
Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
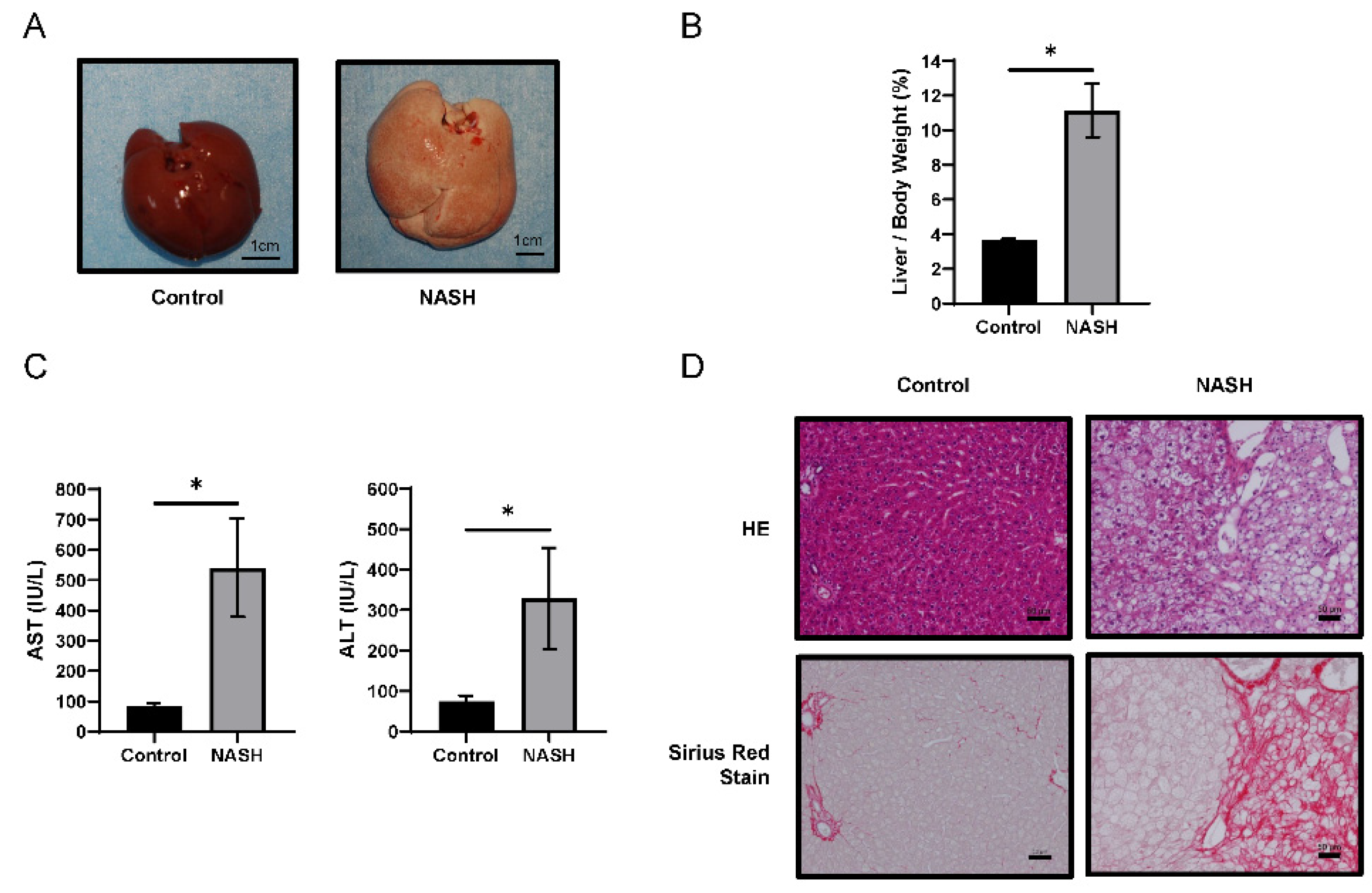
2.1. HFC-Fed Rats Developed NASH after 8 Weeks
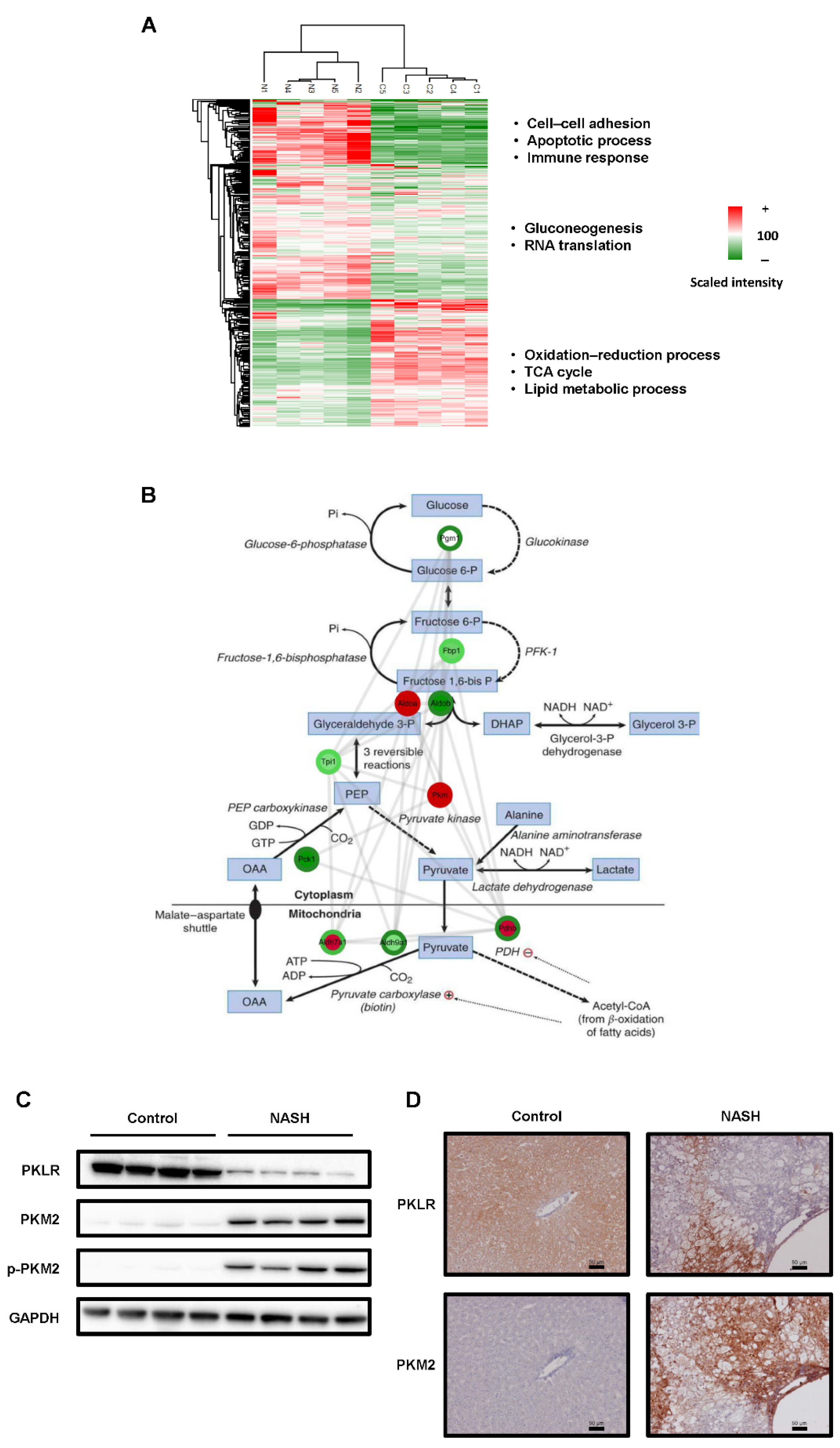
2.2. The Glycolysis Pathway Was Activated and PKM2 Was Upregulated in NASH Tissues
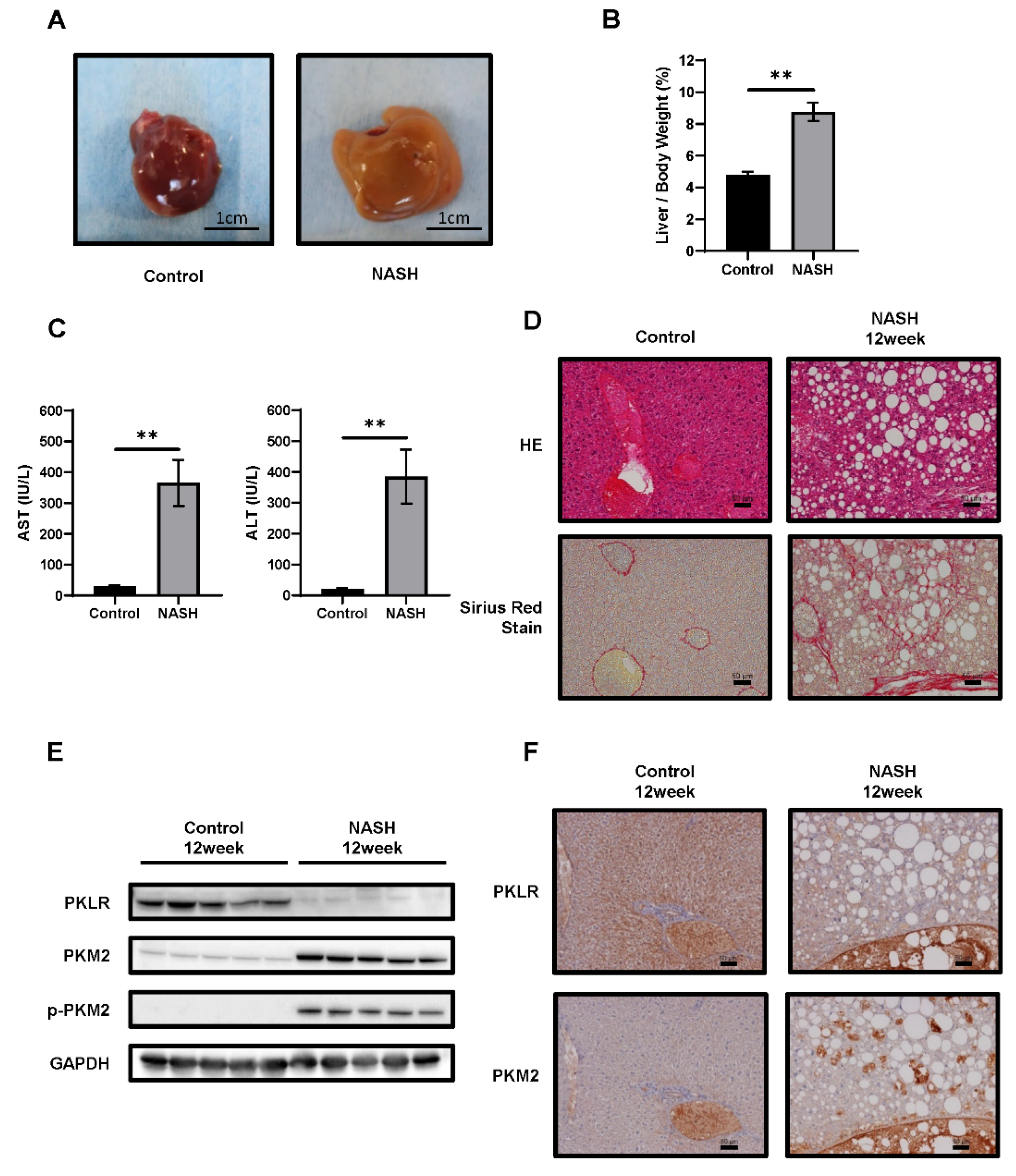
2.3. CDAHFD-Fed Mice Developed NASH
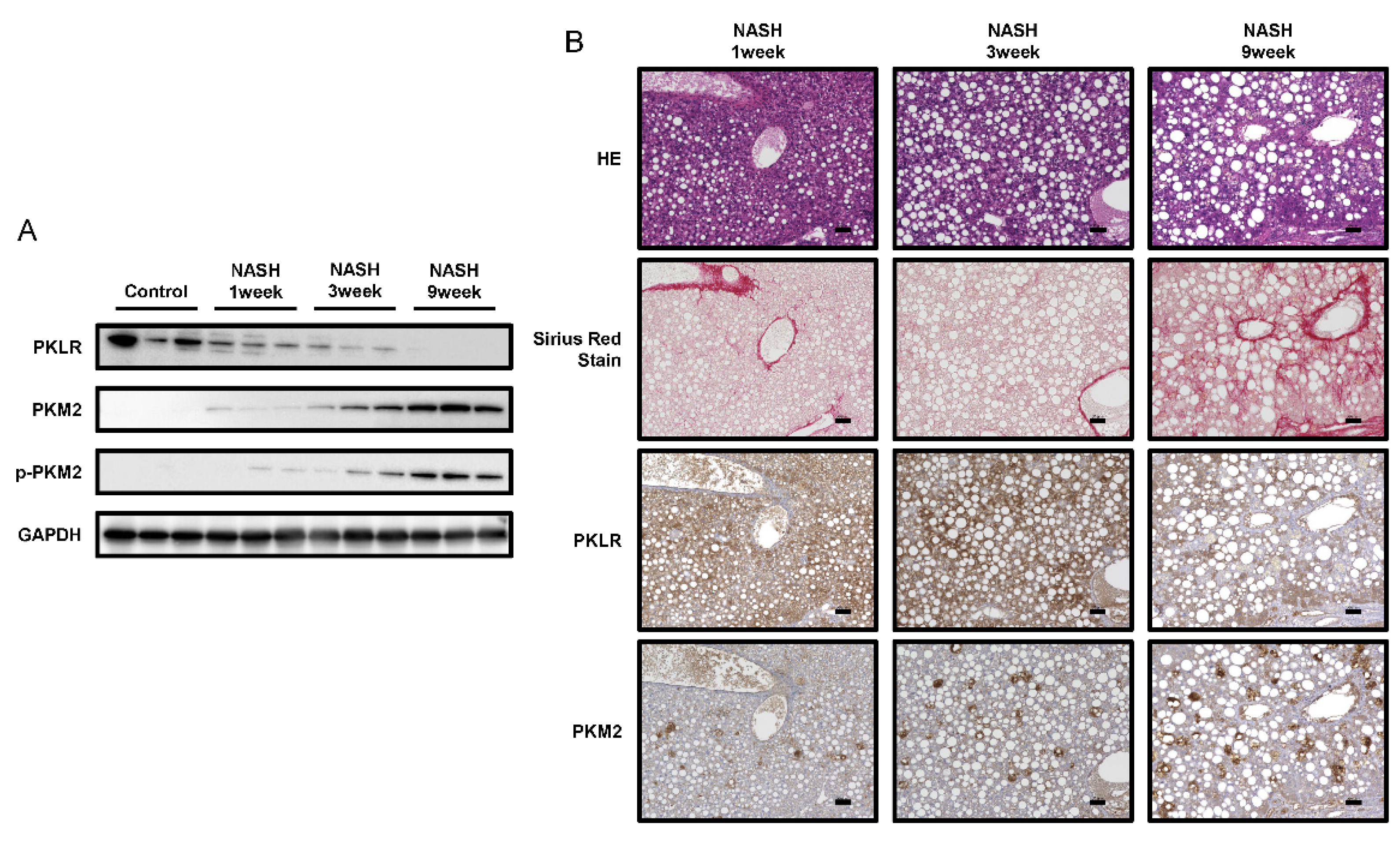
2.4. PKM2 Expression Was Upregulated in the Early Phase of NASH
2.5. The miR-122-5p Targeted PKM2 Directly and Was Downregulated in Kupffer Cells during NASH
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Histological Analysis and Immunohistochemistry (IHC)
4.3. Sample Preparation for Phosphoproteomics
4.4. LC-MS and Phosphoproteomics Analysis
4.5. Western Blotting
4.6. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
4.7. Cell Culture
4.8. Transfection Experiments
4.9. Luciferase Reporter Assay
4.10. Isolation of Kupffer Cells
4.11. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; NASH Clinical Research Network (CRN). Nonalcoholic Fatty Liver Disease (NAFLD) Activity Score and the Histopathologic Diagnosis in NAFLD: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Adams, L.A.; de Lédinghen, V.; Wong, G.L.; Sookoian, S. Noninvasive Biomarkers in NAFLD and NASH—Current Progress and Future Promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef] [PubMed]
- de Alwis, N.M.; Day, C.P. Non-alcoholic Fatty Liver Disease: The Mist Gradually Clears. J. Hepatol. 2008, 48 (Suppl. 1), S104–S112. [Google Scholar] [CrossRef] [PubMed]
- Yatsuji, S.; Hashimoto, E.; Tobari, M.; Taniai, M.; Tokushige, K.; Shiratori, K. Clinical Features and Outcomes of Cirrhosis Due to Non-alcoholic Steatohepatitis Compared with Cirrhosis Caused by Chronic Hepatitis C. J. Gastroenterol. Hepatol. 2009, 24, 248–254. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 Splice Isoform of Pyruvate Kinase Is Important for Cancer Metabolism and Tumour Growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Taniguchi, K.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Ito, Y.; Otsuki, Y.; Uno, B.; Uchiyama, K.; et al. MicroRNA-124 Inhibits Cancer Cell Growth through PTB1/PKM1/PKM2 Feedback Cascade in Colorectal Cancer. Cancer Lett. 2015, 363, 17–27. [Google Scholar] [CrossRef]
- Sugiyama, T.; Taniguchi, K.; Matsuhashi, N.; Tajirika, T.; Futamura, M.; Takai, T.; Akao, Y.; Yoshida, K. MiR-133b Inhibits Growth of Human Gastric Cancer Cells by Silencing Pyruvate Kinase Muscle-Splicer Polypyrimidine Tract-Binding Protein 1. Cancer Sci. 2016, 107, 1767–1775. [Google Scholar] [CrossRef]
- Takai, T.; Yoshikawa, Y.; Inamoto, T.; Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Shinohara, H.; Kumazaki, M.; Tsujino, T.; et al. A Novel Combination RNAi toward Warburg Effect by Replacement with miR-145 and Silencing of PTBP1 Induces Apoptotic Cell Death in Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 179. [Google Scholar] [CrossRef]
- Taniguchi, K.; Ito, Y.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakagawa, Y.; Sugiyama, T.; Futamura, M.; Otsuki, Y.; et al. Organ-Specific PTB1-Associated microRNAs Determine Expression of Pyruvate Kinase Isoforms. Sci. Rep. 2015, 5, 8647. [Google Scholar] [CrossRef]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-Associated microRNA-1 and -133b Suppress the Warburg Effect in Colorectal Tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, L.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The Glycolytic Enzyme PKM2 Bridges Metabolic and Inflammatory Dysfunction in Coronary Artery Disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific microRNAs from Mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a Mammalian Liver-Specific microRNA, is Processed from hcr mRNA and May Downregulate the High Affinity Cationic Amino Acid Transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic Steatohepatitis Is Associated with Altered Hepatic microRNA Expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in Exosomes Indicate Hepatocyte Injury and Inflammation in Alcoholic, Drug-Induced, and Inflammatory Liver Diseases. Hepatology 2012, 56, 1946–1957. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Jin, D.; Tashiro, K.; Komeda, K.; Masubuchi, S.; Hirokawa, F.; Hayashi, M.; Takai, S.; Uchiyama, K. Chymase Inhibitor Prevents the Development and Progression of Non-alcoholic Steatohepatitis in Rats Fed a High-Fat and High-Cholesterol Diet. J. Pharmacol. Sci. 2017, 134, 139–146. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Jin, D.; Tashiro, K.; Masubuchi, S.; Ozeki, M.; Hirokawa, F.; Hayashi, M.; Takai, S.; Uchiyama, K. A Novel Hamster Nonalcoholic Steatohepatitis Model Induced by a High-Fat and High-Cholesterol Diet. Exp. Anim. 2018, 67, 239–247. [Google Scholar] [CrossRef]
- Kitamori, K.; Naito, H.; Tamada, H.; Kobayashi, M.; Miyazawa, D.; Yasui, Y.; Sonoda, K.; Tsuchikura, S.; Yasui, N.; Ikeda, K.; et al. Development of Novel Rat Model for High-Fat and High-Cholesterol Diet-Induced Steatohepatitis and Severe Fibrosis Progression in SHRSP5/Dmcr. Environ. Health Prev. Med. 2012, 17, 173–182. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Sugito, N.; Shinohara, H.; Kuranaga, Y.; Inomata, Y.; Komura, K.; Uchiyama, K.; Akao, Y. Organ-Specific microRNAs (MIR122, 137, and 206) Contribute to Tissue Characteristics and Carcinogenesis by Regulating Pyruvate Kinase M1/2 (PKM) Expression. Int. J. Mol. Sci. 2018, 19, 1276. [Google Scholar] [CrossRef] [PubMed]
- Susor, W.A.; Rutter, W.J. Some Distinctive Properties of Pyruvate Kinase Purified from Rat Liver. Biochem. Biophys. Res. Commun. 1968, 30, 14–20. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An Improved Mouse Model That Rapidly Develops Fibrosis in Non-alcoholic Steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Ji, Q.; Xia, W.; Li, L.; Bai, J.; Ni, R.; Qin, Y. Pyruvate Kinase M2 Regulates Apoptosis of Intestinal Epithelial Cells in Crohn’s Disease. Dig. Dis. Sci. 2015, 60, 393–404. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.; et al. Pyruvate Kinase M2 Regulates HIF-1α Activity and IL-1β Induction and is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular Carcinoma in a Mouse Model Fed a Choline-Deficient, L-Amino Acid-Defined, High-Fat Diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef]
- Dixon, L.J.; Barnes, M.; Tang, H.; Pritchard, M.T.; Nagy, L.E. Kupffer Cells in the Liver. Compr. Physiol. 2013, 3, 785–797. [Google Scholar]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer Cells in Host Defense and Liver Disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Choi, S.; Diehl, A.M. Role of Inflammation in Nonalcoholic Steatohepatitis. Curr. Opin. Gastroenterol. 2005, 21, 702–707. [Google Scholar] [CrossRef]
- Maher, J.J.; Leon, P.; Ryan, J.C. Beyond Insulin Resistance: Innate Immunity in Nonalcoholic Steatohepatitis. Hepatology 2008, 48, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages Sequentially Change Their Functional Phenotype in Response to Changes in Microenvironmental Influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer Cells Promote M1 Kupffer Cell Apoptosis: A Protective Mechanism against Alcoholic and Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Smith, K. Liver disease: Kupffer Cells Regulate the Progression of ALD and NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 503. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Wang, C.; Ma, C.; Gong, L.; Guo, Y.; Fu, K.; Zhang, Y.; Zhou, H.; Li, Y. Macrophage Polarization and Its Role in Liver Disease. Front. Immunol. 2021, 12, 803037. [Google Scholar] [CrossRef]
- Ding, T.; Xu, J.; Wang, F.; Shi, M.; Zhang, Y.; Li, S.P.; Zheng, L. High Tumor-Infiltrating Macrophage Density Predicts Poor Prognosis in Patients with Primary Hepatocellular Carcinoma after Resection. Hum. Pathol. 2009, 40, 381–389. [Google Scholar] [CrossRef]
- Li, Y.W.; Qiu, S.J.; Fan, J.; Gao, Q.; Zhou, J.; Xiao, Y.S.; Xu, Y.; Wang, X.Y.; Sun, J.; Huang, X.W. Tumor-Infiltrating Macrophages Can Predict Favorable Prognosis in Hepatocellular Carcinoma after Resection. J. Cancer Res. Clin. Oncol. 2009, 135, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Cappello, P.; Nguyen, A.V.; Ralainirina, N.; Hardamon, C.R.; Foubert, P.; Schmid, M.C.; Sun, P.; Mose, E.; Bouvet, M.; et al. Macrophage PI3Kγ Drives Pancreatic Ductal Adenocarcinoma Progression. Cancer. Discov. 2016, 6, 870–885. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a Molecular Switch That Controls Immune Suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef]
- Xu, F.; Guo, M.; Huang, W.; Feng, L.; Zhu, J.; Luo, K.; Gao, J.; Zheng, B.; Kong, L.D.; Pang, T.; et al. Annexin A5 Regulates Hepatic Macrophage Polarization via Directly Targeting PKM2 and Ameliorates NASH. Redox. Biol. 2020, 36, 101634. [Google Scholar] [CrossRef]
- Navarro, C.D.C.; Francisco, A.; Figueira, T.R.; Ronchi, J.A.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. Dichloroacetate Reactivates Pyruvate-Supported Peroxide Removal by Liver Mitochondria and Prevents NAFLD Aggravation in NAD(P)+ Transhydrogenase-Null Mice Consuming a High-Fat Diet. Eur. J. Pharmacol. 2022, 917, 174750. [Google Scholar] [CrossRef] [PubMed]
- Saed, C.T.; Tabatabaei Dakhili, S.A.; Ussher, J.R. Pyruvate Dehydrogenase as a Therapeutic Target for Nonalcoholic Fatty Liver Disease. ACS Pharmacol. Transl. Sci. 2021, 4, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.Y.; Tso, S.C.; Chuang, J.L.; Gui, W.J.; Lou, M.; Sharma, G.; Khemtong, C.; Qi, X.; Wynn, R.M.; Chuang, D.T. Targeting Hepatic Pyruvate Dehydrogenase Kinases Restores Insulin Signaling and Mitigates ChREBP-Mediated Lipogenesis in Diet-Induced Obese Mice. Mol. Metab. 2018, 12, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 Targets Pyruvate Kinase M2 and Affects Metabolism of Hepatocellular Carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, M.; Tu, J.; Pang, L.; Cai, W.; Liu, X. MicroRNA-122 Affects Cell Aggressiveness and Apoptosis by Targeting PKM2 in Human Hepatocellular Carcinoma. Oncol. Rep. 2015, 34, 2054–2064. [Google Scholar] [CrossRef]
- Taniguchi, K.; Uchiyama, K.; Akao, Y. PTBP1-Targeting microRNAs Regulate Cancer-Specific Energy Metabolism through the Modulation of PKM1/M2 Splicing. Cancer Sci. 2021, 112, 41–50. [Google Scholar] [CrossRef]
- Nakamura, M.; Kanda, T.; Sasaki, R.; Haga, Y.; Jiang, X.; Wu, S.; Nakamoto, S.; Yokosuka, O. MicroRNA-122 Inhibits the Production of Inflammatory Cytokines by Targeting the PKR Activator PACT in Human Hepatic Stellate Cells. PLoS ONE 2015, 10, e0144295. [Google Scholar] [CrossRef][Green Version]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and Diabetic db/db Mice Develop Marked Liver Fibrosis in a Model of Nonalcoholic Steatohepatitis: Role of Short-Form Leptin Receptors and Osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Horie, Y.; Suzuki, A. Hepatocyte-Specific Pten-Deficient Mice as a Novel Model for Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Hepatol. Res. 2005, 33, 161–166. [Google Scholar] [CrossRef] [PubMed]
- de Lima, V.M.; de Oliveira, C.P.; Sawada, L.Y.; Barbeiro, H.V.; de Mello, E.S.; Soriano, F.G.; Alves, V.A.; Caldwell, S.H.; Carrilho, F.J. Yo Jyo Hen Shi Ko, A Novel Chinese Herbal, Prevents Nonalcoholic Steatohepatitis in ob/ob Mice Fed a High Fat or Methionine-Choline-Deficient Diet. Liver Int. 2007, 27, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S. Regulation of Oxidative Phosphorylation in the Mammalian Cell. Am. J. Physiol. 1990, 258, C377–C389. [Google Scholar] [CrossRef]
- Li, Y.; Ren, Q.; Zhu, L.; Li, Y.; Li, J.; Zhang, Y.; Zheng, G.; Han, T.; Sun, S.; Feng, F. Involvement of Methylation of MicroRNA-122, -125b and -106b in Regulation of Cyclin G1, CAT-1 and STAT3 Target Genes in Isoniazid-Induced Liver Injury. BMC Pharmacol. Toxicol. 2018, 19, 11. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of miR-122 Expression in Liver Cancer Correlates with Suppression of the Hepatic Phenotype and Gain of Metastatic Properties. Oncogene 2009, 28, 3526–3536. [Google Scholar] [CrossRef]
- Xu, H.; He, J.H.; Xiao, Z.D.; Zhang, Q.Q.; Chen, Y.Q.; Zhou, H.; Qu, L.H. Liver-Enriched Transcription Factors Regulate microRNA-122 that Targets CUTL1 during Liver Development. Hepatology 2010, 52, 1431–1442. [Google Scholar] [CrossRef]
- Li, Z.Y.; Xi, Y.; Zhu, W.N.; Zeng, C.; Zhang, Z.Q.; Guo, Z.C.; Hao, D.L.; Liu, G.; Feng, L.; Chen, H.Z.; et al. Positive Regulation of Hepatic miR-122 Expression by HNF4α. J. Hepatol. 2011, 55, 602–611. [Google Scholar] [CrossRef]
- Pasing, Y.; Colnoe, S.; Hansen, T. Proteomics of Hydrophobic Samples: Fast, Robust and Low-Cost Workflows for Clinical Approaches. Proteomics 2017, 17, 1500462. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-Throughput and High-Sensitivity Phosphoproteomics with the EasyPhos Platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Achour, B.; Al-Majdoub, Z.; Rostami-Hodjegan, A.; Barber, J. GASP and FASP are Complementary for LC-MS/MS Proteomic Analysis of Drug-Metabolizing Enzymes and Transporters in Pig Liver. Proteomics 2018, 18, e1800200. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Jung, H.J.; Hyung, S.W.; Kim, H.; Lee, D.; Lee, C.; Yu, M.H.; Lee, S.W. Postexperiment Monoisotopic Mass Filtering and Refinement (PE-MMR) of Tandem Mass Spectrometric Data Increases Accuracy of Peptide Identification in LC/MS/MS. Mol. Cell. Proteom. 2008, 7, 1124–1134. [Google Scholar] [CrossRef]
- Taus, T.; Köcher, T.; Pichler, P.; Paschke, C.; Schmidt, A.; Henrich, C.; Mechtler, K. Universal and Confident Phosphorylation Site Localization using phosphoRS. J. Proteome Res. 2011, 10, 5354–5362. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Strick, P.L.; Kim, C.C. Input to Primate Motor Cortex from Posterior Parietal Cortex (area 5). I. Demonstration by Retrograde Transport. Brain Res. 1978, 157, 325–330. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wada, S.I.; Ito, Y.; Hayashi, J.; Inomata, Y.; Lee, S.W.; Tanaka, T.; Komura, K.; Akao, Y.; Urata, H.; et al. α-aminoisobutyric acid-Containing Amphipathic Helical Peptide-cyclic RGD Conjugation as a Potential Drug Delivery System for microRNA Replacement Therapy in vitro. Mol. Pharm. 2019, 16, 4542–4550. [Google Scholar] [CrossRef]
- Kitani, H.; Sakuma, C.; Takenouchi, T.; Sato, M.; Yoshioka, M.; Yamanaka, N. Establishment of c-myc-Immortalized Kupffer Cell Line from a C57BL/6 Mouse Strain. Results Immunol. 2014, 4, 68–74. [Google Scholar] [CrossRef]
- Bourgognon, M.; Klippstein, R.; Al-Jamal, K.T. Kupffer Cell Isolation for Nanoparticle Toxicity Testing. J. Vis. Exp. 2015, 102, e52989. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA gene name (ID) | miR-122 (406906) |
| Target gene name (ID) | PKM (5315) |
| Species name (ID) | Homo sapiens |
| Genomic location of MTI Nucleotide sequence | 5′-ACACTCCA |
| Location | 15:72199548-72199555 |
| Location within a part of a gen | 520-527 (location within 3’UTR) |
| Existence of precious report (searching with miRTarBase) | Yes |
| Methods for experimental validation in this study | Luciferase reporter assay Western blot analysis |
| Experimental materials used in this study | HuH-7 and KUP5 cell lines |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inomata, Y.; Oh, J.-W.; Taniguchi, K.; Sugito, N.; Kawaguchi, N.; Hirokawa, F.; Lee, S.-W.; Akao, Y.; Takai, S.; Kim, K.-P.; et al. Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 23, 5230. https://doi.org/10.3390/ijms23095230
Inomata Y, Oh J-W, Taniguchi K, Sugito N, Kawaguchi N, Hirokawa F, Lee S-W, Akao Y, Takai S, Kim K-P, et al. Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences. 2022; 23(9):5230. https://doi.org/10.3390/ijms23095230
Chicago/Turabian StyleInomata, Yosuke, Jae-Won Oh, Kohei Taniguchi, Nobuhiko Sugito, Nao Kawaguchi, Fumitoshi Hirokawa, Sang-Woong Lee, Yukihiro Akao, Shinji Takai, Kwang-Pyo Kim, and et al. 2022. "Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis" International Journal of Molecular Sciences 23, no. 9: 5230. https://doi.org/10.3390/ijms23095230
APA StyleInomata, Y., Oh, J.-W., Taniguchi, K., Sugito, N., Kawaguchi, N., Hirokawa, F., Lee, S.-W., Akao, Y., Takai, S., Kim, K.-P., & Uchiyama, K. (2022). Downregulation of miR-122-5p Activates Glycolysis via PKM2 in Kupffer Cells of Rat and Mouse Models of Non-Alcoholic Steatohepatitis. International Journal of Molecular Sciences, 23(9), 5230. https://doi.org/10.3390/ijms23095230