
HIV-Tat Exacerbates the Actions of Atazanavir, Efavirenz, and Ritonavir on Cardiac Ryanodine Receptor (RyR2)

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Effects of ABC, ATV, BIC, EFV, TDF, RPV, and RTV on Equilibrium [3H]ryanodine Binding to RyR2
2.2. Effects of Pretreating RyR2 with HIV-Tat on Equilibrium [3H]ryanodine Binding to ATV, RTV, and EFV
2.3. Effects of ATV, EFV, RTV, and HIV-Tat on Ca2+ Homeostasis in Primary Rat Cardiac Myocytes
2.4. Effects of ATV. EFV, RTV, and HIV-Tat on the Gating and Conductance of RyR2
2.4.1. Effects of ATV on RyR2
2.4.2. Effects of EFV on RyR2
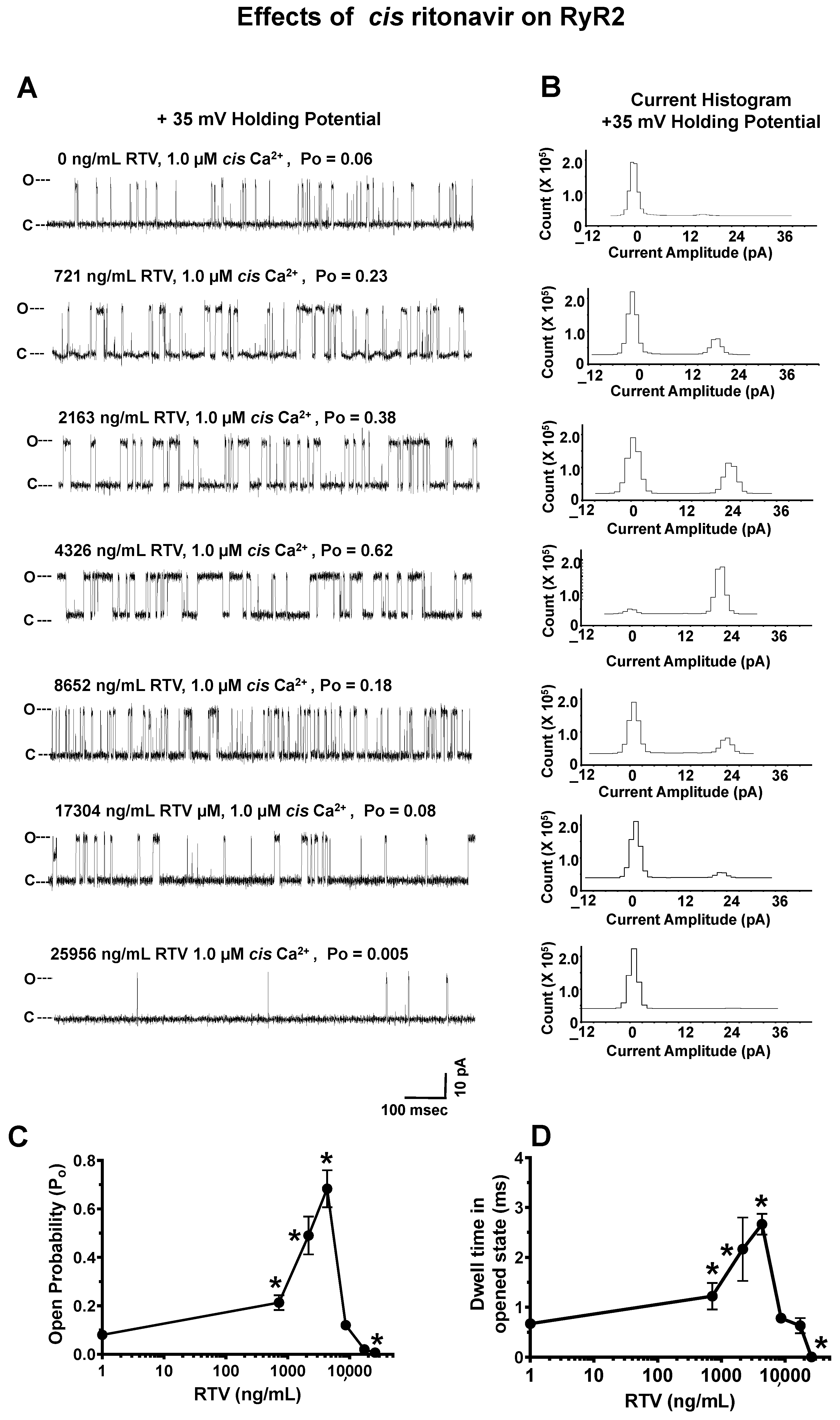
2.4.3. Effects of RTV on RyR2
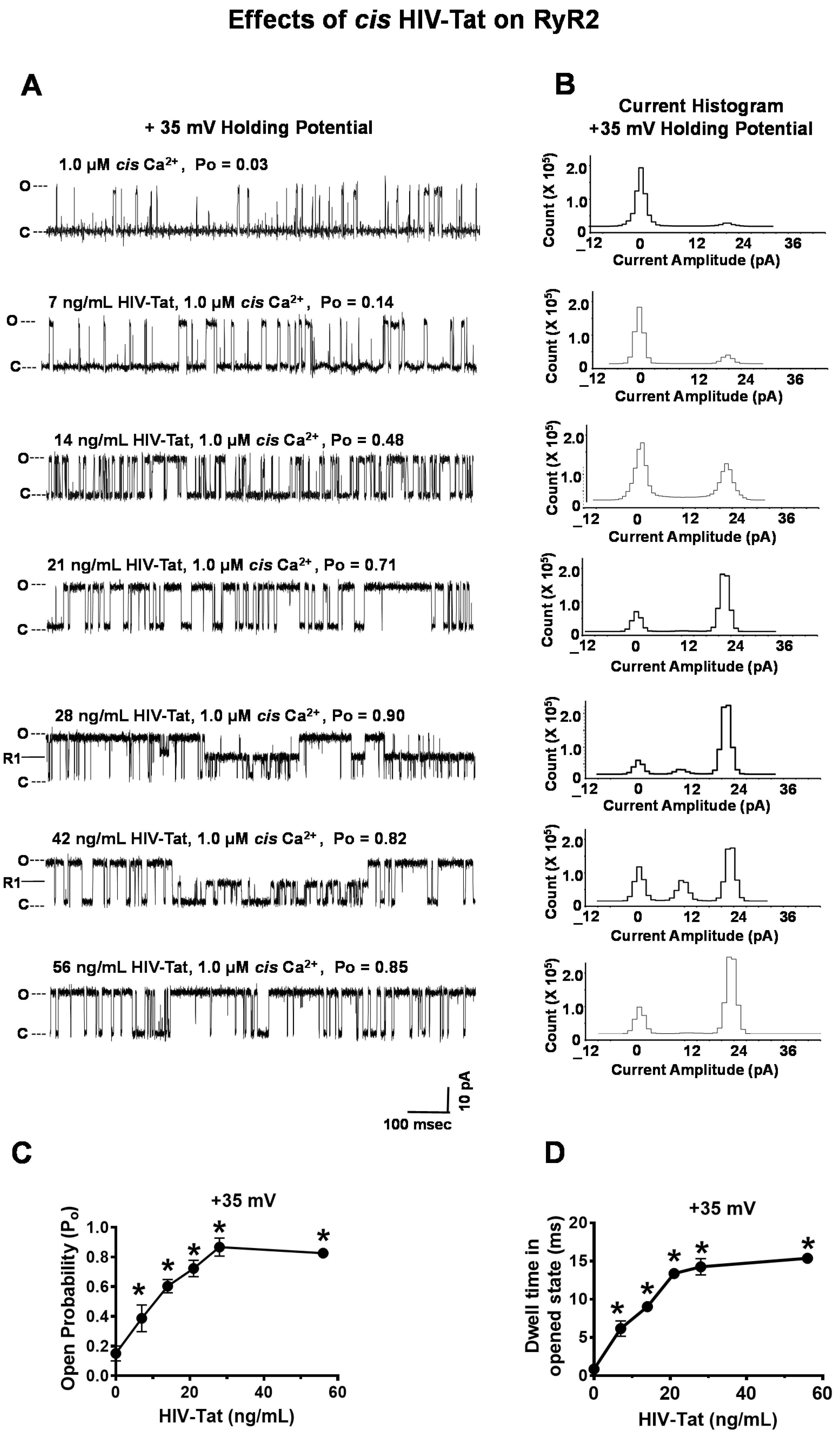
2.4.4. Effects of HIV-Tat on RyR2
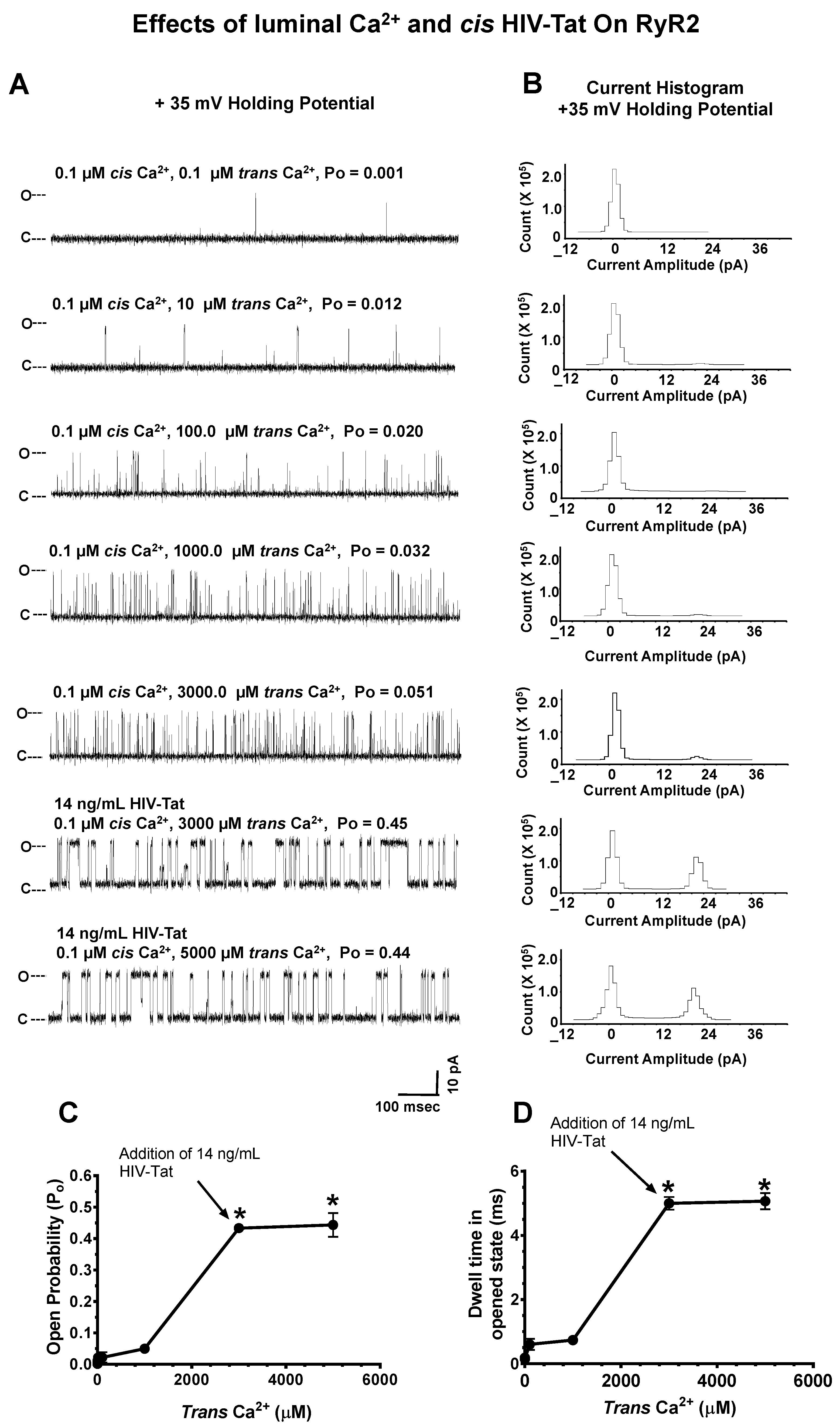
2.4.5. Effects of Luminal Ca2+ on HIV-Tat Actions on RyR2
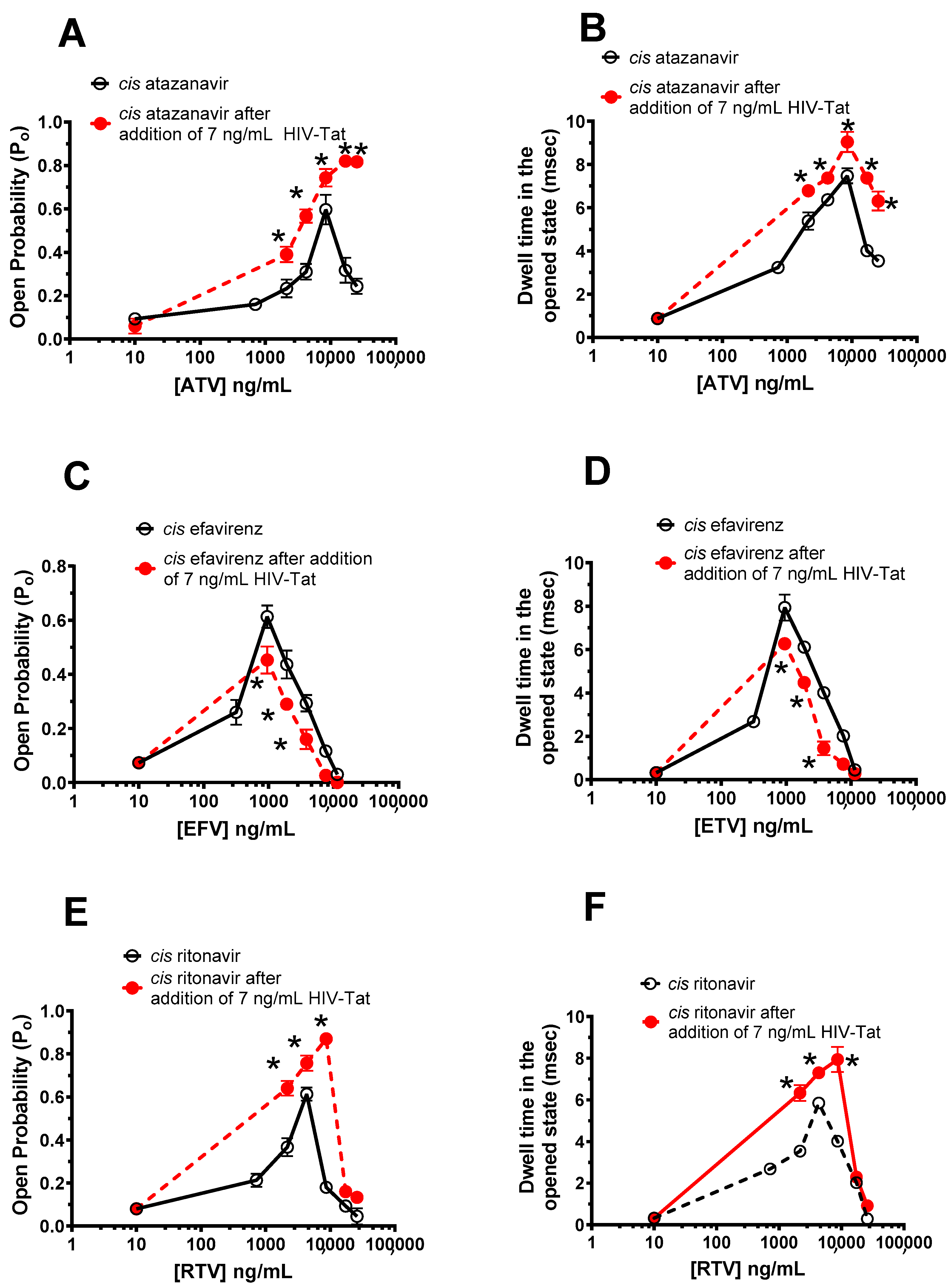
2.5. Effects of HIV-Tat Pretreatment on the Actions of ATV, EFV, and RTV on RyR2
2.6. In Silico Molecular Docking of HIV-Tat on RyR2
3. Discussion
4. Materials and Methods
4.1. Antibodies, Reagents, and Drugs
4.2. Preparation of Stock Solutions of HIV-Tat, EFV, ATV and RTV
4.3. Preparation SR Vesicles and Enrichment of RyR2 from Bovine Hearts
4.4. Ability of HIV-Tat, ATV, EFV, and RTV to Bind to Modulate RyR2
- (i)
- Effects of ATV, EFV, RTV, and HIV-Tat on the gating and conductance of RyR2. ATV (0–25,344 ng/mL), EFV (0–11,376 ng/mL), RTV (0–25,956 ng/mL), or HIV-Tat (7–52 ng/mL) was cumulatively added to the cis chamber of the bilayer (cytoplasmic side of RyR2). After each dose, the chamber was vigorously stirred for 15 s, and three 2-min recordings were obtained at + 35 mV holding potential. All recordings were carried out at room temperature (23–25 °C). Data acquisition was performed using commercially available instruments and software (Axopatch 1D, Digidata 1322A and pClamp 10.0, Axon Instruments, Burlingame, CA, USA). Electrical signals were filtered at 2 kHz, digitized at 10 kHz, and analyzed as described previously [68,69]. Data were obtained using pClamp 10.0 (Axon Instruments, Burlingame, CA, USA) and GraphPrism 7.0 (La Jolla, CA, USA).
- (ii)
- Effects of HIV-Tat pretreatment on the ATV, EFV, and RTV effects on RyR2. HIV-Tat (7 ng/mL) was added to the cis chamber after a single RyR2 was incorporated into the bilayer. After vigorously stirring for 15 s, three 2-min-long recordings at +35 mV were used to obtain activity. Thereafter, ATV (0–25,344 ng/mL), EFV (0–11,376 ng/mL), or RTV (0–25,956 ng/mL) was added to the cis chamber in cumulative amounts. After the addition of each dose of the antiretroviral agent, the cis chamber was vigorously stirred for 15 s, and three 2-min-long recordings at +35 mV were obtained.
4.5. Effects of HIV-Tat, ATV, EFV, and RTV on Intracellular Ca2+ Homeostasis in Primary Rat Ventricular Myocytes
4.6. In Silico Molecular Docking of HIV-Tat on RyR2
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brouillette, J.; Cyr, S.; Fiset, C. Mechanisms of Arrhythmia and Sudden Cardiac Death in Patients With HIV Infection. Can. J. Cardio. 2019, 35, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, B.; Gergis, K.; Baral, N.; Rauniyar, R.; Adhikari, G. Sudden Cardiac Death and Sudden Cardiac Arrest in Patients with Human Immunodeficiency Virus: A Systematic Review. Cureus 2021, 13, e13764. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, M.S.; Duncan, M.S.; Alcorn, C.; Chang, C.H.; Kundu, S.; Mumpuni, A.; Smith, E.K.; Loch, S.; Bedigian, A.; Vittinghoff, E.; et al. HIV Infection and the Risk of World Health Organization-Defined Sudden Cardiac Death. J. Am. Heart Assoc. 2021, 10, e021268. [Google Scholar] [CrossRef] [PubMed]
- Monsuez, J.J.; Lopez-Sublet, M. Prevention of Sudden Cardiac Death in Persons Living with HIV Infection. Curr. HIV Res. 2022, 20, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Narla, V.A. Sudden cardiac death in HIV-infected patients: A contemporary review. Clin. Cardiol. 2021, 44, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Alvi, R.M.; Neilan, A.M.; Tariq, N.; Hassan, M.O.; Awadalla, M.; Zhang, L.; Afshar, M.; Rokicki, A.; Mulligan, C.P.; Triant, V.A.; et al. The Risk for Sudden Cardiac Death Among Patients Living With Heart Failure and Human Immunodeficiency Virus. JACC Heart Fail. 2019, 7, 759–767. [Google Scholar] [CrossRef]
- Reinsch, N.; On behalf of HIV HEART Study Group and the Heinz Nixdorf Recall Investigative Group; Arendt, M.; Geisel, M.H.; Schulze, C.; Holzendorf, V.; Warnke, A.; Neumann, T.; Brockmeyer, N.H.; Schadendorf, D.; et al. Prolongation of the QTc interval in HIV-infected individuals compared to the general population. Infection 2017, 45, 659–667. [Google Scholar] [CrossRef]
- Liu, J.; Shah, S.K.; Basu-Ray, I.; Garcia-Diaz, J.; Khalid, K.; Saeed, M. QT prolongation in HIV-positive patients: Review article. Indian Heart J. 2019, 71, 434–439. [Google Scholar] [CrossRef]
- Wu, K.C.; Zhang, L.; A Haberlen, S.; Ashikaga, H.; Brown, T.T.; Budoff, M.J.; D’Souza, G.; A Kingsley, L.; Palella, F.J.; Margolick, J.B.; et al. Predictors of electrocardiographic QT interval prolongation in men with HIV. Heart 2019, 105, 559–565. [Google Scholar] [CrossRef]
- Tseng, Z.H.; Moffatt, E.; Kim, A.; Vittinghoff, E.; Ursell, P.; Connolly, A.; Olgin, J.E.; Wong, J.K.; Hsue, P.Y. Sudden Cardiac Death and Myocardial Fibrosis, Determined by Autopsy, in Persons with HIV. N. Engl. J. Med. 2021, 384, 2306–2316. [Google Scholar] [CrossRef]
- Moyers, B.S.; Secemsky, E.A.; Vittinghoff, E.; Wong, J.K.; Havlir, D.V.; Hsue, P.Y.; Tseng, Z.H. Effect of left ventricular dysfunction and viral load on risk of sudden cardiac death in patients with human immunodeficiency virus. Am. J. Cardiol. 2014, 113, 1260–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, A.D.; Graff, C.; Nielsen, J.B.; Thomsen, M.T.; Høgh, J.; Benfield, T.; Gerstoft, J.; Køber, L.; Kofoed, K.F.; Nielsen, S.D. De novo electrocardiographic abnormalities in persons living with HIV. Sci. Rep. 2021, 11, 20750. [Google Scholar] [CrossRef] [PubMed]
- Es-Salah-Lamoureux, Z.; Jouni, M.; Malak, O.A.; Belbachir, N.; Al Sayed, Z.R.; Gandon-Renard, M.; Lamirault, G.; Gauthier, C.; Baró, I.; Charpentier, F. HIV-Tat induces a decrease in IKr and IKsvia reduction in phosphatidylinositol-(4,5)-bisphosphate availability. J. Mol. Cell Cardiol. 2016, 99, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.R.; Best, A.M.; Pade, P.A.; McCance-Katz, E.F. Effect of buprenorphine and antiretroviral agents on the QT interval in opioid-dependent patients. Ann. Pharmacother. 2006, 40, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.-L.; Liu, H.-B.; Sun, B.; Zhang, Y.; Li, Q.; Hu, C.-W.; Zhu, J.-X.; Gong, D.-M.; Teng, X.; Zhang, Q.; et al. HIV Tat protein inhibits hERG K+ channels: A potential mechanism of HIV infection induced LQTs. J. Mol. Cell Cardiol. 2011, 51, 876–880. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Rybniker, J.; Zoghi, S.; Faetkenheuer, G.; Michels, G.; Erdmann, E.; Von Bergwelt-Baildon, M.; Kochanek, M. Acquired Long QT Syndrome and Torsade de Pointes Associated with HIV Infection. Case Rep. Med. 2010, 2010, 278427. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Arora, R.; Jawad, E. HIV protease inhibitors induced prolongation of the QT Interval: Electrophysiology and clinical implications. Am. J. Ther. 2010, 17, e193–e201. [Google Scholar] [CrossRef]
- Puri, R.; Roberts-Thomson, K.C.; Young, G.D. HIV and Long QT syndrome--cause or coincidence. Int. J. Cardiol. 2009, 133, e9–e10. [Google Scholar] [CrossRef]
- Anson, B.D.; Weaver, J.G.; Ackerman, M.J.; Akinsete, O.; Henry, K.; January, C.T.; Badley, A.D. Blockade of HERG channels by HIV protease inhibitors. Lancet 2005, 365, 682–686. [Google Scholar] [CrossRef]
- Chastain, D.B.; Veve, M.P.; Wagner, J.L. Abnormal QTc syndrome in HIV-infected patients: A systematic review of prevalence and risk factors. Antivir. Ther. 2019, 24, 459–465. [Google Scholar] [CrossRef]
- Eyawo, O.; Brockman, G.; Goldsmith, C.H.; Hull, M.W.; A Lear, S.; Bennett, M.; Guillemi, S.; Franco-Villalobos, C.; Adam, A.; Mills, E.J.; et al. Risk of myocardial infarction among people living with HIV: An updated systematic review and meta-analysis. BMJ Open 2019, 9, e025874. [Google Scholar] [CrossRef] [PubMed]
- Olalla, J.; Pombo, M. Commentary to Editor on Sudden Cardiac Death and Myocardial Fibrosis in Persons with HIV. N. Engl. J. Med. 2021, 385, e50. Available online: https://pubmed.ncbi.nlm.nih.gov/34614340/ (accessed on 18 December 2022). [PubMed]
- Tseng, T.H.; Hsue, P.; Moffatt, E. Response to Commentary to Editor on Sudden Cardiac Death and Myocardial Fibrosis in Persons with HIV. N. Engl. J. Med. 2021, 385, e50. Available online: https://pubmed.ncbi.nlm.nih.gov/34614341/ (accessed on 18 December 2022).
- Simpson, T.F.; Salazar, J.; Vittinghoff, E.; Probert, J.; Iwahashi, A.; Olgin, J.E.; Ursell, P.; Hart, A.; Moffatt, E.; Tseng, Z.H. Association of QT-prolonging medications with risk of autopsy-defined causes of sudden death. JAMA Intern. Med. 2020, 180, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Myerson, M.; Kaplan-Lewis, E.; Poltavskiy, E.; Ferris, D.; Bang, H. Prolonged QTc in HIV-Infected Patients: A Need for Routine ECG Screening. J. Int. Assoc. Provid. AIDS Care 2019, 18, 2325958219833926. [Google Scholar] [CrossRef] [Green Version]
- Shavadia, J.; Shah, R.; Yonga, G.; Patel, R.; Stebbing, J.; Nelson, M. The influence of antiretroviral therapy on the QTc interval in an African cohort. Clin. Infect. Dis. 2012, 54, 448–449. [Google Scholar] [CrossRef] [Green Version]
- Morita, N.; Mandel, W.J.; Kobayashi, Y.; Karagueuzian, H.S. Cardiac fibrosis as a determinant of ventricular tachyarrhythmias. J. Arrhythm. 2014, 30, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Ono, N.; Hayashi, H.; Kawase, A.; Lin, S.-F.; Li, H.; Weiss, J.N.; Chen, P.-S.; Karagueuzian, H.S. Spontaneous atrial fibrillation initiated by triggered activity near the pulmonary veins in aged rats subjected to glycolytic inhibition. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H639–H648. [Google Scholar] [CrossRef]
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.-F.; Chen, P.-S.; Xie, L.-H.; Chen, F.; et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1594–H1605. [Google Scholar] [CrossRef] [Green Version]
- Dash, P.K.; Alomar, F.A.; Cox, J.L.; McMillan, J.; Hackfort, B.T.; Makarov, E.; Morsey, B.; Fox, H.S.; Gendelman, H.E.; Gorantla, S.; et al. A Link Between Methylglyoxal and Heart Failure During HIV-1 Infection. Front. Cardiovasc. Med. 2021, 8, 792180. [Google Scholar] [CrossRef]
- Armoundas, A.A. Mechanism of abnormal sarcoplasmic reticulum calcium release in canine left-ventricular myocytes results in cellular alternans. IEEE Trans. Biomed. Eng. 2009, 56, 220–228. [Google Scholar] [CrossRef]
- Wilson, L.D.; Rosenbaum, D.S. Mechanisms of arrythmogenic cardiac alternans. Europace 2007, 9 (Suppl 6), 77–82. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Sun, B.; Vallmitjana, A.; Mi, T.; Guo, W.; Ni, M.; Wang, R.; Guo, A.; Duff, H.J.; Gillis, A.M.; et al. Suppression of ryanodine receptor function prolongs Ca2+ release refractoriness and promotes cardiac alternans in intact hearts. Biochem. J. 2016, 473, 3951–3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Wei, J.; Zhong, X.; Guo, W.; Yao, J.; Wang, R.; Vallmitjana, A.; Benitez, R.; Hove-Madsen, L.; Chen, S.W. The cardiac ryanodine receptor, but not sarcoplasmic reticulum Ca(2+)-ATPase, is a major determinant of Ca(2+) alternans in intact mouse hearts. J. Biol. Chem. 2018, 293, 13650–13661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Yao, J.; Belke, D.; Guo, W.; Zhong, X.; Sun, B.; Wang, R.; Paul Estillore, J.; Vallmitjana, A.; Benitez, R.; et al. Ca(2+)-CaM Dependent Inactivation of RyR2 Underlies Ca(2+) Alternans in Intact Heart. Circ. Res. 2021, 128, e63–e83. [Google Scholar] [CrossRef] [PubMed]
- Benitah, J.P.; Perrier, R.; Mercadier, J.J.; Pereira, L.; Gomez, A.M. RyR2 and Calcium Release in Heart Failure. Front. Physiol. 2021, 12, 734210. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Mongillo, M.; Bellinger, A.; Lindegger, N.; Chen, B.-X.; Hsueh, W.; Reiken, S.; Wronska, A.; Drew, L.J.; Ward, C.W.; et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Investig. 2008, 118, 2230–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoote, M.; Williams, A.J. The cardiac ryanodine receptor (calcium release channel): Emerging role in heart failure and arrhythmia pathogenesis. Cardiovasc. Res. 2002, 56, 359–372. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, D.; Fill, M. Pernicious attrition and inter-RyR2 CICR current control in cardiac muscle. J. Mol. Cell Cardiol. 2013, 58, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Blayney, L.M.; Lai, F.A. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol. Ther. 2009, 123, 151–177. [Google Scholar] [CrossRef] [Green Version]
- Kurebayashi, N.; Murayama, T. RyR2 mutation-linked arrhythmogenic diseases and its therapeutic strategies. Nihon Yakurigaku Zasshi 2020, 155, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-T.; Valdivia, C.R.; Gurrola, G.B.; Powers, P.P.; Willis, B.C.; Moss, R.L.; Jalife, J.; Valdivia, H.H. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc. Natl. Acad. Sci. USA 2015, 112, E1669–E1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paavola, J.; Viitasalo, M.; Laitinen-Forsblom, P.J.; Pasternack, M.; Swan, H.; Tikkanen, I.; Toivonen, L.; Kontula, K.; Laine, M. Mutant ryanodine receptors in catecholaminergic polymorphic ventricular tachycardia generate delayed afterdepolarizations due to increased propensity to Ca2+ waves. Eur. Heart J. 2007, 28, 1135–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnir, A.; Wajsberg, B.; Marks, A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, D.H.; Chen, S.R. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J. Physiol. 2009, 587 Pt 13, 3113–3115. [Google Scholar] [CrossRef] [PubMed]
- Salvage, S.C.; Gallant, E.M.; Fraser, J.A.; Huang, C.L.; Dulhunty, A.F. Flecainide Paradoxically Activates Cardiac Ryanodine Receptor Channels under Low Activity Conditions: A Potential Pro-Arrhythmic Action. Cells 2021, 10, 2101. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, G.K.; Pastromas, S.; Tzeis, S. Flecainide: Current status and perspectives in arrhythmia management. World J. Cardiol. 2015, 7, 76–85. [Google Scholar] [CrossRef]
- Bannister, M.L.; MacLeod, K.T.; George, C.H. Moving in the right direction: Elucidating the mechanisms of interaction between flecainide and the cardiac ryanodine receptor. Br. J. Pharmacol. 2022, 179, 2558–2563. [Google Scholar] [CrossRef]
- Norman, J.P.; Perry, S.W.; Reynolds, H.M.; Kiebala, M.; Bentley, K.L.D.M.; Trejo, M.; Volsky, D.J.; Maggirwar, S.B.; Dewhurst, S.; Masliah, E.; et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE 2008, 3, e3731. [Google Scholar] [CrossRef]
- Alomar, F.A.; Tian, C.; Dash, P.K.; McMillan, J.M.; Gendelman, H.E.; Gorantla, S.; Bidasee, K.R. Efavirenz, atazanavir, and ritonavir disrupt sarcoplasmic reticulum Ca(2+) homeostasis in skeletal muscles. Antiviral. Res. 2021, 187, 104975. [Google Scholar] [CrossRef]
- Perry, S.W.; Barbieri, J.; Tong, N.; Polesskaya, O.; Pudasaini, S.; Stout, A.; Lu, R.; Kiebala, M.; Maggirwar, S.B.; Gelbard, H.A. Human immunodeficiency virus-1 Tat activates calpain proteases via the ryanodine receptor to enhance surface dopamine transporter levels and increase transporter-specific uptake and Vmax. J. Neurosci. 2010, 30, 14153–14164. [Google Scholar] [CrossRef] [PubMed]
- WHO. Update of Recommendations on First- and Second-Line Antiretroviral Regimens Policy Brief; Reference Number: WHO/CDS/HIV/19.15; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Humerickhouse, R.A.; Bidasee, K.R.; Gerzon, K.; Emmick, J.T.; Kwon, S.; Sutko, J.L.; Ruest, L.; Besch, H.R. High affinity C10-Oeq ester derivatives of ryanodine. Activator-selective agonists of the sarcoplasmic reticulum calcium release channel. J. Biol. Chem. 1994, 269, 30243–30253. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Shao, C.H.; Moore, C.J.; Kutty, S.; Walseth, T.; DeSouza, C.; Bidasee, K.R. Gain of function of cardiac ryanodine receptor in a rat model of type 1 diabetes. Cardiovasc. Res. 2011, 91, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.W.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef]
- Fowler, E.D.; Zissimopoulos, S. Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease. Biomolecules 2022, 12, 1030. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, F. Ryanodine Receptor Structure and Function in Health and Disease. Subcell Biochem. 2018, 87, 329–352. [Google Scholar] [PubMed] [Green Version]
- Gonano, L.A.; Jones, P.P. FK506-binding proteins 12 and 12.6 (FKBPs) as regulators of cardiac Ryanodine Receptors: Insights from new functional and structural knowledge. Channels 2017, 11, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, Z.; Masumiya, H.; Wang, R.; Jiang, D.; Li, F.; Wagenknecht, T.; Chen, S.R.W. Three-dimensional localization of divergent region 3 of the ryanodine receptor to the clamp-shaped structures adjacent to the FKBP binding sites. J. Biol. Chem. 2003, 278, 14211–14218. [Google Scholar] [CrossRef] [Green Version]
- Lehnart, S.E.; Wehrens, X.H.; Marks, A.R. Calstabin deficiency, ryanodine receptors, and sudden cardiac death. Biochem. Biophys. Res. Commun. 2004, 322, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Kaftan, E.; Marks, A.R.; Ehrlich, B.E. Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ. Res. 1996, 78, 990–997. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, T.; Brillantes, A.M.; Timerman, A.P.; Fleischer, S.; Erdjument-Bromage, H.; Tempst, P.; Marks, A.R. FK506 binding protein associated with the calcium release channel (ryanodine receptor). J. Biol. Chem. 1992, 267, 9474–9477. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Liu, X.; Gong, Y.; Zhang, P.; Qiang, S.; Zhao, Q.; Guo, R.; Qian, Y.; Wang, L.; Zhu, L.; et al. Pathogenic mechanism of a catecholaminergic polymorphic ventricular tachycardia causing-mutation in cardiac calcium release channel RyR2. J. Mol. Cell Cardiol. 2018, 117, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.; Pollard, K.A. Drugs of Abuse: Sympathomimetics. Crit. Care Clin. 2021, 37, 487–499. [Google Scholar] [CrossRef]
- Zhao, Y.T.; Valdivia, C.R.; Gurrola, G.B.; Hernandez, J.J.; Valdivia, H.H. Arrhythmogenic mechanisms in ryanodine receptor channelopathies. Sci. China Life Sci. 2015, 58, 54–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wei, J.; Guo, W.; Sun, B.; Estillore, J.P.; Wang, R.; Yoruk, A.; Roston, T.M.; Sanatani, S.; Wilde, A.A.; et al. Human RyR2 (Ryanodine Receptor 2) Loss-of-Function Mutations: Clinical Phenotypes and In Vitro Characterization. Circ. Arrhythm. Electrophysiol. 2021, 14, e010013. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Xu, L.; Meissner, G.; Besch, H.R., Jr. Diketopyridylryanodine has three concentration-dependent effects on the cardiac calcium-release channel/ryanodine receptor. J. Biol. Chem. 2003, 278, 14237–14248. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.H.; Tian, C.; Ouyang, S.; Moore, C.J.; Alomar, F.; Nemet, I.; D’Souza, A.; Nagai, R.; Kutty, S.; Rozanski, G.J.; et al. Carbonylation induces heterogeneity in cardiac ryanodine receptor function in diabetes mellitus. Mol. Pharmacol. 2012, 82, 383–399. [Google Scholar] [CrossRef] [Green Version]
- Shao, C.H.; Rozanski, G.J.; Patel, K.P.; Bidasee, K.R. Dyssynchronous (non-uniform) Ca2+ release in myocytes from streptozotocin-induced diabetic rats. J. Mol. Cell Cardiol. 2007, 42, 234–246. [Google Scholar] [CrossRef]




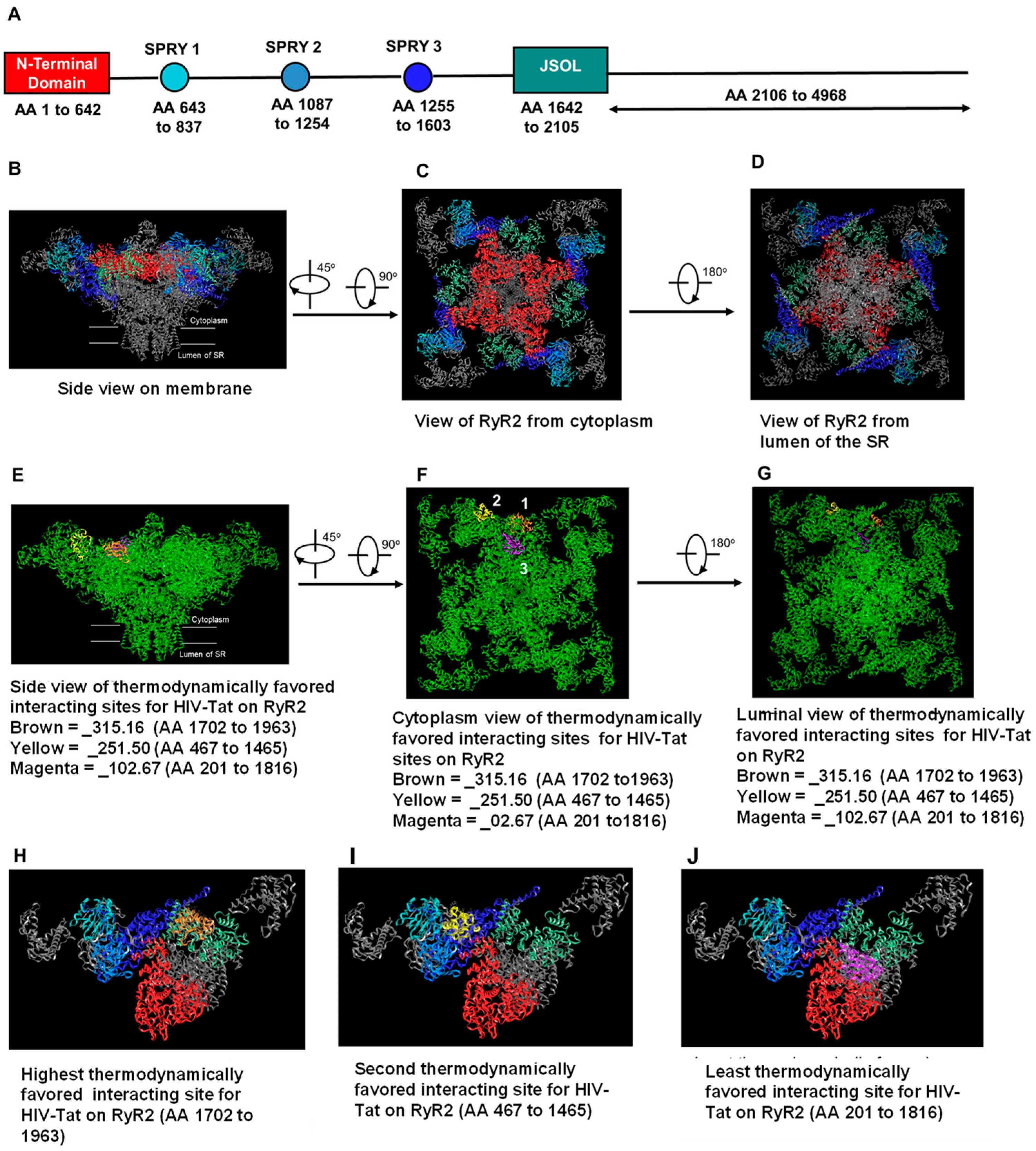

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RyR2 Residues | HIV-Tat Residues | Distance (A) | Specific Interactions |
|---|---|---|---|
| A:Lys 1963 | B:Asp 279 | 2.3 | 1× hydrogen bonding (hb) |
| A:Arg 1960 | B:Asp 279 | 2.3 | 1× salt bridge |
| A:His 1920 | B:Glh 278 | 1.9 | 1× hb |
| A:Arg 1919 | B:Asp 279 | 2.3 | 1× hb |
| A:Asp 1915 | B:Arg 340 | 2.2 | 2× hb, 1× salt bridge |
| A:Tyr 1912 | B:Gln 341 | 1.8 | 1× hb |
| A:Ser 1869 | B:Ash 312 | 1.8 | 1× hb |
| A:Tyr 1826 | B:Phe 277 | 2.4 | 1× hb |
| A:Ile 1822 | B:Arg 309 | 2.2 | 1× hb |
| A:Phe 1782 | B:Phe 307 | 2.7 | 1× pi stack |
| A:Arg 1758 | B:Pro 281 | 2.2 | 1× hb |
| A:Lys 1744 | B:Asp 352 | 2.3 | 1× hb, 1× salt bridge |
| A:Lys 1744 | B:Arg 261 | 2.2 | 1× hb |
| A:Asn 1743 | B:Asp 352 | 1.8 | 1× hb |
| A:Tyr 1702 | B:Arg 264 | 2.2 | 1× hb |
| RyR2 Residues | HIV-Tat Residues | Distance (A) | Specific Interactions |
|---|---|---|---|
| A:Val 1465 | B:Arg 309 | 2.0 | 1× hydrogen bonding (hb) |
| A:Arg 1463 | B:Glh 278 | 2.2 | 1× hb |
| A:Arg 1383 | B:Asp 279 | 2.1 | 1× hb |
| A:Lys 1380 | B:Asp 308 | 2.3 | 1× hb, 1× salt bridge |
| A:Glu 1335 | B:Asn 285 | 2.1 | 1× hb |
| A:Met 1249 | B:Arg 292 | 1.9 | 1× hb |
| A:Ile 1247 | B:Gln 300 | 2.4 | 1× hb |
| A:Asp 1246 | B:Gln 300 | 2.5 | 1× hb |
| A:Val 1161 | B:Arg 302 | 2.2 | 2× hb |
| A:Ile 659 | B:Lys 297 | 2.4 | 1× hb |
| A:Arg 647 | B:Glu 286 | 2.2 | 1× hb, 1× salt bridge |
| A:Arg 647 | B:Asp 280 | 2.9 | 1× salt bridge |
| RyR2 Residues | HIV-Tat Residues | Distance (A) | Specific Interactions |
|---|---|---|---|
| A:Phe 1816 | B:Arg 302 | 1.7 | 2× hydrogen bonding (hb) |
| A:Glu 1815 | B:Arg 292 | 2.4 | 1× hb, 1× salt bridge |
| A:Ser 1779 | B:Arg 292 | 2.2 | 1× hb |
| A:Tyr 1776 | B:Glu 289 | 2.0 | 1× hb |
| A:Lys 1744 | B:Asp 308 | 2.5 | 1× salt bridge |
| A:Phe 1739 | B:Leu 282 | 2.0 | 1× hb |
| A:Ser 554 | B:Ash 279 | 1.6 | 2× hb |
| A:Ser 554 | B:Glu 278 | 2.1 | 1× hb |
| A:Arg 520 | B:Asp 276 | 2.2 | 1× hb |
| A:Phe 514 | B:Glu 278 | 1.8 | 1× hb |
| A:Arg 480 | B:Gly 338 | 1.8 | 1× hb |
| A:Arg 480 | B:Met 271 | 2.2 | 1× hb |
| A:Gln 476 | B:Glu 286 | 2.3 | 1× hb |
| A:Phe 429 | B:Trp 336 | 2.4 | 1× pi stack |
| A:Ser 326 | B:Lys 297 | 1.8 | 1× hb |
| A:Ser 326 | B:Tyr 327 | 1.8 | 1× hb |
| A:Lys 325 | B:Gln 300 | 2.1 | 1× hb |
| A:Trp 212 | B:Tyr 327 | 2.0 | 1× hb |
| A:Leu 201 | B:Arg 335 | 2.2 | 1× hb |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alomar, F.A.; Tian, C.; Bidasee, S.R.; Venn, Z.L.; Schroder, E.; Palermo, N.Y.; AlShabeeb, M.; Edagwa, B.J.; Payne, J.J.; Bidasee, K.R. HIV-Tat Exacerbates the Actions of Atazanavir, Efavirenz, and Ritonavir on Cardiac Ryanodine Receptor (RyR2). Int. J. Mol. Sci. 2023, 24, 274. https://doi.org/10.3390/ijms24010274
Alomar FA, Tian C, Bidasee SR, Venn ZL, Schroder E, Palermo NY, AlShabeeb M, Edagwa BJ, Payne JJ, Bidasee KR. HIV-Tat Exacerbates the Actions of Atazanavir, Efavirenz, and Ritonavir on Cardiac Ryanodine Receptor (RyR2). International Journal of Molecular Sciences. 2023; 24(1):274. https://doi.org/10.3390/ijms24010274
Chicago/Turabian StyleAlomar, Fadhel A., Chengju Tian, Sean R. Bidasee, Zachary L. Venn, Evan Schroder, Nicholas Y. Palermo, Mohammad AlShabeeb, Benson J. Edagwa, Jason J. Payne, and Keshore R. Bidasee. 2023. "HIV-Tat Exacerbates the Actions of Atazanavir, Efavirenz, and Ritonavir on Cardiac Ryanodine Receptor (RyR2)" International Journal of Molecular Sciences 24, no. 1: 274. https://doi.org/10.3390/ijms24010274