
Pancreatic Ductal Cell-Derived Extracellular Vesicles Are Effective Drug Carriers to Enhance Paclitaxel’s Efficacy in Pancreatic Cancer Cells through Clathrin-Mediated Endocytosis




Abstract
:1. Introduction
2. Results
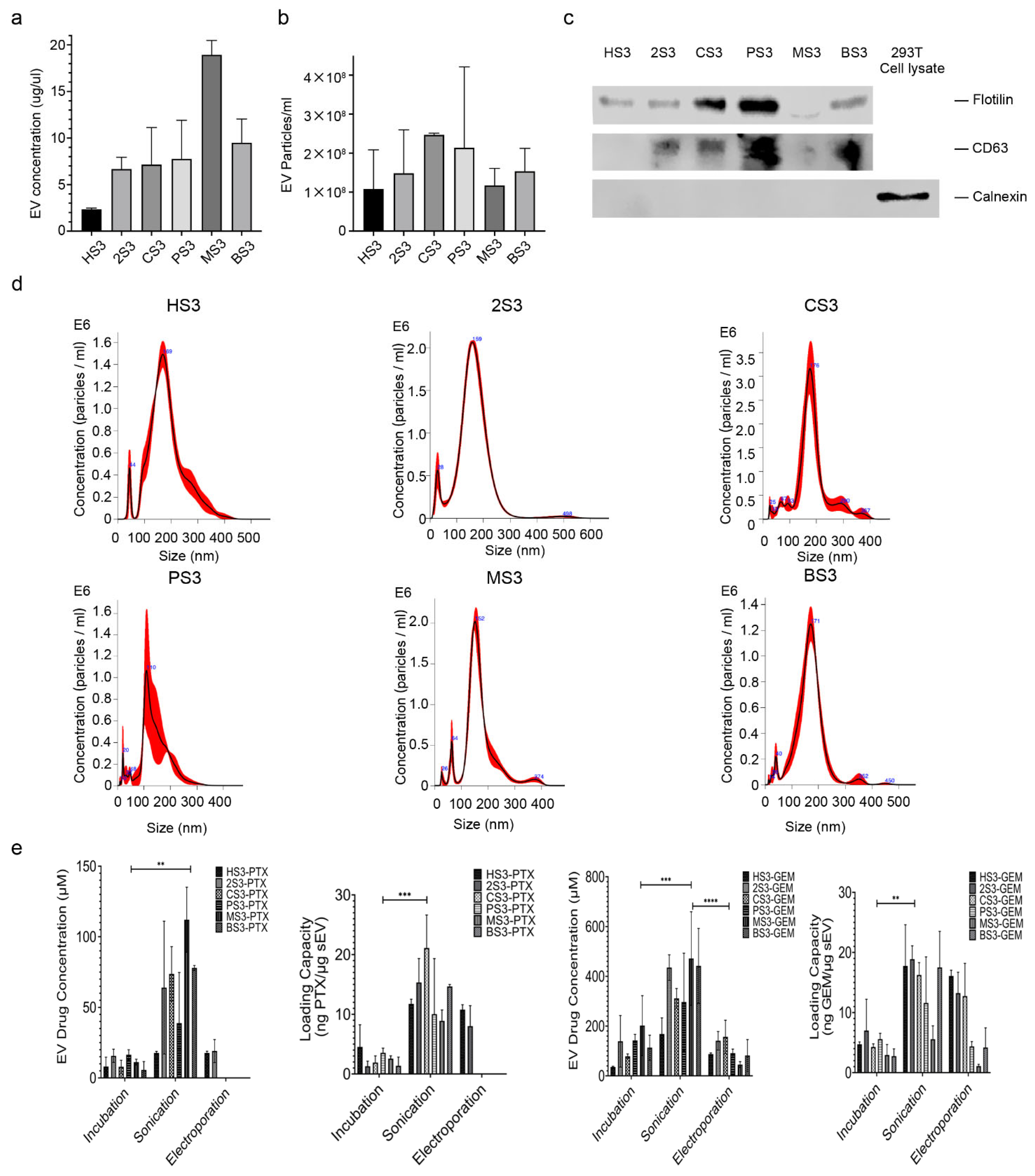
2.1. Characterization of sEVs and Drug Encapsulation
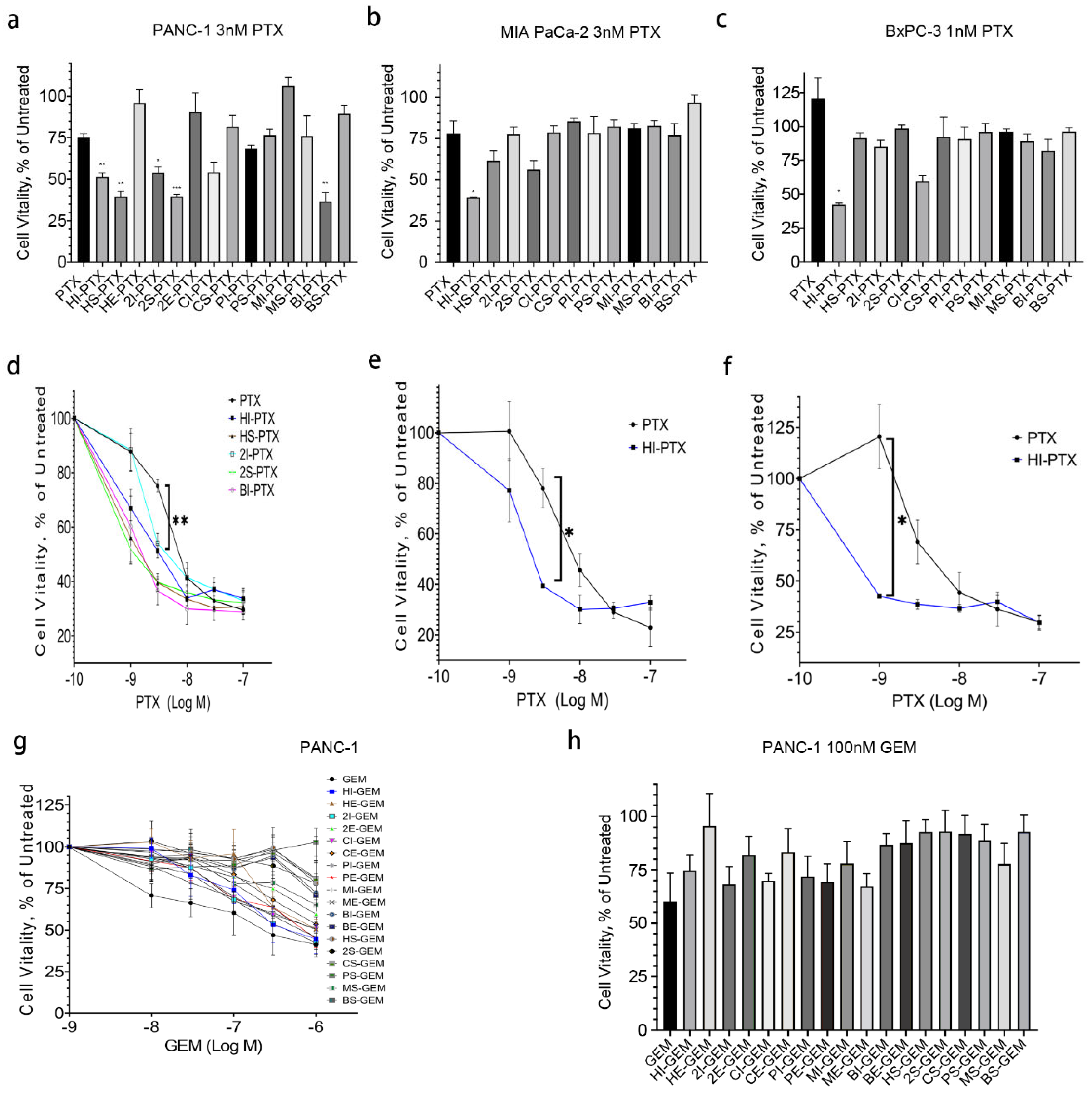
2.2. HPNE sEV-PTX Derived from Incubation (HI-PTX) Is most Efficacious in Killing Pancreatic Cancer Cells
2.3. Uptake of HI-PTX in PANC-1 Cells
2.4. HI-PTX’S Uptake and Cytotoxicity Is Associated with Clathrin-Mediated Endocytosis
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tas, F.; Sen, F.; Keskin, S.; Kilic, L.; Yildiz, I. Prognostic factors in metastatic pancreatic cancer: Older patients are associated with reduced overall survival. Mol. Clin. Oncol. 2013, 1, 788–792. [Google Scholar] [CrossRef] [Green Version]
- National Comprehensive Cancer Network Pancreatic Cancer (Version 1, 2021). Available online: https://www.nccn.org/professionals/physician_gls/pdf/pancreatic.pdf (accessed on 6 August 2021).
- Mosquera, J.; Garcia, I.; Liz-Marzan, L.M. Cellular Uptake of Nanoparticles versus Small Molecules: A Matter of Size. ACC Chem. Res. 2018, 51, 2305–2313. [Google Scholar] [CrossRef] [Green Version]
- Grantab, R.; Sivananthan, S.; Tannock, I.F. The penetration of anticancer drugs through tumor tissue as a function of cellular adhesion and packing density of tumor cells. Cancer Res. 2006, 66, 1033–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Witzigmann, D.; Balasubramanian, V.; Huwyler, J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J. Control Release 2015, 200, 138–157. [Google Scholar] [CrossRef]
- Vishnu, P.; Roy, V. Safety and Efficacy of nab-Paclitaxel in the Treatment of Patients with Breast Cancer. Breast Cancer 2011, 5, 53–65. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A. Pre-clinical immunotoxicity studies of nanotechnology-formulated drugs: Challenges, considerations and strategy. J. Control Release 2015, 220, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Giannakou, C.; Park, M.V.; de Jong, W.H.; van Loveren, H.; Vandebriel, R.J.; Geertsma, R.E. A comparison of immunotoxic effects of nanomedicinal products with regulatory immunotoxicity testing requirements. Int. J. Nanomed. 2016, 11, 2935–2952. [Google Scholar] [CrossRef] [Green Version]
- Hannon, G.; Lysaght, J.; Liptrott, N.J.; Prina-Mello, A. Immunotoxicity Considerations for Next Generation Cancer Nanomedicines. Adv. Sci. 2019, 6, 1900133. [Google Scholar] [CrossRef] [Green Version]
- Baj-Krzyworzeka, M.; Szatanek, R.; Weglarczyk, K.; Baran, J.; Urbanowicz, B.; Branski, P.; Ratajczak, M.Z.; Zembala, M. Tumour-derived microvesicles carry several surface determinants and mRNA of tumour cells and transfer some of these determinants to monocytes. Cancer Immunol. Immunother. 2006, 55, 808–818. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Miekus, K.; Kucia, M.; Zhang, J.; Reca, R.; Dvorak, P.; Ratajczak, M.Z. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006, 20, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Bastos, N.; Ruivo, C.F.; da Silva, S.; Melo, S.A. Exosomes in cancer: Use them or target them? Semin. Cell Dev. Biol. 2018, 78, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Drummen, G.P.; Kuroda, M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. Int. J. Mol. Sci. 2016, 17, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Sun, H.; Burrola, S.; Wu, J.; Ding, W.Q. Extracellular Vesicles in the Development of Cancer Therapeutics. Int. J. Mol. Sci. 2020, 21, 6097. [Google Scholar] [CrossRef]
- Xu, Y.F.; Xu, X.; Gin, A.; Nshimiyimana, J.D.; Mooers, B.H.M.; Caputi, M.; Hannafon, B.N.; Ding, W.Q. SRSF1 regulates exosome microRNA enrichment in human cancer cells. Cell Commun. Signal 2020, 18, 130. [Google Scholar] [CrossRef]
- Ding, W.Q.; Liu, B.; Vaught, J.L.; Palmiter, R.D.; Lind, S.E. Clioquinol and docosahexaenoic acid act synergistically to kill tumor cells. Mol. Cancer 2006, 5, 1864–1872. [Google Scholar] [CrossRef] [Green Version]
- Hannafon, B.N.; Carpenter, K.J.; Berry, W.L.; Janknecht, R.; Dooley, W.C.; Ding, W.Q. Exosome-mediated microRNA signaling from breast cancer cells is altered by the anti-angiogenesis agent docosahexaenoic acid (DHA). Mol. Cancer 2015, 14, 133. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Rosendale, M.; Campbell, R.E.; Perrais, D. pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J. Cell Biol. 2014, 207, 419–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cureton, D.K.; Massol, R.H.; Whelan, S.P.; Kirchhausen, T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 2010, 6, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Majeed, S.R.; Vasudevan, L.; Chen, C.Y.; Luo, Y.; Torres, J.A.; Evans, T.M.; Sharkey, A.; Foraker, A.B.; Wong, N.M.; Esk, C.; et al. Clathrin light chains are required for the gyrating-clathrin recycling pathway and thereby promote cell migration. Nat. Commun. 2014, 5, 3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolte-’t Hoen, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanchanapally, R.; Deshmukh, S.K.; Chavva, S.R.; Tyagi, N.; Srivastava, S.K.; Patel, G.K.; Singh, A.P.; Singh, S. Drug-loaded exosomal preparations from different cell types exhibit distinctive loading capability, yield, and antitumor efficacies: A comparative analysis. Int. J. Nanomed. 2019, 14, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, C.; Du, Z.; Zhang, H.; Feng, Y.; Qi, Y.; Zheng, Y.; Liu, J.; Wang, J. Endocytic pathway inhibition attenuates extracellular vesicle-induced reduction of chemosensitivity to bortezomib in multiple myeloma cells. Theranostics 2021, 11, 2364–2380. [Google Scholar] [CrossRef]
- Schindler, C.; Collinson, A.; Matthews, C.; Pointon, A.; Jenkinson, L.; Minter, R.R.; Vaughan, T.J.; Tigue, N.J. Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. PLoS ONE 2019, 14, e0214545. [Google Scholar] [CrossRef] [Green Version]
- Nedeva, C.; Mathivanan, S. Engineering Extracellular Vesicles for Cancer Therapy. Subcell. Biochem. 2021, 97, 375–392. [Google Scholar]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chen, P.; Dong, Y.; Xie, H.; Wang, Y.; Soto, F.; Ma, P.; Feng, X.; Du, W.; Liu, B.F. Designer exosomes enabling tumor targeted efficient chemo/gene/photothermal therapy. Biomaterials 2021, 276, 121056. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, W.; Zhang, L.; Ban, L.; Chen, P.; Du, W.; Feng, X.; Liu, B.F. Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells. ACS Appl. Mater. Interfaces 2017, 9, 27441–27452. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, Y.; Li, Y.W.; Li, W.; Cheng, K.; Qian, Y.; Xu, G.Q.; Zhang, X.S.; Hu, L.; Chen, P.; et al. Designer Exosomes for Active Targeted Chemo-Photothermal Synergistic Tumor Therapy. Adv. Funct. Mater. 2018, 28, 1707360. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, W.; Chen, X.; Wang, Q.; Li, C.; Chen, Q.; Zhang, Y.; Lu, Y.; Ding, X.; Jiang, C. Bone marrow mesenchymal stem cells-derived exosomes for penetrating and targeted chemotherapy of pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Hu, X.B.; Cai, J.X.; Xiang, D.X. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020, 101, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Wang, M.; Liu, H.; Liu, X.; Situ, A.; Wu, B.; Ji, Z.; Chang, C.H.; Nel, A.E. Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice. ACS Nano 2015, 9, 3540–3557. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Guo, S.; Zhang, J.; Kim, W.Y.; Huang, L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv. Funct. Mater. 2014, 24, 6601–6611. [Google Scholar] [CrossRef]
- Xu, Y.F.; Xu, X.; Bhandari, K.; Gin, A.; Rao, C.V.; Morris, K.T.; Hannafon, B.N.; Ding, W.Q. Isolation of extra-cellular vesicles in the context of pancreatic adenocarcinomas: Addition of one stringent filtration step improves recovery of specific microRNAs. PLoS ONE 2021, 16, e0259563. [Google Scholar] [CrossRef]
- Singh, R.; Shakya, A.K.; Naik, R.; Shalan, N. Stability-indicating HPLC determination of gemcitabine in pharmaceutical formulations. Int. J. Anal. Chem. 2015, 2015, 862592. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Vigano, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence | Target |
|---|---|---|
| si_CLTB91 | sense: GCCCAGCUAUGUGACUUCATT | |
| antisense: UGAAGUCACAUAGCUGGGCCA | Clathrin light chain | |
| si_CLTB92 | sense: CCUCCUCUCAGUCUACUCATT | |
| antisense: UGAGUAGACUGAGAGGAGGCG | Clathrin light chain | |
| si_CLTB93 | sense: GAACAAGUAGAGAAGAACATT | |
| antisense: UGUUCUUCUCUACUUGUUCAC | Clathrin light chain | |
| si_CAV1 | sense: GCUUCCUGAUUGAGAUUCATT | |
| antisense: UGAAUCUCAAUCAGGAAGCTC | Caveolin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Bhandari, K.; Burrola, S.; Wu, J.; Ding, W.-Q. Pancreatic Ductal Cell-Derived Extracellular Vesicles Are Effective Drug Carriers to Enhance Paclitaxel’s Efficacy in Pancreatic Cancer Cells through Clathrin-Mediated Endocytosis. Int. J. Mol. Sci. 2022, 23, 4773. https://doi.org/10.3390/ijms23094773
Sun H, Bhandari K, Burrola S, Wu J, Ding W-Q. Pancreatic Ductal Cell-Derived Extracellular Vesicles Are Effective Drug Carriers to Enhance Paclitaxel’s Efficacy in Pancreatic Cancer Cells through Clathrin-Mediated Endocytosis. International Journal of Molecular Sciences. 2022; 23(9):4773. https://doi.org/10.3390/ijms23094773
Chicago/Turabian StyleSun, Haoyao, Kritisha Bhandari, Stephanie Burrola, Jinchang Wu, and Wei-Qun Ding. 2022. "Pancreatic Ductal Cell-Derived Extracellular Vesicles Are Effective Drug Carriers to Enhance Paclitaxel’s Efficacy in Pancreatic Cancer Cells through Clathrin-Mediated Endocytosis" International Journal of Molecular Sciences 23, no. 9: 4773. https://doi.org/10.3390/ijms23094773
APA StyleSun, H., Bhandari, K., Burrola, S., Wu, J., & Ding, W.-Q. (2022). Pancreatic Ductal Cell-Derived Extracellular Vesicles Are Effective Drug Carriers to Enhance Paclitaxel’s Efficacy in Pancreatic Cancer Cells through Clathrin-Mediated Endocytosis. International Journal of Molecular Sciences, 23(9), 4773. https://doi.org/10.3390/ijms23094773