 were both provided as an indication of the fit quality.
HAp–Th (Hanning window = 2.7–11 Å
−1, fit range = 1–4 Å). The model used for phases and amplitude calculations was a Th
4(PO
4)
4P
2O
7 crystallographic structure [
26] where the coordination sphere of Th was composed of six monodentate and one bidentate phosphate. The first coordination sphere was adjusted with three contributions of oxygen atoms sharing the same Debye–Waller factor. The total number of oxygen atoms was set to nine atoms as the average for Th coordination. Two contributions of one and six phosphorus atoms were included in the fitting procedure. The triple Th–O–P and quadruple Th–O–P–O paths for the monodentate phosphorus contribution were also considered. The multiple scattering paths shared the same Debye–Waller and distance correction factors.
PEI-MP–Th and PEI-MP–Th–HAp (Hanning window = 2.7–10 Å
−1, fit range = 1–4 Å). The model used for phases and amplitude calculations was described elsewhere [
32]. The fitting procedure was the same as that used to fit previous EXAFS data of PEI-MP–Th (but synthesized differently, as described in Lahrouch et al. [
17]). The first coordination sphere was fitted with a single scattering path of nine (fixed) oxygen atoms. A single scattering path of phosphorus atoms and the corresponding quadruple scattering path Th–O–P–O were also included in the fitting procedure. The addition of a single scattering path of chlorine atoms significantly improved the quality of the fit, as already observed elsewhere [
17].
4. Conclusions
We showed herein, on the basis of strongly supported literature data and previous work we performed on actinide complexation with chelating polymers based on a PEI scaffold, that PEI-MP, i.e., the polyphosphonate analog of DTPA, can be considered as serious candidate for decontamination of Pu(IV) specifically targeted to the bones. Firstly, the PEI-MP, synthetized from a branched PEI (25 kDa), used without mass fractionation, did not show any toxicity toward bones constitutive cells, SAOS-2 and MLO-A5, unlike DTPA, which substantially decreased the viability at a concentration of 10 mM. Secondly, we demonstrated, using thermogravimetric analysis, that PEI-MP had a 15-fold higher affinity than DTPA toward hydroxyapatite, which constitutes the major part of the mineral bone matrix. We then successfully prepared hydroxyapatite contaminated with 0.7% Th(IV) (used as a Pu(IV) surrogate) and demonstrated through EXAFS experiments the full incorporation of this actinide into the bone mimicking matrix. Under the tested conditions, an optimum chelate concentration of 6.3 mM was sufficient to achieve maximum Th extraction with both compounds (DTPA and PEI-MP). However, the PEI-MP was able, in this case, to extract twice the amount of Th(IV), 29% versus 17%, than the gold standard DTPA. Additionally, when a kinetic study was performed over a 21 day period, this difference continued to increase, and the decontamination with the PEI-MP reached almost 50% at day 21, whereas it remained constant at 20% from day 1 with DTPA. Subsequent treatment with a fresh dose even increased the final level to almost 65%. In a more general context of actinide contamination, targeting the biological sites (lungs, liver, bones) that are involved in the sequestering of a substantial proportion of these radioelements could constitute a real advance in the field. This polymeric strategy, thus, provides a first, but nonetheless interesting complementary approach to the chelating therapy currently used based on Ca-/Zn-DTPA. Future studies aimed at tuning the kinetics and limiting the depletion of endogen cations are currently in progress.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}