


RNA-Seq Analysis Identifies Differentially Expressed Genes in the Longissimus dorsi of Wagyu and Chinese Red Steppe Cattle

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
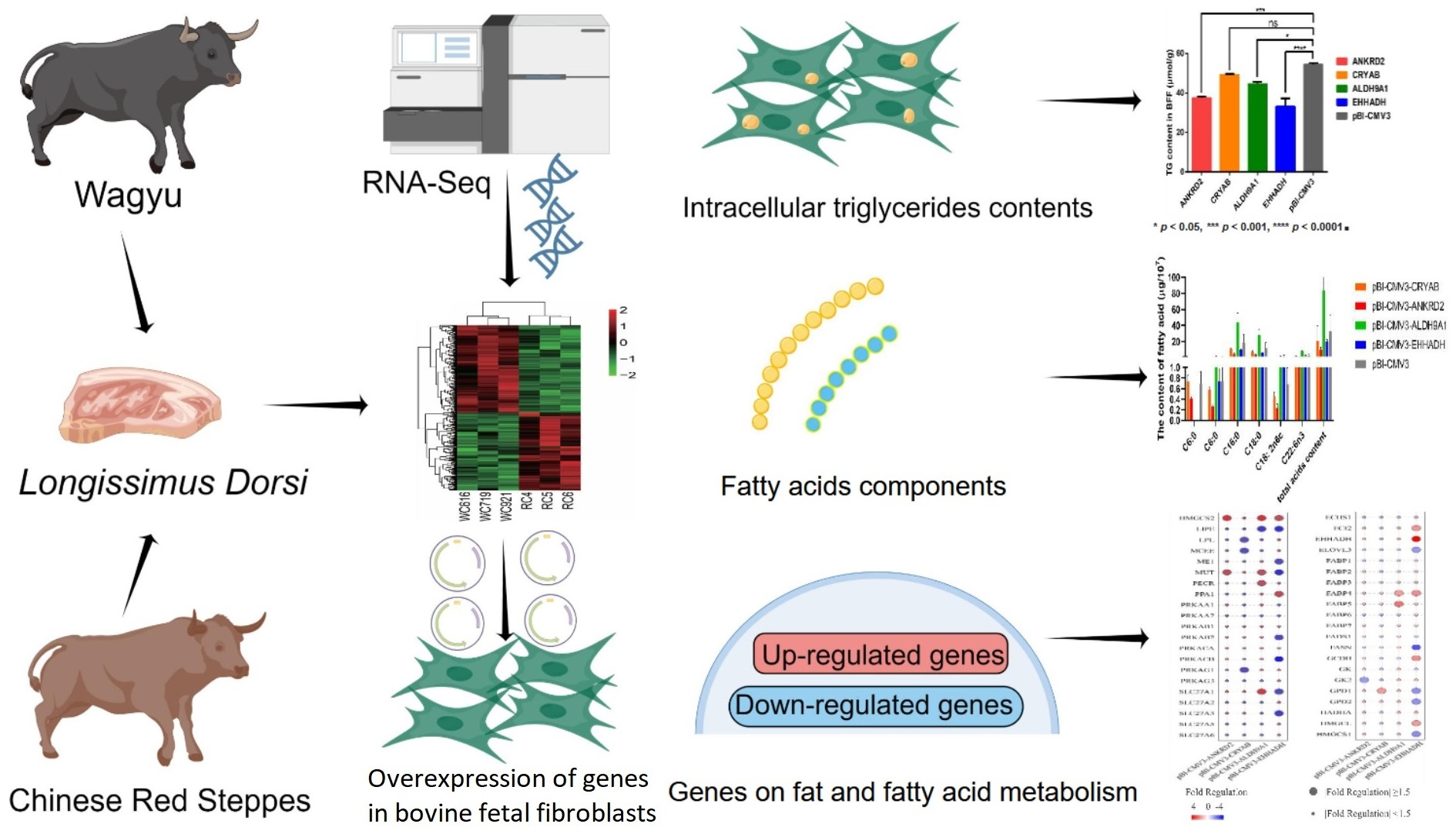
2.1. Analysis of DEGs in Longissimus dorsi Muscle between Cattle Breeds
2.2. DEG Participation in Biological Processes Related to Lipid Metabolism
2.3. Prediction Analysis of Genes Interaction Network
2.4. Validation of Candidate DEGs Expression Levels
2.5. Effects of Candidate Genes on Triglyceride Content in Bovine Fetal Fibroblasts (BFFs)
2.6. Effects of Candidate Genes on Bovine Intracellular Fatty Acids
2.7. Regulation of Genes Related to Lipid and Fatty Acid Metabolism by Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Animals and Longissimus dorsi Sample
4.2. RNA Extraction and RNA Sequencing
4.3. Sequencing Data Analysis
4.4. Gene Ontology Enrichment and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis
4.5. Real-Time Quantitative PCR Analysis
4.6. Construction of Candidate Gene Overexpression Vector
4.7. Cell Culture and Transfection
4.8. Analysis of the Triglyceride Contents in BFFs
4.9. Fatty Acids (FAs) Extraction and Content Analysis in BFFs by GC
4.10. Analysis of Lipid Metabolism by RT2 Profiler PCR Array
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, G. Important Roles of Dietary Taurine, Creatine, Carnosine, Anserine and 4-Hydroxyproline in Human Nutrition and Health. Amino Acids 2020, 52, 329–360. [Google Scholar] [CrossRef] [Green Version]
- King, D.A.; Wheeler, T.L.; Shackelford, S.D.; Koohmaraie, M. Fresh Meat Texture and Tenderness. In Improving the Sensory and Nutritional Quality of Fresh Meat; Elsevier: Cambridge, UK, 2009; pp. 61–88. [Google Scholar]
- Nafikov, R.A.; Beitz, D.C. Carbohydrate and Lipid Metabolism in Farm Animals. J. Nutr. 2007, 137, 702–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Cui, Y.; Gu, X. Genome-Wide DNA Methylation Profiles Changes Associated with Constant Heat Stress in Pigs as Measured by Bisulfite Sequencing. Sci. Rep. 2016, 6, 27507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, T.P.; Zhang, L. Application of whole transcriptomics in animal husbandry. Yi Chuan 2019, 41, 193–205. [Google Scholar] [PubMed]
- Song, C.; Huang, Y.; Yang, Z.; Ma, Y.; Chaogetu, B.; Zhuoma, Z.; Chen, H. RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle. Animals 2019, 9, 1077. [Google Scholar] [CrossRef] [Green Version]
- Simielli Fonseca, L.F.; dos Santos Silva, D.B.; Jovino Gimenez, D.F.; Baldi, F.; Ferro, J.A.; Loyola Chardulo, L.A.; de Albuquerque, L.G. Gene Expression Profiling and Identification of Hub Genes in Nellore Cattle with Different Marbling Score Levels. Genomics 2020, 112, 873–879. [Google Scholar] [CrossRef]
- McCabe, M.; Waters, S.; Morris, D.; Kenny, D.; Lynn, D.; Creevey, C. RNA-Seq Analysis of Differential Gene Expression in Liver from Lactating Dairy Cows Divergent in Negative Energy Balance. BMC Genom. 2012, 13, 193. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.M.; Chitwood, J.L.; Ross, P.J. RNA-Seq Profiling of Single Bovine Oocyte Transcript Abundance and Its Modulation by Cytoplasmic Polyadenylation. Mol. Reprod. Dev. 2015, 82, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Qin, L.; Yu, H.; Jiang, P.; Xia, L.; Gao, Z.; Yang, R.; Zhao, Y.; Yu, X.; Zhao, Z. Comprehensive Analysis of MiRNAs and Target MRNAs between Immature and Mature Testis Tissue in Chinese Red Steppes Cattle. Animals 2021, 11, 3024. [Google Scholar] [CrossRef]
- Keel, B.N.; Zarek, C.M.; Keele, J.W.; Kuehn, L.A.; Snelling, W.M.; Oliver, W.T.; Freetly, H.C.; Lindholm-Perry, A.K. RNA-Seq Meta-Analysis Identifies Genes in Skeletal Muscle Associated with Gain and Intake across a Multi-Season Study of Crossbred Beef Steers. BMC Genom. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Park, H.-S.; Kim, W.; Yoon, D.; Seo, S. Comparison of Metabolic Network between Muscle and Intramuscular Adipose Tissues in Hanwoo Beef Cattle Using a Systems Biology Approach. Int. J. Genom. 2014, 2014, 679437. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Liu, Q.; Welborn, M.; Wang, L.; Li, Y.; Deng, B.; McMillin, K.; Fu, X. Differential Gene Expression of Fibro/Adipogenic Progenitors between Wagyu and Brahman Cattle: A Possible Contribution to Their Different Meat Quality. J. Anim. Sci. 2021, 99, 301. [Google Scholar] [CrossRef]
- Huang, W.; Guo, Y.; Du, W.; Zhang, X.; Li, A.; Miao, X. Global Transcriptome Analysis Identifies Differentially Expressed Genes Related to Lipid Metabolism in Wagyu and Holstein Cattle. Sci. Rep. 2017, 7, 5278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motoyama, M.; Sasaki, K.; Watanabe, A. Wagyu and the Factors Contributing to Its Beef Quality: A Japanese Industry Overview. Meat Sci. 2016, 120, 10–18. [Google Scholar] [CrossRef]
- Berry, D.P.; Conroy, S.; Hegarty, P.J.; Evans, R.D.; Pabiou, T.; Judge, M.M. Inter-Animal Genetic Variability Exist in Organoleptic Properties of Prime Beef Meat. Meat Sci 2021, 173, 108401. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Scollan, N.D.; Dannenberger, D.; Nuernberg, K.; Richardson, I.; MacKintosh, S.; Hocquette, J.-F.; Moloney, A.P. Enhancing the Nutritional and Health Value of Beef Lipids and Their Relationship with Meat Quality. Meat Sci. 2014, 97, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Aaslyng, M.D.; Meinert, L. Meat Flavour in Pork and Beef—From Animal to Meal. Meat Sci. 2017, 132, 112–117. [Google Scholar] [CrossRef]
- Dodson, M.V.; Hausman, G.J.; Guan, L.; Du, M.; Rasmussen, T.P.; Poulos, S.P.; Mir, P.; Bergen, W.G.; Fernyhough, M.E.; McFarland, D.C.; et al. Skeletal Muscle Stem Cells from Animals I. Basic Cell Biology. Int. J. Biol. Sci. 2010, 6, 465–474. [Google Scholar] [CrossRef]
- Sumida, T.; Hino, N.; Kawachi, H.; Matsui, T.; Yano, H. Expression of Agouti Gene in Bovine Adipocytes. Anim. Sci. J. 2004, 75, 49–51. [Google Scholar] [CrossRef]
- Albrecht, E.; Komolka, K.; Kuzinski, J.; Maak, S. Agouti Revisited: Transcript Quantification of the ASIP Gene in Bovine Tissues Related to Protein Expression and Localization. PLoS ONE 2012, 7, e35282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voisin, S.; Almen, M.S.; Zheleznyakova, G.Y.; Lundberg, L.; Zarei, S.; Castillo, S.; Eriksson, F.E.; Nilsson, E.K.; Blueher, M.; Boettcher, Y.; et al. Many Obesity-Associated SNPs Strongly Associate with DNA Methylation Changes at Proximal Promoters and Enhancers. Genome Med. 2015, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Komolka, K.; Ponsuksili, S.; Albrecht, E.; Kühn, C.; Wimmers, K.; Maak, S. Gene Expression Profile of Musculus Longissimus Dorsi in Bulls of a Charolais × Holstein F 2 -Cross with Divergent Intramuscular Fat Content. Genom. Data 2016, 7, 131–133. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Liao, G.; DeGraff, L.M.; Gerrish, K.; Bortner, C.D.; Garantziotis, S.; Jetten, A.M. CD44 Plays a Critical Role in Regulating Diet-Induced Adipose Inflammation, Hepatic Steatosis, and Insulin Resistance. PLoS ONE 2013, 8, e58417. [Google Scholar] [CrossRef] [PubMed]
- Schmits, R.; Filmus, J.; Gerwin, N.; Senaldi, G.; Kiefer, F.; Kundig, T.; Wakeham, A.; Shahinian, A.; Catzavelos, C.; Rak, J.; et al. CD44 Regulates Hematopoietic Progenitor Distribution, Granuloma Formation, and Tumorigenicity. Blood 1997, 90, 2217–2233. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.K.; Wulur, I.; Alfonso, Z.; Hedrick, M.H. Fat Tissue: An Underappreciated Source of Stem Cells for Biotechnology. Trends Biotechnol. 2006, 24, 150–154. [Google Scholar] [CrossRef]
- Kodama, K.; Horikoshi, M.; Toda, K.; Yamada, S.; Hara, K.; Irie, J.; Sirota, M.; Morgan, A.A.; Chen, R.; Ohtsu, H.; et al. Expression-Based Genome-Wide Association Study Links the Receptor CD44 in Adipose Tissue with Type 2 Diabetes. Proc. Natl. Acad. Sci. USA 2012, 109, 7049–7054. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Xia, L.; Jin, Z.; Ali, S.; Wang, M.; Li, X.; Yang, R.; Fang, X.; Zhao, Z. New Function of the CD44 Gene: Lipid Metabolism Regulation in Bovine Mammary Epithelial Cells. J. Dairy Sci. 2020, 103, 6661–6671. [Google Scholar] [CrossRef]
- Reizes, O.; Benoit, S.C.; Clegg, D.J. The Role of Syndecans in the Regulation of Body Weight and Synaptic Plasticity. Int. J. Biochem. Cell Biol. 2008, 40, 28–45. [Google Scholar] [CrossRef]
- Strader, A.D.; Reizes, O.; Woods, S.C.; Benoit, S.C.; Seeley, R.J. Mice Lacking the Syndecan-3 Gene Are Resistant to Diet-Induced Obesity. J. Clin. Investig. 2004, 114, 1354–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüring, A.N.; Lutz, F.; Tüttelmann, F.; Gromoll, J.; Kiesel, L.; Götte, M. Role of Syndecan-3 Polymorphisms in Obesity and Female Hyperandrogenism. J. Mol. Med. (Berl.) 2009, 87, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Z.; Wang, Q.; Zhang, C.-L.; Fang, X.-T.; Song, E.-L.; Chen, H. Genetic Variants in SDC3 Gene Are Significantly Associated with Growth Traits in Two Chinese Beef Cattle Breeds. Anim. Biotechnol. 2016, 27, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Bower, N.I.; Reverter, A.; Tan, S.H.; De Jager, N.; Wang, R.; McWilliam, S.M.; Cafe, L.M.; Greenwood, P.L.; Lehnert, S.A. Gene Expression Patterns during Intramuscular Fat Development in Cattle. J. Anim. Sci. 2009, 87, 119–130. [Google Scholar] [CrossRef]
- Park, J.-J.; Berggren, J.R.; Hulver, M.W.; Houmard, J.A.; Hoffman, E.P. GRB14, GPD1, and GDF8 as Potential Network Collaborators in Weight Loss-Induced Improvements in Insulin Action in Human Skeletal Muscle. Physiol. Genom. 2006, 27, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Poulain-Godefroy, O.; Lecoeur, C.; Pattou, F.; Fruhbeck, G.; Froguel, P. Inflammation Is Associated with a Decrease of Lipogenic Factors in Omental Fat in Women. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2008, 295, R1–R7. [Google Scholar] [CrossRef]
- Ishiguro, N.; Baba, T.; Ishida, T.; Takeuchi, K.; Osaki, M.; Araki, N.; Okada, E.; Takahashi, S.; Saito, M.; Watanabe, M.; et al. Carp, a Cardiac Ankyrin-Repeated Protein, and Its New Homologue, Arpp, Are Differentially Expressed in Heart, Skeletal Muscle, and Rhabdomyosarcomas. Am. J. Pathol. 2002, 160, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Lehti, T.M.; Silvennoinen, M.; Kivelä, R.; Kainulainen, H.; Komulainen, J. Effects of Streptozotocin-Induced Diabetes and Physical Training on Gene Expression of Titin-Based Stretch-Sensing Complexes in Mouse Striated Muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E533–E542. [Google Scholar] [CrossRef] [Green Version]
- Vila-Brau, A.; Luisa De Sousa-Coelho, A.; Mayordomo, C.; Haro, D.; Marrero, P.F. Human HMGCS2 Regulates Mitochondrial Fatty Acid Oxidation and FGF21 Expression in HepG2 Cell Line. J. Biol. Chem. 2011, 286, 20423–20430. [Google Scholar] [CrossRef] [Green Version]
- Ros-Freixedes, R.; Gol, S.; Pena, R.N.; Tor, M.; Ibanez-Escriche, N.; Dekkers, J.C.M.; Estany, J. Genome-Wide Association Study Singles Out SCD and LEPR as the Two Main Loci Influencing Intramuscular Fat Content and Fatty Acid Composition in Duroc Pigs. PLoS ONE 2016, 11, e0152496. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Miyazawa, M.; Kamei, A.; Abe, K.; Kojima, T. Ameliorative Effects of Mulberry (Morus alba L.) Leaves on Hyperlipidemia in Rats Fed a High-Fat Diet: Induction of Fatty Acid Oxidation, Inhibition of Lipogenesis, and Suppression of Oxidative Stress. Biosci. Biotechnol. Biochem. 2010, 74, 2385–2395. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Shimano, H.; Yahagi, N.; Kato, T.; Atsumi, A.; Yamamoto, T.; Inoue, N.; Ishikawa, M.; Okada, S.; Ishigaki, N.; et al. Crucial Role of a Long-Chain Fatty Acid Elongase, Elovl6, in Obesity-Induced Insulin Resistance. Nat. Med. 2007, 13, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, W.; Zhang, Z.; Fan, Y.; Xie, X.; Ai, H.; Ma, J.; Xiao, S.; Huang, L.; Ren, J. Genome-Wide Association Analyses for Fatty Acid Composition in Porcine Muscle and Abdominal Fat Tissues. PLoS ONE 2013, 8, e65554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Fang, X.; Zhao, Z.; Yu, X.; Sun, B.; Yu, H.; Yang, R. The Effect of Short/Branched yangChain Acyl-Coenzyme A Dehydrogenase Gene on Triglyceride Synthesis of Bovine Mammary Epithelial Cells. Arch. Anim. Breed. 2018, 61, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Iqbal, A.; Wang, M.; Li, X.; Fang, X.; Yu, H.; Zhao, Z. Transcriptomic Analysis of Short/Branched-Chain Acyl-Coenzyme a Dehydrogenase Knocked Out BMECs Revealed Its Regulatory Effect on Lipid Metabolism. Front. Vet. Sci. 2021, 8, 744287. [Google Scholar] [CrossRef]
- Qi, C.; Zhu, Y.; Pan, J.; Usuda, N.; Maeda, N.; Yeldandi, A.V.; Rao, M.S.; Hashimoto, T.; Reddy, J.K. Absence of Spontaneous Peroxisome Proliferation in Enoyl-CoA Hydratase/l-3-Hydroxyacyl-CoA Dehydrogenase-Deficient Mouse Liver: Further support for the role of fatty acyl coa oxidase in pparα ligand metabolism. J. Biol. Chem. 1999, 274, 15775–15780. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Sun, D.; Zhang, S.; Wang, S.; Wu, X.; Zhang, Q.; Liu, L.; Li, Y.; Qiao, L. Genome Wide Association Study Identifies 20 Novel Promising Genes Associated with Milk Fatty Acid Traits in Chinese Holstein. PLoS ONE 2014, 9, e96186. [Google Scholar] [CrossRef] [Green Version]
- Bruce, C.R.; Brolin, C.; Turner, N.; Cleasby, M.E.; van der Leij, F.R.; Cooney, G.J.; Kraegen, E.W. Overexpression of Carnitine Palmitoyltransferase I in Skeletal Muscle in Vivo Increases Fatty Acid Oxidation and Reduces Triacylglycerol Esterification. Am. J. Physiol.-Endocrinol. Metab. 2007, 292, E1231–E1237. [Google Scholar] [CrossRef] [Green Version]
- Schreurs, M.; Kuipers, F.; van der Leij, F.R. Regulatory Enzymes of Mitochondrial Beta-Oxidation as Targets for Treatment of the Metabolic Syndrome. Obes. Rev. 2010, 11, 380–388. [Google Scholar] [CrossRef]
- van der Leij, F.R.; Bloks, V.W.; Grefhorst, A.; Hoekstra, J.; Gerding, A.; Kooi, K.; Gerbens, F.; te Meerman, G.; Kulpers, F. Gene Expression Profiling in Livers of Mice after Acute Inhibition of Beta-Oxidation. Genomics 2007, 90, 680–689. [Google Scholar] [CrossRef]
- Fang, X.; Zhao, Z.; Jiang, P.; Yu, H.; Xiao, H.; Yang, R. Identification of the Bovine HSL Gene Expression Profiles and Its Association with Fatty Acid Composition and Fat Deposition Traits. Meat Sci. 2017, 131, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Denis, S.; Argmann, C.A.; Jia, Y.; Ferdinandusse, S.; Reddy, J.K.; Wanders, R.J.A. Peroxisomal L-Bifunctional Enzyme (Ehhadh) Is Essential for the Production of Medium-Chain Dicarboxylic Acids. J. Lipid Res. 2012, 53, 1296–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. A Study of the Regulatory Mechanism of the CB1/PPARγ2/PLIN1/HSL Pathway for Fat Metabolism in Cattle. Front. Genet. 2021, 12, 631187. [Google Scholar] [CrossRef]
- Lampidonis, A.D.; Rogdakis, E.; Voutsinas, G.E.; Stravopodis, D.J. The Resurgence of Hormone-Sensitive Lipase (HSL) in Mammalian Lipolysis. Gene 2011, 477, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated Genome Annotation and Pathway Identification Using the KEGG Orthology (KO) as a Controlled Vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Lu, C.; Yang, R.; Liu, B.; Li, Z.; Shen, B.; Yan, S.; Zhang, Y.; Zhang, L.; Zhao, Z. Establishment of Two Types of Mammary Epithelial Cell Lines from Chinese Holstein Dairy Cow. J. Anim. Vet. Adv. 2012, 11, 1166–1172. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | EnsenbleID | Wagyu | Chinese Red Steppe | Log2FC | FDR |
|---|---|---|---|---|---|
| CLDN7 | ENSBTAG00000019448 | 38.11 | 0.68 | 5.80 | 0.00 |
| ENSBTAG00000023318 | ENSBTAG00000023318 | 15.24 | 0.68 | 4.48 | 0.00 |
| ACTC1 | ENSBTAG00000005714 | 1727.76 | 92.25 | 4.23 | 0.04 |
| CCL22 | ENSBTAG00000017718 | 17.49 | 1.36 | 3.68 | 0.00 |
| HUNK | ENSBTAG00000020762 | 29.78 | 2.39 | 3.64 | 0.00 |
| FBN3 | ENSBTAG00000000595 | 23.37 | 3.07 | 2.93 | 0.02 |
| KCNC3 | ENSBTAG00000039935 | 15.62 | 2.39 | 2.71 | 0.01 |
| RFX2 | ENSBTAG00000017661 | 195.96 | 30.82 | 2.67 | 0.00 |
| RIMBP2 | ENSBTAG00000009797 | 14.83 | 2.40 | 2.63 | 0.00 |
| CDS1 | ENSBTAG00000045787 | 10.62 | 1.72 | 2.62 | 0.03 |
| Gene | EnsenbleID | Wagyu | Chinese Red Steppe | Log2FC | FDR |
|---|---|---|---|---|---|
| ASIP | ENSBTAG00000034077 | 0.00 | 165.14 | −20.00 | 0.00 |
| CLRN2 | ENSBTAG00000003357 | 0.00 | 133.14 | −20.00 | 0.00 |
| ENSBTAG00000021906 | ENSBTAG00000021906 | 0.00 | 40.38 | −20.00 | 0.00 |
| PRM1 | ENSBTAG00000021493 | 0.00 | 25.86 | −20.00 | 0.00 |
| HBB | ENSBTAG00000038748 | 1.01 | 134.73 | −7.06 | 0.00 |
| DNER | ENSBTAG00000016063 | 2.17 | 93.40 | −5.42 | 0.00 |
| IRX5 | ENSBTAG00000004838 | 1.95 | 82.41 | −5.40 | 0.00 |
| HP | ENSBTAG00000006354 | 0.31 | 8.25 | −4.74 | 0.00 |
| FBP1 | ENSBTAG00000009733 | 6.89 | 121.92 | −4.15 | 0.04 |
| MPZ | ENSBTAG00000033835 | 15.76 | 214.42 | −3.77 | 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Yang, R.; Lu, X.; Liu, Y.; He, W.; Li, Y.; Yu, H.; Qin, L.; Cao, Y.; Zhao, Z.; et al. RNA-Seq Analysis Identifies Differentially Expressed Genes in the Longissimus dorsi of Wagyu and Chinese Red Steppe Cattle. Int. J. Mol. Sci. 2023, 24, 387. https://doi.org/10.3390/ijms24010387
Li G, Yang R, Lu X, Liu Y, He W, Li Y, Yu H, Qin L, Cao Y, Zhao Z, et al. RNA-Seq Analysis Identifies Differentially Expressed Genes in the Longissimus dorsi of Wagyu and Chinese Red Steppe Cattle. International Journal of Molecular Sciences. 2023; 24(1):387. https://doi.org/10.3390/ijms24010387
Chicago/Turabian StyleLi, Guanghui, Runjun Yang, Xin Lu, Yue Liu, Wei He, Yue Li, Haibin Yu, Lihong Qin, Yang Cao, Zhihui Zhao, and et al. 2023. "RNA-Seq Analysis Identifies Differentially Expressed Genes in the Longissimus dorsi of Wagyu and Chinese Red Steppe Cattle" International Journal of Molecular Sciences 24, no. 1: 387. https://doi.org/10.3390/ijms24010387