3.1. Chemistry
Reagents and starting materials were obtained from commercial sources (Merck Life Science srl, Milan, Italy). Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. All reactions were monitored by thin-layer chromatography (TLC) using commercial plates pre-coated with Merck silica gel 60 F-254. Visualisation was performed by UV fluorescence (λmax = 254 nm) or by staining with iodine or potassium permanganate. Chromatographic separations were performed on a silica gel column by gravity chromatography (Kieselgel 40, 0.063–0.200 mm; Merck), flash chromatography (Kieselgel 40, 0.040–0.063 mm; Merck), silica gel preparative TLC (Kieselgel 60 F254, 20 × 20 cm, 2 mm). Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. Compounds were named following IUPAC rules, as applied by Beilstein-Institut AutoNom 2000 (4.01.305) or CA Index Name. All melting points were determined on a microscope hot stage Büchi apparatus and are uncorrected. The identity and purity of intermediates and final compounds were ascertained through 1H-NMR, 13C-NMR, and TLC chromatography. Monodimensional spectra 1H-NMR and 13C-NMR were registered by a 400 MHz field through Avance 400 apparatus (Bruker Biospin Version 002 with SGU). Chemical shifts (d) are in parts per million (ppm) approximated by the nearest 0.01 ppm, using the solvent as internal standard. Coupling constants (J) are in Hz, they were calculated by Top Spin 3.1 and approximated by 0.1 Hz. Data are reported as follows: chemical shift, multiplicity (exch, exchange; br, broad; s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; or a combination of those, e.g, dd), integral, assignments, and coupling constant. Mass spectra (m/z) were recorded on a ESI-MS triple quadrupole (Varian 1200 L) system, in positive ion mode, by infusing a 10 mg/L solution of each analyte dissolved in a mixture of mQ H2O:acetonitrile 1:1 v/v. All new compounds possess a purity ≥ 95%; microanalyses indicated by the symbols of the elements were performed with a Perkine-Elmer 260 elemental analyzer for C, H, and N, and they were within ±0.4% of the theoretical values.
3.1.1. 5-(Benzylamino)naphthalene-2-Sulfonic Acid (3a)
A suspension of 0.89 mmol of 5-aminonaphthalene-2-sulfonic acid (
1), which is commercially available, and 1.79 mmol of Na
2CO
3 in 5 mL of dry DMF was stirred at room temperature for 30 min. Then, 1.33 mmol of benzyl bromide (
2a) were added and the mixture was heated at reflux for 2–3 h. After cooling, the pH was checked and adjusted to 6–7 and the solvent was evaporated. The crude product was purified by flash column chromatography using CH
2Cl
2/MeOH/CH
3COOH 80:20:2 as eluent to obtain the desired compound as white powder [
24]. Yield = 86%; mp > 300 °C (EtOH/H
2O).
1H-NMR (400 MHz, DMSO-d
6) δ 4.48 (s, 2H, CH
2-Ph), 6.36 (d, 1H, Ar, J = 6.4 Hz), 6.93 (exch br s, 1H, NH), 7.05–7.10 (m, 1H, Ar), 7.15–7.25 (m, 2H, Ar), 7.28–7.35 (m, 2H, Ar), 7.39–7.45 (m, 2H, Ar), 7.60 (d, 1H, Ar, J = 8.8 Hz), 7.95 (s, 1H, Ar), 8.18 (d, 1H, Ar, J = 8.0 Hz).
13C-NMR (100 MHz, DMSO-d
6) δ 48.4 (CH
2), 119.8 (CH), 121.5 (CH), 123.2 (CH), 125.1 (C), 126.5 (CH), 126.7 (CH), 128.2 (CH), 128.5 (CH), 133.2 (C), 134.1 (C), 139.9 (C), 141.0 (C). ESI-MS calcd. for C
17H
15NO
3S, 313.08; found:
m/
z 314.08 [M + H]
+. Anal. C
17H
15NO
3S (C, H, N).
3.1.2. General Procedure for Synthesis of Compounds 3b–f
A total of 0.90 mmol of 5-aminonaphthalene-2-sulfonic acid (1) and 1.80 mmol of sodium hydride (60% dispersion in mineral oil) were dissolved in 5 mL of dry DMF. After 30 min in stirring at room temperature, 1.35 mmol of appropriate phenyllalkyl bromide (2b–f), which is commercially available, were added and the mixture was stirred at 90 °C for 3–4 h. After cooling, the pH was checked and adjusted to 6–7 and the solvent was evaporated. The crude products were purified by flash column chromatography using CH2Cl2/MeOH/CH3COOH 80:20:2 as eluent.
5-(Phenethylamino)naphthalene-2-sulfonic acid (3b). Yield = 60%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 2.98 (t, 2H, NHCH2CH2Ph, J = 7.6 Hz), 3.40 (q, 2H, NHCH2CH2Ph, J = 7.0 Hz), 6.21 (t, 1H, NH, J = 4.8 Hz), 6.55 (d, 1H, Ar, J = 7.6 Hz), 7.11 (d, 1H, Ar, J = 8.0 Hz), 7.17–7.22 (m, 1H, Ar), 7.26–7.32 (m, 5H, Ar), 7.55 (dd, 1H, Ar, J1 = 8.8 Hz, J2 = 1.2 Hz), 7.94 (s, 1H, Ar), 8.06 (d, 1H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 34.7 (CH2), 45.3 (CH2), 104.5 (CH), 116.6 (CH), 121.9 (CH), 121.9 (CH), 123.2 (C), 125.0 (CH), 126.6 (CH), 128.1 (CH), 128.8 (CH), 129.2 (CH), 129.5 (CH), 133.6 (C), 140.4 (C), 144.1 (C), 144.2 (C). ESI-MS calcd. for C18H17NO3S, 327.09; found: m/z 328.10 [M + H]+. Anal. C18H17NO3S (C, H, N).
5-[(3-Phenylpropyl)amino]naphthalene-2-sulfonic acid (3c). Yield = 46%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 1.98 (quin, 2H, NHCH2CH2CH2Ph, J = 7.2 Hz), 2.72 (t, 2H, NHCH2CH2CH2Ph, J = 7.6 Hz), 3.17 (q, 2H, NHCH2CH2CH2Ph, J = 6.2 Hz), 6.10 (t, 1H, NH, J = 4.8 Hz), 6.42 (d, 1H, Ar, J = 7.6 Hz), 7.08 (d, 2H, Ar, J = 8.0 Hz), 7.16–7.29 (m, 5H, Ar), 7.54 (d, 1H, Ar, J = 8.8 Hz), 7.92 (s, 1H, Ar), 8.09 (d, 1H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 30.4 (CH2), 33.4 (CH2), 43.3 (CH2), 103.8 (CH), 116.3 (CH), 121.8 (CH), 122.1 (CH), 123.1 (C), 125.2 (CH), 126.2 (CH), 127.7 (CH), 128.7 (CH), 128.8 (CH), 133.7 (C), 142.4 (C), 144.6 (C), 145.6 (C). ESI-MS calcd. for C19H19NO3S, 341.11; found: m/z 342.11 [M + H]+. Anal. C19H19NO3S (C, H, N).
5-[(4-Phenylbutyl)amino]naphthalene-2-sulfonic acid (3d). Yield = 63%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 1.60–1.70 (m, 4H, NHCH2CH2CH2CH2Ph), 2.57–2.62 (m, 2H, NHCH2CH2CH2CH2Ph), 3.10–3.20 (m, 2H, NHCH2CH2CH2CH2Ph), 6.14 (exch br s, 1H, NH), 6.45 (d, 1H, Ar, J = 7.2 Hz), 7.07 (d, 1H, Ar, J = 7.6 Hz), 7.12–7.22 (m, 6H, Ar), 7.56 (d, 1H, Ar, J = 8.4 Hz), 7.96 (s, 1H, Ar), 8.13 (d, 1H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 28.2 (CH2), 29.2 (CH2), 35.4 (CH2), 43.5 (CH2), 103.9 (CH), 116.0 (CH), 121.9 (CH), 122.0 (CH), 123.1 (C), 124.9 (CH), 126.1 (CH), 127.8 (CH), 128.7 (CH), 128.8 (CH), 133.7 (C), 142.6 (C), 144.6 (C), 145.0 (C). ESI-MS calcd. for C20H21NO3S, 355.12; found: m/z 356.13 [M + H]+. Anal. C20H21NO3S (C, H, N).
5-[(5-Phenylpentyl)amino]naphthalene-2-sulfonic acid (3e). Yield = 67%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 1.35–1.45 (m, 2H, NHCH2CH2CH2CH2CH2Ph), 1.59 (quin, 2H, NHCH2CH2CH2CH2CH2Ph, J = 7.2 Hz), 1.68 (quin, 2H, NHCH2CH2CH2CH2CH2Ph, J = 7.2 Hz), 2.55 (t, 2H, NHCH2CH2CH2CH2CH2Ph, J = 7.6 Hz), 3.13 (t, 2H, NHCH2CH2CH2CH2CH2Ph, J = 7.2 Hz), 6.09 (exch br s, 1H, NH), 6.45 (d, 1H, Ar, J = 7.2 Hz), 7.07 (d, 1H, Ar, J = 8.0 Hz), 7.10–7.26 (m, 6H, Ar), 7.55 (d, 1H, Ar, J = 8.4 Hz), 7.95 (s, 1H, Ar), 8.11 (d, 1H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 26.7 (CH2), 28.2 (CH2), 31.2 (CH2), 35.5 (CH2), 43.4 (CH2), 104.3 (CH), 116.2 (CH), 121.8 (CH), 122.0 (CH), 123.2 (C), 125.0 (CH), 126.1 (CH), 128.1 (CH), 128.7 (CH), 133.5 (C), 142.7 (C), 144.0 (C), 144.4 (C). ESI-MS calcd. for C21H23NO3S, 369.14; found: m/z 370.14 [M + H]+. Anal. C21H23NO3S (C, H, N).
5-[(4-(Methoxycarbonyl)benzyl)amino]naphthalene-2-sulfonic acid (3f). Yield = 20%; mp > 300 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 3.79 (s, 3H, OCH3), 4.54 (d, 2H, NHCH2Ph, J = 5.2 Hz), 6.27 (d, 1H, Ar, J = 7.2 Hz), 7.03 (t, 1H, NH, J = 5.6 Hz), 7.06–7.13 (m, 1H, Ar), 7.47–7.52 (m, 3H, Ar), 7.60 (d, 1H, Ar, J = 8.8 Hz), 7.87 (d, 2H, Ar, J = 8.0 Hz), 7.94 (s, 1H, Ar), 8.17 (d, 1H, Ar, J = 8.8 Hz). ESI-MS calcd. for C19H17NO5S, 371.41; found: m/z 372.09 [M + H]+. Anal. C19H17NO5S (C, H, N).
3.1.3. 4-[(6-Sulfonaphthalen-1-yl)aminomethyl]benzoic Acid (4)
A mixture of 0.20 mmol of compound 3f, 40% NaOH (3.2 mL) and a little amount of EtOH 96% was stirred at reflux for 3 h. After cooling and maintaining the reaction in an ice bath, HCl 6N was added up to acid pH and the precipitate was filtered off. The acid derivative was purified by flash column chromatography using CH2Cl2/MeOH/CH3COOH 80:20:2 as eluent. Yield = 73%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 4.53 (d, 2H, NHCH2Ph, J = 6.0 Hz), 6.22 (d, 1H, Ar, J = 8.4 Hz), 7.26 (t, 1H, NH, J = 5.6 Hz), 7.45 (d, 3H, Ar, J = 8.0 Hz), 7.68 (d, 1H, Ar, J = 8.8 Hz), 7.85 (d, 2H, Ar, J = 8.0 Hz), 8.00 (s, 1H, Ar), 8.25 (d, 2H, Ar, J = 9.6 Hz), 12.76 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 48.4 (CH2), 119.8 (CH), 123.3 (CH), 125.0 (CH), 126.5 (CH), 126.8 (CH), 128.2 (CH), 128.6 (CH), 130.1 (CH), 133.2 (C), 134.1 (C), 141.1 (C), 145.1 (C), 169.3 (C). ESI-MS calcd. for C18H15NO5S, 357.07; found: m/z 358.07 [M + H]+. Anal. C18H15NO5S (C, H, N).
3.1.4. 5-(Benzyl(methyl)amino)naphthalene-2-sulfonic Acid (5)
A total of 0.32 mmol of compound 3a and 0.64 mmol of sodium hydride (60% dispersion in mineral oil) were dissolved in 4 mL of dry DMF. After 30 min in stirring, 1.28 mmol of methyl iodide was added and the mixture was stirred at 50–60 °C for 5 h. After cooling, ice-cold water (15 mL) was added and the pH was adjusted to 6–7. The suspension was extracted with CH2Cl2 (3 × 15 mL), dried on sodium sulfate and evaporated. The crude product was purified by flash column chromatography using CH2Cl2/MeOH/NH3 80:20:2 as eluent. Yield = 95%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 2.70 (s, 3H, CH3), 4.24 (s, 2H, CH2-Ph), 7.12 (d, 1H, Ar, J = 7.2 Hz), 7.23 (d, 1H, Ar, J = 6.8 Hz), 7.28–7.39 (m, 5H, Ar), 7.60 (d, 1H, Ar, J = 8.0 Hz), 7.69 (d, 1H, Ar, J = 8.8 Hz), 8.08 (s, 1H, Ar), 8.20 (d, 1H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 42.3 (CH3), 58.4 (CH2), 116.0 (CH), 124.2 (CH), 124.5 (CH), 125.7 (CH), 127.9 (CH), 127.9 (CH), 128.5 (CH), 131.3 (C), 134.8 (C), 138.7 (C), 141.1 (C), 151.1 (C). ESI-MS calcd. for C18H17NO3S, 327.09; found: m/z 328.10 [M + H]+. Anal. C18H17NO3S (C, H, N).
3.1.5. General Procedure for Synthesis of Compounds 7a–g
A total of 1.31 mmol of acid intermediates
6a–
g (6a [
26], 6b [
27], 6c [
28], 6d,f [
29], 6g [
30]) were dissolved in 13 mL of dry DMF, under N
2 flow. Then, 1.31 mmol of hydroxybenzotriazole (HOBt), 2.62 mmol of triethylamine and 1.96 mmol of 5-aminonaphthalene-2-sulfonic acid (1) were added. The mixture was maintained in an ice bath and 1.31 mmol of DCC was added. After 15 min in the ice bath, the mixture was heated at 60 °C for 48 h. Finally, after cooling, the solvent was evaporated and the crude products were purified by flash column chromatography using CH
2Cl
2/MeOH/CH
3COOH 90:10:1 then 80:20:2 as eluents.
5-[4-(N,N-diethylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7a). Yield = 20%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 1.05 (t, 6H, N(CH2CH3)2, J = 7.0 Hz), 3.20 (q, 4H, N(CH2CH3)2, J = 7.2 Hz), 7.55 (t, 1H, Ar, J = 7.8 Hz), 7.61 (d, 1H, Ar, J = 6.8 Hz), 7.72 (dd, 1H, Ar, J1 = 7.2 Hz, J2 = 1.6 Hz), 7.90–7.96 (m, 4H, Ar), 8.18 (d, 1H, Ar, J = 0.8 Hz), 8.24 (d, 2H, Ar, J = 8.0 Hz), 10.68 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 14.6 (CH3), 42.4 (CH2), 123.5 (CH), 124.5 (CH), 124.8 (CH), 124.9 (CH), 126.4 (CH), 127.3 (CH), 127.6 (CH), 129.2 (C), 129.4 (CH), 133.5 (C), 133.8 (C), 138.5 (C), 142.8 (C), 146.1 (C), 165.6 (C). ESI-MS calcd. for C21H22N2O6S2, 462.09; found: m/z 463.10 [M + H]+. Anal. C21H22N2O6S2 (C, H, N).
5-[4-(N,N-dipropylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7b). Yield = 46%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 0.81 (t, 6H, N(CH2CH2CH3)2, J = 7.2 Hz), 1.47 (sex, 4H, N(CH2CH2CH3)2, J = 7.2 Hz), 3.05 (t, 4H, N(CH2CH2CH3)2, J = 7.2 Hz), 7.54 (t, 1H, Ar, J = 6.8 Hz), 7.55–7.60 (m, 1H, Ar), 7.70 (d, 1H, Ar, J = 8.4 Hz), 7.90–8.00 (m, 4H, Ar), 8.17 (s, 1H, Ar), 8.23 (d, 2H, J = 7.6 Hz), 10.65 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 11.5 (CH3), 20.0 (CH2), 52.4 (CH2), 119.9 (CH), 121.5 (CH), 123.3 (CH), 126.5 (CH), 127.5 (CH), 127.6 (CH), 127.3 (CH), 127.6 (CH), 133.2 (C), 137.5 (C), 140.5 (C), 141.0 (C), 143.8 (C), 146.1 (C), 164.7 (C). ESI-MS calcd. for C23H26N2O6S2, 490.12; found: m/z 491.13 [M + H]+. Anal. C23H26N2O6S2 (C, H, N).
5-[4-(N,N-dibutylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7c). Yield = 46%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 0.88 (t, 6H, N(CH2CH2CH2CH3)2, J = 7.2 Hz), 1.22–1.31 (m, 4H, N(CH2CH2CH2CH3)2), 1.43–1.50 (m, 4H, N(CH2CH2CH2CH3)2), 3.10 (t, 4H, N(CH2CH2CH2CH3)2, J = 7.4 Hz), 7.56 (t, 1H, Ar, J = 7.6 Hz), 7.62 (d, 1H, Ar, J = 7.2 Hz), 7.72 (d, 1H, Ar, J = 8.8 Hz), 7.90–8.00 (m, 4H, Ar), 8.18 (s, 1H, Ar), 8.26 (d, 2H, Ar, J = 8.0 Hz), 10.66 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 14.0 (CH3), 19.7 (CH2), 30.9 (CH2), 48.2 (CH2), 123.5 (CH), 124.4 (CH), 124.9 (CH), 126.5 (CH), 127.4 (CH), 127.7 (CH), 129.2 (CH), 129.3 (CH), 133.4 (C), 133.6 (C), 138.4 (C), 142.3 (C), 145.9 (C), 165.6 (C). ESI-MS calcd. for C25H30N2O6S2, 518.15; found: m/z 519.16 [M + H]+. Anal. C25H30N2O6S2 (C, H, N).
5-[4-(N,N-dibenzylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7d). Yield = 31%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 4.34 (s, 4H, N(CH2Ph)2), 7.08–7.21 (m, 10H, Ar), 7.56 (t, 1H, Ar, J = 7.6 Hz), 7.63 (d, 1H, Ar, J = 7.2 Hz), 7.73 (d, 1H, Ar, J = 8.4 Hz), 7.94 (t, 2H, Ar, J = 9.0 Hz), 8.05 (d, 2H, Ar, J = 8.4 Hz), 8.18 (s, 1H, Ar), 8.27 (d, 2H, Ar, J = 8.0 Hz), 10.69 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 51.9 (CH2), 123.5 (CH), 124.9 (CH), 126.2 (CH), 126.5 (CH), 127.9 (CH), 128.7 (CH), 129.1 (CH), 129.3 (C), 129.4 (CH), 133.5 (C), 133.9 (C), 136.5 (C), 138.7 (C), 142.4 (C), 146.0 (C), 165.5 (C). ESI-MS calcd. for C31H26N2O6S2, 586.12; found: m/z 587.13 [M + H]+. Anal. C31H26N2O6S2 (C, H, N).
5-[4-(N,N-diphenethylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7e). Yield = 20%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 2.78 (t, 4H, N(CH2CH2Ph)2, J = 7.8 Hz), 3.38 (t, 4H, N(CH2CH2Ph)2, J = 7.8 Hz), 7.21–7.31 (m, 10H, Ar), 7.54 (t, 1H, Ar, J = 7.8 Hz), 7.61 (d, 1H, Ar, J = 7.2 Hz), 7.70 (dd, 1H, Ar, J1 = 8.8 Hz, J2 = 1.2 Hz), 7.93 (t, 2H, Ar, J = 8.8 Hz), 7.99 (d, 2H, Ar, J = 8.4 Hz), 8.17 (s, 1H, Ar), 8.24 (d, 2H, Ar, J = 8.4 Hz), 10.65 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 35.3 (CH2), 50.1 (CH2), 123.4 (CH), 124.5 (CH), 124.7 (CH), 124.8 (CH), 126.4 (CH), 126.9 (CH), 127.5 (CH), 127.6 (CH), 128.9 (CH), 129.2 (C), 129.3 (CH), 129.5 (CH), 133.5 (C), 133.8 (C), 138.7 (C), 138.9 (C), 142.2 (C), 146.3 (C), 165.5 (C). ESI-MS calcd. for C33H30N2O6S2, 614.15; found: m/z 615.16 [M + H]+. Anal. C33H30N2O6S2 (C, H, N).
5-[4-(N-benzyl-N-phenethylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7f). Yield = 27%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 2.55 (t, 2H, NCH2CH2Ph, J = 8.0 Hz), 3.29 (t, 2H, NCH2CH2Ph, J = 8.0 Hz), 4.43 (s, 2H, NCH2Ph), 7.00 (d, 2H, Ar, J = 6.8 Hz), 7.16–7.24 (m, 3H, Ar), 7.30–7.38 (m, 5H, Ar), 7.56 (t, 1H, Ar, J = 7.8 Hz), 7.62 (d, 1H, Ar, J = 7.2 Hz), 7.72 (d, 1H, Ar, J = 8.8 Hz), 7.94 (t, 2H, Ar, J = 8.8 Hz), 8.05 (d, 2H, Ar, J = 8.4 Hz), 8.18 (s, 1H, Ar), 8.27 (d, 2H, Ar, J = 8.0 Hz), 10.70 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 34.9 (CH2), 49.9 (CH2), 51.9 (CH2), 123.4 (CH), 124.5 (CH), 124.8 (CH), 124.9 (CH), 126.4 (CH), 126.8 (CH), 127.5 (CH), 128.2 (CH), 128.8 (CH), 128.9 (CH), 129.0 (CH), 129.2 (C), 129.5 (CH), 133.5 (C), 133.9 (C), 137.2 (C), 138.6 (C), 138.7 (C), 142.3 (C), 146.2 (C), 165.5 (C). ESI-MS calcd. for C32H28N2O6S2, 600.14; found: m/z 601.14 [M + H]+. Anal. C32H28N2O6S2 (C, H, N).
5-[4-(N-phenethylsulfamoyl)benzamido]naphthalene-2-sulfonic acid (7g). Yield = 21%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 2.65–2.75 (m, 2H, NCH2CH2Ph), 3.00–3.10 (m, 2H, NCH2CH2Ph), 7.10–7.20 (m, 2H, Ar), 7.27 (d, 2H, Ar, J = 6.8 Hz), 7.56 (t, 1H, Ar, J = 7.2 Hz), 7.61 (d, 1H, Ar, J = 7.8 Hz), 7.71 (d, 2H, SO2NH + 1H Ar, J = 8.0 Hz), 7.90–8.00 (m, 5H, Ar), 8.18 (s, 1H, Ar), 8.23 (d, 2H, Ar, J = 6.8 Hz), 10.63 (exch br s, 1H, NH). 13C-NMR (100 MHz, DMSO-d6) δ 35.8 (CH2), 44.6 (CH2), 123.4 (CH), 124.5 (CH), 124.6 (CH), 124.8 (CH), 126.4 (CH), 126.7 (CH), 127.1 (CH), 127.6 (CH), 128.8 (CH), 129.1 (CH), 129.2 (CH), 133.5 (C), 133.9 (C), 138.4 (C), 139.1 (C), 143.4 (C), 146.3 (C), 165.6 (C). ESI-MS calcd. for C25H22N2O6S2, 510.09; found: m/z 511.10 [M + H]+. Anal. C25H22N2O6S2 (C, H, N).
3.1.6. 5-Benzamidonaphthalene-2-sulfonic Acid (8)
A total of 0.45 mmol of 5-aminonaphthalene-2-sulfonic acid (
1) was dissolved in water (2 mL) and the pH wad adjusted to 3-4. To the stirred solution, 0.63 mmol of benzoyl chloride dissolved in 1 mL of toluene was slowly added. The reaction mixture was kept at a constant pH of 4 by automatic addition of a 2 M Na
2CO
3 solution at room temperature for 6 h. After separating the toluene from the water phase, pH was adjusted to 2.0 with HCl 6N and the aqueous phase was extracted with diethyl ether (4 × 15 mL). The organic phase was removed, the aqueous phase was neutralised (pH = 7) with 2M Na
2CO
3 solution and then evaporated under vacuum. The crude product was purified by recrystallisation from methanol to obtain the desired compound [
25].
3.1.7. Sodium 5-[4-(N,N-dibutylsulfamoyl)benzamido]naphthalene-2-sulfonate (9)
To a fresh solution of sodium ethylate (0.193 mmol of Na° in 1 mL of absolute ethanol), 0.193 mmol of compound 7c was added. The mixture was stirred at room temperature for 1 h. The solvent was evaporated affording the corresponding sodium salt. Yield = 99%. 1H-NMR (400 MHz, D2O-d2) δ 0.65–0.90 (m, 6H, N(CH2CH2CH2CH3)2), 1.10–1.29 (m, 4H, N(CH2CH2CH2CH3)2), 1.30–1.50 (m, 4H, N(CH2CH2CH2CH3)2), 3.00–3.25 (m, 4H, N(CH2CH2CH2CH3)2), 7.40–7.60 (m, 2H, Ar), 7.65–7.80 (m, 2H, Ar), 7.80–8.15 (m, 5H, Ar), 8.20–8.40 (m, 1H, Ar). Anal. C25H29N2NaO6S2 (C, H, N).
3.1.8. General Procedure for Synthesis of Compounds 11 and 12
A mixture of 1.19 mmol of ethyl 4-sulfamoyl benzoate
10 [
31] and 2.38 mmol of anhydrous K
2CO
3 in 20 mL of dry DMF was stirred at room temperature for 30 min. Then 5.58 mmol of 2-phenethyl bromide (
2b) or methyl 4-(bromomethyl)benzoate (
2f), respectively, were added and the mixture was heated at 80 °C for 4 h. After cooling, the solvent was evaporated and cold H
2O was added. The suspension was extracted with ethyl acetate (3 × 15 mL) and the collected organic phase was recovered, dried over sodium sulfate and evaporated in vacuum. The crude product was purified by flash column chromatography using cyclohexane/ethyl acetate 1:2 (for 11) or 3:1 (for 12) as eluent.
Ethyl 4-(N,N-diphenethylsulfamoyl)benzoate (11). Yield = 12%; mp = 84–87 °C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 1.41 (t, 3H, COOCH2CH3, J = 7.0 Hz), 2.83 (t, 4H, N(CH2CH2Ph)2, J = 7.8 Hz), 3.40 (t, 4H, N(CH2CH2Ph)2, J = 7.8 Hz), 4.41 (q, 2H, COOCH2CH3, J = 7.0 Hz), 7.13 (d, 4H, Ar, J = 7.2 Hz), 7.21–7.30 (m, 6H, Ar), 7.83 (d, 2H, Ar, J = 8.0 Hz), 8.13 (d, 2H, Ar, J = 8.0 Hz). ESI-MS calcd. for C25H27NO4S, 437.55; found: m/z 438.17 [M + H]+. Anal. C25H27NO4S (C, H, N).
Dimethyl 4,4’-{{{[4-(ethoxycarbonyl)phenyl]sulfonyl}azanediyl}bis(methylene)}dibenzoate (12). Yield = 66%; mp = 141–144 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 1.45 (t, 3H, COOCH2CH3, J = 7.0 Hz), 3.90 (s, 6H, 2 × COOCH3), 4.38 (s, 4H, N(CH2PhCOOCH3)2), 4.45 (q, 2H, COOCH2CH3, J = 7.2 Hz), 7.09 (d, 4H, Ar, J = 8.4 Hz), 7.88 (d, 4H, Ar, J = 8.4 Hz), 7.93 (d, 2H, Ar, J = 8.4 Hz), 8.20 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C27H27NO8S, 525.57; found: m/z 526.15 [M + H]+. Anal. C27H27NO8S (C, H, N).
3.1.9. General Procedure for Synthesis of Compounds 6e and 13
A mixture of 0.72 mmol of compounds 11 or 12, 40% NaOH (19.5 mL) and 0.5 mL of EtOH 96% was stirred at reflux for 2 h. After cooling and maintaining the reaction in ice bath, HCl 6M was added up to acid pH and the precipitate was filtered off to obtain the desired product.
4-(N,N-Diphenethylsulfamoyl)benzoic acid (6e). Yield = 42%; mp = 167–170 °C dec. (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 2.72 (t, 4H, N(CH2CH2Ph)2, J = 7.4 Hz), 3.33 (t, 4H, N(CH2CH2Ph)2, J = 7.4 Hz), 7.25–7.28 (m, 4H, Ar), 7.87 (d, 2H, Ar, J = 8.0 Hz), 8.06 (d, 2H, Ar, J = 8.0 Hz), 13.35 (exch br s, 1H, COOH). ESI-MS calcd. for C23H23NO4S, 409.50; found: m/z 410.14 [M + H]+. Anal. C23H23NO4S (C, H, N).
4,4′-{{[(4-Carboxyphenyl)sulfonyl]azanediyl}bis(methylene)}dibenzoic acid (13). Yield = 99%; mp > 300 °C (EtOH/H2O). 1H-NMR (400 MHz, DMSO-d6) δ 4.41 (s, 4H, N(CH2PhCOOH)2), 7.19 (d, 4H, Ar, J = 7.6 Hz), 7.72 (d, 4H, Ar, J = 7.6 Hz), 7.99 (d, 2H, Ar, J = 8.0 Hz), 8.11 (d, 2H, Ar, J = 8.0 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 52.2 (CH2), 127.8 (CH2), 128.7 (CH), 124.4 (CH), 129.6 (CH), 130.4 (CH), 130.7 (CH), 135.7 (C), 141.5 (C), 142.8 (C), 166.7 (C), 167.4 (C). ESI-MS calcd. for C23H19NO8S, 469.08; found: m/z 470.09 [M + H]+. Anal. C23H19NO8S (C, H, N).
3.1.10. General Procedure for Synthesis of Compounds 15a–c and 16a–c
A mixture of 0.64 mmol of 5-amino-1-(4-sulfamoylphenyl)-1H-pyrazole-4-carboxylate
14 [
33] and 1.28 mmol of anhydrous K
2CO
3 in 5 mL of dry DMF was stirred at room temperature for 30 min. Then, 0.96 mmol of appropriate alkyl halide (iodoethane, 1-bromopropane or 1-bromobutane) were added and the mixture was heated at 80 °C for 4 h (for the synthesis of
16a–
c an excess (3 equiv.) of alkyl halide was used). After cooling, the solvent was evaporated and the crude product was purified by flash column chromatography using cyclohexane/ethyl acetate 2:1 as eluent.
Ethyl 5-amino-1-(4-(N-ethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (15a). Yield = 37%; mp = 166–168 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 1.02 (t, 3H, SO2NH(CH2CH3), J = 7.2 Hz), 1.28 (t, 3H, COOCH2CH3, J = 7.0 Hz), 2.83 (quin, 2H, SO2NH(CH2CH3), J = 7.2 Hz), 4.23 (q, 2H, COOCH2CH3, J = 7.0 Hz), 6.56 (exch br s, 2H, NH2), 7.68 (t, 1H, SO2NH, J = 5.8 Hz), 7.77 (s, 1H, pyrazole), 7.80 (d, 2H, Ar, J = 8.8 Hz), 7.92 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C14H18N4O4S, 338.10; found: m/z 339.11 [M + H]+. Anal. C14H18N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N-propylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (15b). Yield = 61%; mp = 123–126 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 0.81 (t, 3H, SO2NH(CH2CH2CH3), J = 7.4 Hz), 1.24 (t, 3H, COOCH2CH3, J = 7.2 Hz), 1.33–1.42 (m, 2H, SO2NH(CH2CH2CH3)), 2.71 (q, 2H, SO2NH(CH2CH2CH3), J = 6.8 Hz), 4.19 (q, 2H, COOCH2CH3, J = 7.2 Hz), 6.53 (exch br s, 2H, NH2), 7.68 (t, 1H, SO2NH, J = 5.6 Hz), 7.76 (d, 3H, 2H Ar + 1H pyrazole, J = 8.8 Hz), 7.88 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C15H20N4O4S, 352.12; found: m/z 353.12 [M + H]+. Anal. C15H20N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N-butylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (15c). Yield = 14%; mp = 130–132 °C (EtOH/H2O). 1H-NMR (400 MHz, CDCl3) δ 0.86 (t, 3H, NHCH2CH2CH2CH3, J = 7.2 Hz), 1.27–1.38 (m, 5H, 3H COOCH2CH3 + 2H NHCH2CH2CH2CH3), 1.45 (quin, 2H, NHCH2CH2CH2CH3, J = 7.2 Hz), 2.97 (t, 2H, NHCH2CH2CH2CH3, J = 7.0 Hz), 4.30 (q, 2H, COOCH2CH3, J = 7.0 Hz), 7.74 (d, 2H, Ar, J = 8.0 Hz), 7.82 (s, 1H, pyrazole), 7.98 (d, 2H, Ar, J = 7.6 Hz). ESI-MS calcd. for C16H22N4O4S, 366.14; found: m/z 367.14 [M + H]+. Anal. C16H22N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N,N-diethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (16a). Yield = 30%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 1.08 (t, 6H, SO2N(CH2CH3)2, J = 7.2 Hz), 1.28 (t, 3H, COOCH2CH3, J = 7.0 Hz), 3.22 (q, 4H, SO2N(CH2CH3)2, J = 7.2 Hz), 4.23 (q, 2H, COOCH2CH3, J = 7.0 Hz), 6.58 (exch br s, 2H, NH2), 7.80 (d, 3H, 2H Ar + 1H pyrazole, J = 6.8 Hz), 7.92 (d, 2H, Ar, J1 = 6.8 Hz and J2 = 2.0 Hz). ESI-MS calcd. for C16H22N4O4S, 366.14; found: m/z 367.14 [M + H]+. Anal. C16H22N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N,N-dipropylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (16b). Yield = 14%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 0.81 (t, 6H, SO2N(CH2CH2CH3)2, J = 7.2 Hz), 1.24 (t, 3H, COOCH2CH3, J = 7.2 Hz), 1.47 (sex, 4H, SO2N(CH2CH2CH3)2, J = 7.6 Hz), 3.03 (t, 4H, SO2N(CH2CH2CH3)2, J = 7.6 Hz), 4.19 (q, 2H, COOCH2CH3, J = 7.2 Hz), 6.54 (exch br s, 2H, NH2), 7.76 (d, 3H, 2H Ar + 1H pyrazole, J = 9.2 Hz), 7.89 (d, 2H, Ar, J = 8.8 Hz). ESI-MS calcd. for C18H26N4O4S, 394.17; found: m/z 395.17 [M + H]+. Anal. C18H26N4O4S (C, H, N).
Ethyl 1-(4-(N,N-dipropylsulfamoyl)phenyl)-5-(propylamino)-1H-pyrazole-4-carboxylate (16c). Yield = 33%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 0.66 (t, 3H, NHCH2CH2CH3, J = 7.4 Hz), 0.77 (t, 6H, SO2N(CH2CH2CH3)2, J = 7.4 Hz), 1.24–1.32 (m, 5H, 3H COOCH2CH3 + 2H NHCH2CH2CH3), 1.43 (quin, 4H, SO2N(CH2CH2CH3)2, J = 7.2 Hz), 2.74 (q, 2H, NHCH2CH2CH3, J = 6.8 Hz), 3.05 (t, 4H, SO2N(CH2CH2CH3)2, J = 7.4 Hz), 4.21 (q, 2H, COOCH2CH3, J = 7.2 Hz), 6.25 (t, 1H, NH, J = 6.2 Hz), 7.74 (d, 2H, Ar, J = 8.4 Hz), 7.81 (s, 1H, pyrazole), 7.92 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C21H32N4O4S, 436.21; found: m/z 437.22 [M + H]+. Anal. C21H32N4O4S (C, H, N).
3.1.11. General Procedure for Synthesis of Acid Derivatives 17, 18a–c and 19a–c
A mixture of 0.27 mmol of appropriate ethyl ester derivatives 14, 15a–c or 16a–c, 0.30 mL of NaOH 6N and 1.50 mL of 96% ethanol was stirred at 100 °C for 4 h. After cooling and maintaining the reaction in ice bath, HCl 6M was added up to acid pH and the precipitate was filtered off to obtain the desired acid products which were purified by crystallisation with ethanol.
5-Amino-1-(4-sulfamoylphenyl)-1H-pyrazole-4-carboxylic acid (17). Yield = 48%; mp = 210–211 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 6.48 (exch br s, 2H, NH2), 7.46 (exh br s, 2H, SO2NH2), 7.71 (s, 1H, pyrazole), 7.74 (d, 2H, Ar, J = 8.8 Hz), 7.92 (d, 2H, Ar, J = 8.4 Hz), 12.16 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 96.2 (C), 123.7 (CH), 127.4 (CH), 141.0 (C), 141.7 (CH), 142.8 (C), 150.8 (C), 165.6 (C). ESI-MS calcd. for C10H10N4O4S, 282.04; found: m/z 283.05 [M + H]+. Anal. C10H10N4O4S (C, H, N).
5-Amino-1-(4-(N-ethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (18a). Yield = 75%; mp = 208–210 °C dec. (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 1.03 (t, 3H, SO2NH(CH2CH3), J = 7.2 Hz), 2.83 (quin, 2H, SO2NH(CH2CH3), J = 7.2 Hz), 6.52 (exch br s, 2H, NH2), 7.68 (t, 1H, SO2NH, J = 5.8 Hz), 7.75 (s, 1H, pyrazole), 7.80 (d, 2H, Ar, J = 8.8 Hz), 7.92 (d, 2H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 14.1 (CH3), 37.4 (CH2), 109.0 (C), 123.0 (CH), 127.9 (CH), 140.6 (CH), 142.0 (C), 142.9 (C), 150.2 (C), 164.9 (C). ESI-MS calcd. for C12H14N4O4S, 310.07; found: m/z 311.08 [M + H]+. Anal. C12H14N4O4S (C, H, N).
5-Amino-1-(4-(N-propylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (18b). Yield = 98%; mp = 215–218 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 0.79 (t, 3H, NHCH2CH2CH3, J = 7.6 Hz), 1.38 (sex, 2H, NHCH2CH2CH3, J = 7.2 Hz), 2.71 (q, 2H, NHCH2CH2CH3, J = 6.8 Hz), 6.48 (exch br s, 2H, NH2), 7.31 (exch br s, 1H, SO2NH), 7.72 (s, 1H, pyrazole), 7.76 (d, 2H, Ar, J = 8.4 Hz), 7.88 (d, 2H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 11.5 (CH3), 22.7 (CH2), 44.7 (CH2), 124.0 (2 × CH), 128.6 (2 × CH), 139.2 (C), 141.1 (CH), 141.9 (CH), 150.5 (C), 165.5 (C). ESI-MS calcd. for C13H16N4O4S, 324.09; found: m/z 325.09 [M + H]+. Anal. C13H16N4O4S (C, H, N).
5-Amino-1-(4-(N-butylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (18c). Yield = 92%; mp > 300 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 0.78 (t, 3H, NHCH2CH2CH2CH3, J = 7.0 Hz), 1.20–1.26 (m, 2H, NHCH2CH2CH2CH3), 1.33–1.38 (m, 2H, NHCH2CH2CH2CH3), 2.74 (q, 2H, NHCH2CH2CH2CH3, J = 6.4 Hz), 6.49 (exch br s, 2H, NH2), 7.65 (t, 1H, SO2NH, J = 5.6 Hz), 7.72 (s, 1H, pyrazole), 7.76 (d, 2H, Ar, J = 8.4 Hz), 7.88 (d, 2H, Ar, J = 8.4 Hz), 12.16 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 13.8 (CH3), 19.6 (CH2), 31.4 (CH2), 42.6 (CH2), 123.9 (2 × CH), 128.8 (2 × CH), 139.2 (C), 141.3 (C), 141.9 (CH), 150.6 (C), 165.5 (C). ESI-MS calcd. for C14H18N4O4S, 338.10; found: m/z 339.11 [M + H]+. Anal. C14H18N4O4S (C, H, N).
5-Amino-1-(4-(N,N-diethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (19a). Yield = 47%; mp = 240 °C dec. (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 1.08 (t, 6H, SO2N(CH2CH3)2, J = 7.2 Hz), 3.20 (q, 4H, SO2N(CH2CH3)2, J = 7.2 Hz), 6.52 (exch br s, 2H, NH2), 7.70 (s, 1H, pyrazole), 7.82 (d, 2H, Ar, J = 8.8 Hz), 7.91 (d, 2H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 10.7 (CH3), 38.9 (CH2), 109.0 (C), 123.0 (CH), 127.9 (CH), 137.2 (C), 140.6 (CH), 142.9 (C), 150.2 (C), 164.9 (C). ESI-MS calcd. for C14H18N4O4S, 338.10; found: m/z 339.11 [M + H]+. Anal. C14H18N4O4S (C, H, N).
5-Amino-1-(4-(N,N-dipropylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (19b). Yield = 42%; mp = 167–168 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 0.80 (t, 6H, N(CH2CH2CH3)2, J = 7.4 Hz), 1,48 (quin, 4H, N(CH2CH2CH3)2, J = 7.2 Hz), 3.03 (t, 4H, N(CH2CH2CH3)2, J = 7.4 Hz), 6.50 (exch br s, 2H, NH2), 7.72 (s, 1H, pyrazole), 7.77 (d, 2H, Ar, J = 6.8 Hz), 7.89 (d, 2H, Ar, J = 6.8 Hz), 12.17 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 11.4 (CH3), 22.2 (CH2), 50.3 (CH2), 123.8 (CH), 128.7 (CH), 138.0 (C), 141.6 (C), 142.0 (CH), 165.6 (C). ESI-MS calcd. for C16H22N4O4S, 366.14; found: m/z 367.14 [M + H]+. Anal. C16H22N4O4S (C, H, N).
1-(4-(N,N-dipropylsulfamoyl)phenyl)-5-(propylamino)-1H-pyrazole-4-carboxylic acid (19c). Yield = 49%; mp = 170–172 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 0.66 (t, 3H, NHCH2CH2CH3, J = 7.4 Hz), 0.77 (t, 6H, SO2N(CH2CH2CH3)2, J = 7.2 Hz), 1.28 (quin, 4H, SO2N(CH2CH2CH3)2, J = 6.8 Hz), 1.44 (quin, 2H, NHCH2CH2CH3, J = 7.2 Hz), 2.70–2.75 (m, 2H, NHCH2CH2CH3), 3.05 (t, 4H, SO2N(CH2CH2CH3)2, J = 7.4 Hz), 6.33 (exch br s, 1H, NH), 7.73–7.78 (m, 3H, 2H Ar + 1H pyrazole), 7.91 (d, 2H, Ar, J = 8.4 Hz), 12.23 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 11.1 (CH3), 11.4 (CH3), 21.7 (CH2), 23.1 (CH2), 47.4 (CH2), 49.7 (CH2), 125.4 (2 × CH), 128.4 (2 × CH), 139.2 (C), 142.1 (CH), 142.8 (C), 152.7 (C), 167.4 (C). ESI-MS calcd. for C19H28N4O4S, 408.18; found: m/z 409.19 [M + H]+. Anal. C19H28N4O4S (C, H, N).
3.1.12. General Procedure for Synthesis of Compounds 20–22 and 25–27
Compounds 20–22 and 25–27 were obtained following the same synthetic procedure used for compounds 15a–c and 16a–c but using the appropriate reagent (benzyl bromide for 20–22 and 27 and phenethyl bromide for 25 and 26). The crude compounds were purified by flash chromatography column using cyclohexane/ethyl acetate 2:1 as eluent.
Ethyl 5-amino-1-(4-(N-benzylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (20). Yield = 23%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 1.27 (t, 3H, COOCH2CH3, J = 7.0 Hz), 4.02 (d, 2H, NHCH2Ph, J = 6.0 Hz), 4.22 (q, 2H, COOCH2CH3, J = 7.0 Hz), 6.53 (exch br s, 2H, NH2), 7.25–7.30 (m, 5H, Ar), 7.76 (d, 3H, 2H Ar + 1H pyrazole, J = 8.0 Hz), 7.93 (d, 2H, Ar, J = 8.4 Hz), 8.26 (t, 1H, SO2NH, J = 6.2 Hz). ESI-MS calcd. for C19H20N4O4S, 400.12; found: m/z 401.12 [M + H]+. Anal. C19H20N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N,N-dibenzylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (21). Yield = 42%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 1.27 (t, 3H, COOCH2CH3, J = 6.8 Hz), 4.22 (q, 2H, COOCH2CH3, J = 6.8 Hz), 4.33 (s, 4H, N(CH2Ph)2), 6.57 (exch br s, 2H, NH2), 7.10–7.36 (m, 10H, Ar), 7.80 (d, 3H, 2H Ar + 1H pyrazole, J = 7.2 Hz), 8.00 (d, 2H, Ar, J = 8.0 Hz). ESI-MS calcd. for C26H26N4O4S, 490.17; found: m/z 491.17 [M + H]+. Anal. C26H26N4O4S (C, H, N).
Ethyl 5-(benzylamino)-1-(4-(N,N-dibenzylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (22). Yield = 10%; oil. 1H-NMR (400 MHz, DMSO-d6) δ 1.25 (t, 3H, COOCH2CH3, J = 7.2 Hz), 3.94 (d, 2H, NHCH2Ph, J = 5.6 Hz), 4.22 (q, 2H, COOCH2CH3, J = 7.2 Hz), 4.35 (s, 4H, SO2N(CH2Ph)2), 6.78 (t, 1H, NHCH2Ph, J = 5.6 Hz), 7.10–7.40 (m, 15H, Ar), 7.71 (d, 2H, Ar, J = 8.0 Hz), 7.82 (s, 1H, pyrazole), 8.00 (d, 2H, Ar, J = 8.0 Hz). ESI-MS calcd. for C33H32N4O4S, 580.21; found: m/z 581.22 [M + H]+. Anal. C33H32N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (25). Yield = 35%; mp = 108–111 °C (EtOH). 1H-NMR (400 MHz, CDCl3) δ 1.38 (t, 3H, COOCH2CH3, J = 7.0 Hz), 2.81 (t, 2H, NHCH2CH2Ph, J = 6.8 Hz), 3.28 (q, 2H, NHCH2CH2Ph, J = 6.8 Hz), 4.32 (q, 2H, COOCH2CH3, J = 7.0 Hz), 4.48 (t, 1H, SO2NH, J = 6.0 Hz), 5.43 (exch br s, 2H, NH2), 7.10 (d, 2H, Ar, J = 6.8 Hz), 7.23–7.30 (m, 3H, Ar), 7.50–7.60 (m, 1H, Ar), 7.73 (d, 2H, Ar, J = 8.4 Hz), 7.82 (s, 1H, pyrazole), 7.93 (d, 2H, Ar, J = 8.4 Hz). ESI-MS calcd. for C20H22N4O4S, 414.14; found: m/z 415.14 [M + H]+. Anal. C20H22N4O4S (C, H, N).
Ethyl 5-(phenethylamino)-1-(4-(N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (26). Yield = 13%; oil. 1H-NMR (400 MHz, CDCl3) δ 1.37 (t, 3H, COOCH2CH3, J = 7.0 Hz), 2.77 (t, 2H, SO2NHCH2CH2Ph, J = 7.0 Hz), 2.85–2.90 (m, 2H, NHCH2CH2Ph), 3.41 (t, 2H, NHCH2CH2Ph, J = 7.8 Hz), 3.86 (t, 2H, SO2NHCH2CH2Ph, J = 6.6 Hz), 4.31 (q, 2H, COOCH2CH3, J = 7.0 Hz), 5.47 (exch br s, 1H, NH), 7.15–7.30 (m, 10H, Ar), 7.49 (d, 3H, 2H Ar + 1H pyrazole, J = 7.2 Hz), 7.70 (exch br s, 1H, SO2NH), 7.82 (d, 2H, Ar, J = 7.2 Hz). ESI-MS calcd. for C28H30N4O4S, 518.20; found: m/z 519.20 [M + H]+. Anal. C20H22N4O4S (C, H, N).
Ethyl 5-amino-1-(4-(N-benzyl-N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylate (27). Yield = 65%; mp = 131–134 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 1.26 (t, 3H, COOCH2CH3, J = 7.0 Hz), 2.55 (t, 2H, NCH2CH2Ph, J = 8.0 Hz), 3.27 (t, 2H, NCH2CH2Ph, J = 8.0 Hz), 4.21 (q, 2H, COOCH2CH3, J = 7.0 Hz), 4.40 (s, 2H, NCH2Ph), 6.58 (exch br s, 2H, NH2), 6.99 (d, 2H, Ar, J = 7.2 Hz), 7.10–7.20 (m, 4H, Ar), 7.30–7.40 (m, 4H, Ar), 7.79 (d, 3H, 2H Ar + 1H pyrazole, J = 10.0 Hz), 7.99 (d, 2H, Ar, J = 8.0 Hz). ESI-MS calcd. for C27H28N4O4S, 504.18; found: m/z 505.19 [M + H]+. Anal. C27H28N4O4S (C, H, N).
3.1.13. General Procedure for Synthesis of Acid Derivatives 23, 24 and 28–30
Compounds 23, 24 and 28–30 were obtained following the same alkaline hydrolysis used for acids 17, 18a–c and 19a–c. After cooling and maintaining the reaction in ice bath, HCl 6M was added up to acid pH and the precipitate was filtered off to obtain the desired acid products which were purified by crystallisation with ethanol.
5-Amino-1-(4-(N-benzylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (23). Yield = 40%; mp = 207–210 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 4.02 (d, 2H, NHCH2Ph, J = 6.0 Hz), 6.48 (exch br s, 2H, NH2), 7.25–7.30 (m, 5H, Ar), 7.74 (s, 1H, pyrazole), 7.77 (d, 2H, Ar, J = 7.6 Hz), 7.92 (d, 2H, Ar, J = 7.6 Hz), 8.25 (t, 1H, NHCH2Ph, J = 6.0 Hz), 12.14 (exch br s, 1H, COOH). 13C-NMR (100 MHz, DMSO-d6) δ 46.2 (CH2), 109.0 (CH), 123.0 (CH), 126.7 (CH), 126.9 (CH), 127.9 (CH), 128.5 (CH), 140.6 (CH), 141.6 (CH), 142.2 (C), 142.9 (C), 150.2 (C), 164.9 (C). ESI-MS calcd. for C17H16N4O4S, 372.09; found: m/z 373.09 [M + H]+. Anal. C17H16N4O4S (C, H, N).
5-Amino-1-(4-(N,N-dibenzylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (24). Yield = 20%; mp = 168–170 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 4.32 (s, 4H, SO2N(CH2Ph)2), 6.54 (exch br s, 2H, NH2), 7.05–7.15 (m, 4H, Ar), 7.19–7.25 (m, 6H, Ar), 7.72 (s, 1H, pyrazole), 7.83 (d, 2H, Ar, J = 8.4 Hz), 7.99 (d, 2H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 51.9 (CH2), 109.0 (C), 123.3 (CH), 127.9 (CH), 128.7 (CH), 128.9 (CH), 136.6 (C), 137.4 (C), 140.6 (CH), 142.9 (C), 150.5 (C), 164.9 (C). ESI-MS calcd. for C24H22N4O4S, 462.14; found: m/z 463.14 [M + H]+. Anal. C24H22N4O4S (C, H, N).
5-Amino-1-(4-(N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (28). Yield = 13%; mp = 186–188 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 2.71 (t, 2H, NHCH2CH2Ph, J = 7.4 Hz), 3.00 (q, 2H, NHCH2CH2Ph, J = 7.0 Hz), 6.49 (exch br s, 2H, NH2), 7.15–7.20 (m, 3H, Ar), 7.26 (t, 2H, Ar, J = 7.4 Hz), 7.73 (s, 1H, pyrazole), 7.77 (d, 2H, Ar, J = 8.8 Hz), 7.82 (t, 1H, SO2NH, J = 5.8 Hz), 7.89 (d, 2H, Ar, J = 8.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 35.7 (CH2), 42.4 (CH2), 109.0 (C), 123.8 (CH), 126.7 (CH), 128.4 (CH), 128.8 (CH), 129.1 (CH), 139.1 (C), 141.4 (C), 141.9 (CH), 150.2 (C), 164.9 (C). ESI-MS calcd. for C18H18N4O4S, 386.10; found: m/z 387.11 [M + H]+. Anal. C18H18N4O4S (C, H, N).
5-(Phenethylamino)-1-(4-(N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (29). Yield = 18%; mp = 105–107 °C (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 2.65 (t, 2H, NHCH2CH2Ph, J = 7.2 Hz), 2.77 (t, 2H, NHCH2CH2Ph, J = 7.2 Hz), 2.94 (t, 2H, SO2NHCH2CH2Ph, J = 6.4 Hz), 3.35 (t, 2H, SO2NHCH2CH2Ph, J = 7.6 Hz), 6.50 (exch br s, 1H, NHCH2CH2Ph), 7.10–7.30 (m, 10H, Ar), 7.55–7.62 (m, 3H, 2H Ar + 1H pyrazole), 7.70 (exch br s, 1H, SO2NH), 7.81 (d, 2H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 35.3 (CH2), 35.6 (CH2), 44.4 (CH2), 50.9 (CH2), 109.0 (C), 123.1 (CH), 126.7 (CH), 126.9 (CH), 127.9 (C), 128.8 (CH), 128.9 (CH), 129.1 (CH), 129.2 (CH), 129.7 (CH), 132.9 (CH), 139.4 (C), 142.0 (C), 142.9 (C), 151.2 (C), 164.9 (C). ESI-MS calcd. for C26H26N4O4S, 490.17; found: m/z 491.17 [M + H]+. Anal. C26H26N4O4S, (C, H, N).
5-Amino-1-(4-(N-benzyl-N-phenethylsulfamoyl)phenyl)-1H-pyrazole-4-carboxylic acid (30). Yield = 59%; mp = 263–266 °C dec. (EtOH). 1H-NMR (400 MHz, DMSO-d6) δ 2.55 (t, 2H,NCH2CH2Ph, J = 7.4 Hz), 3.26 (t, 2H, NHCH2CH2Ph, J = 7.6 Hz), 4.39 (s, 2H,NCH2Ph), 6.53 (exch br s, 2H, NH2), 6.99 (d, 2H, Ar, J = 7.2 Hz), 7.15–7.20 (m, 3H, Ar), 7.31–7.36 (m, 5H, Ar), 7.61 (s, 1H, pyrazole), 7.86 (d, 2H, Ar, J = 8.0 Hz), 7.95 (d, 2H, Ar, J = 8.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 34.9 (CH2), 49.9 (CH2), 52.0 (CH2), 103.4 (C), 122.6 (CH), 126.8 (CH), 128.2 (CH), 128.7 (C), 128.9 (CH), 128.9 (CH), 129.0 (CH), 136.6 (C), 137.2 (C), 138.6 (C), 142.8 (CH), 142.9 (C), 149.7 (C), 168.4 (C). ESI-MS calcd. for C25H24N4O4S, 476.15; found: m/z 477.16 [M + H]+. Anal. C25H24N4O4S (C, H, N).


 ,
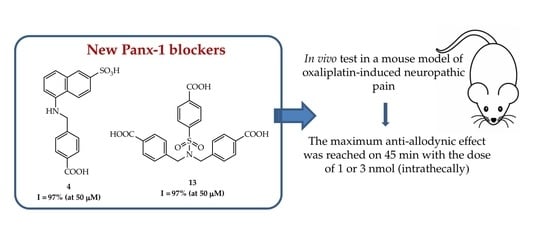
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


