
Design and Synthesis of Conformationally Flexible Scaffold as Bitopic Ligands for Potent D3-Selective Antagonists
 , ,
, ,
Abstract
:1. Introduction
2. Results
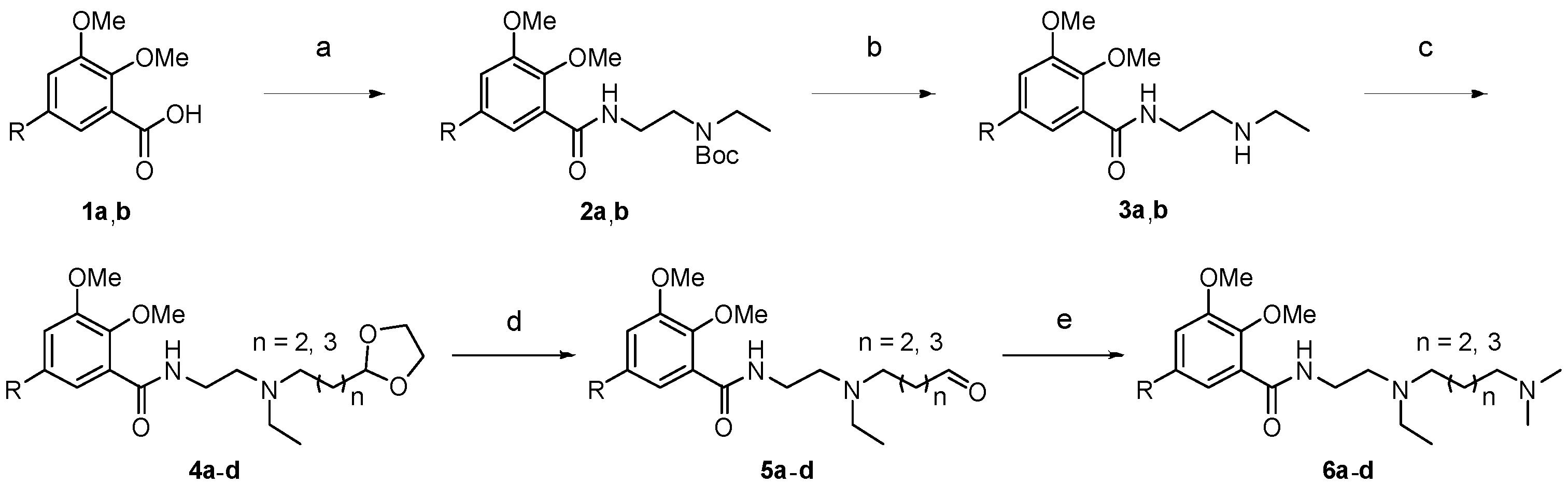
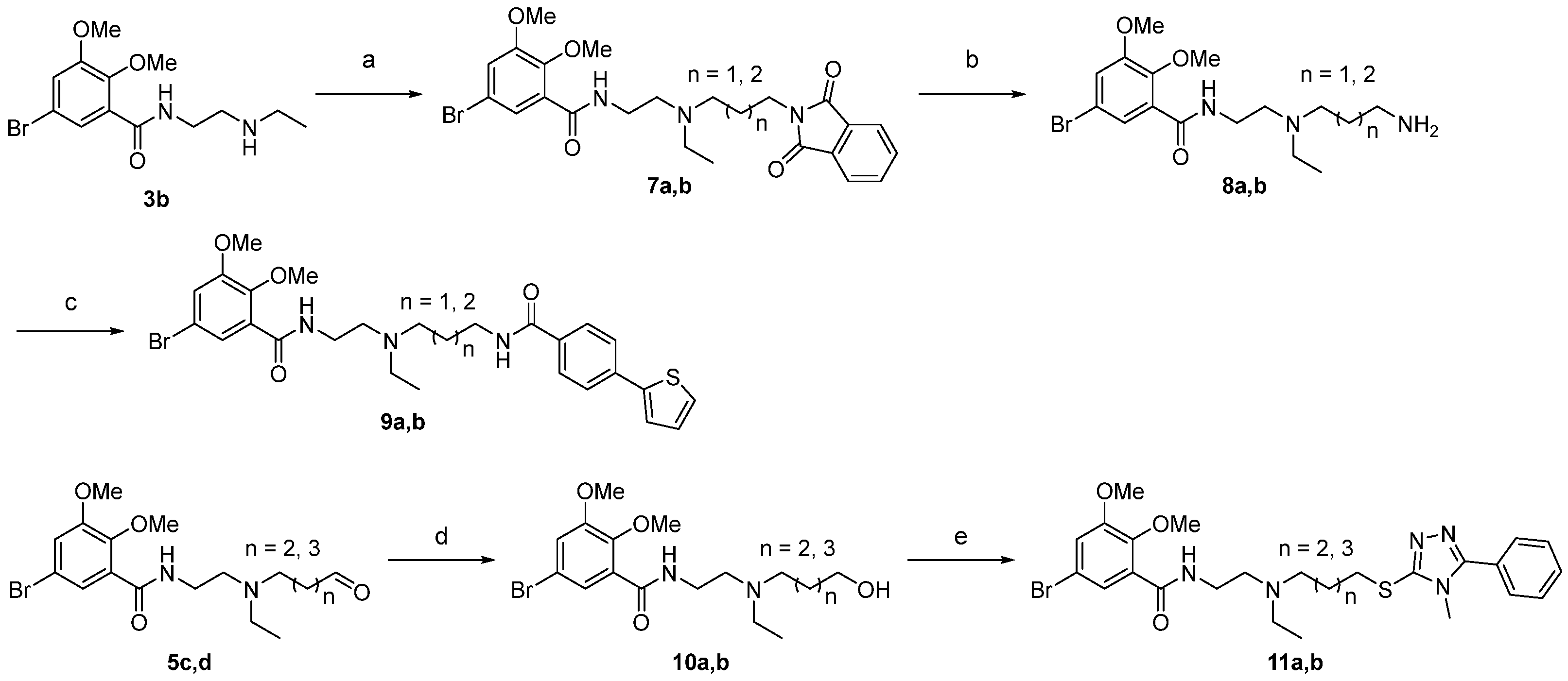
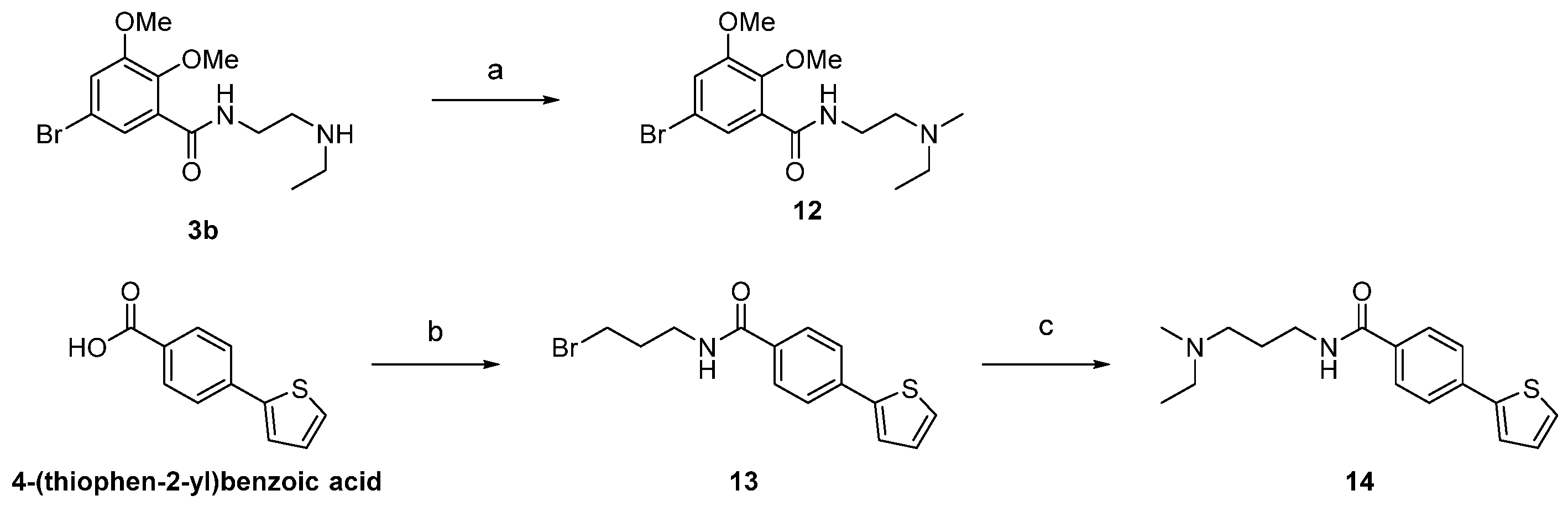
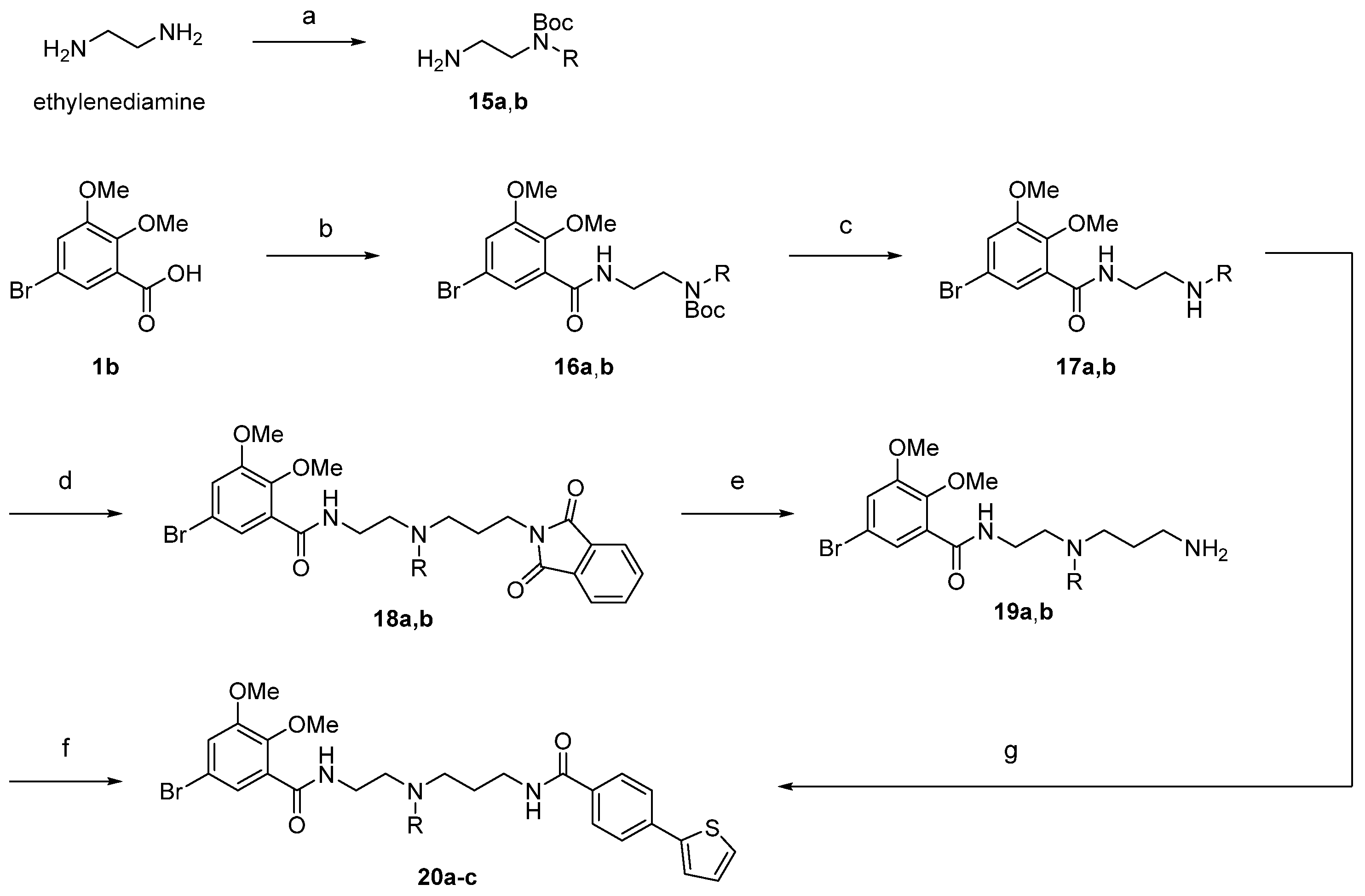
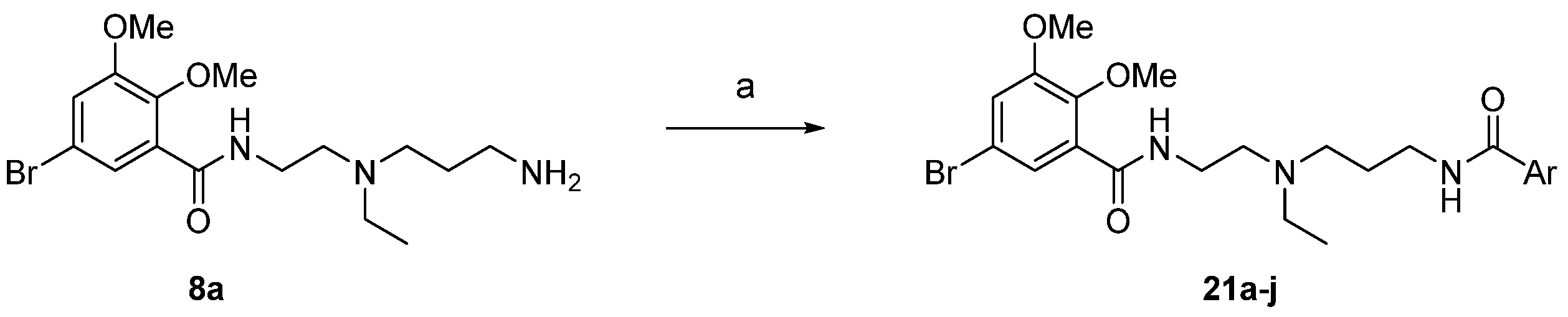
2.1. Chemistry
2.2. SAR Study
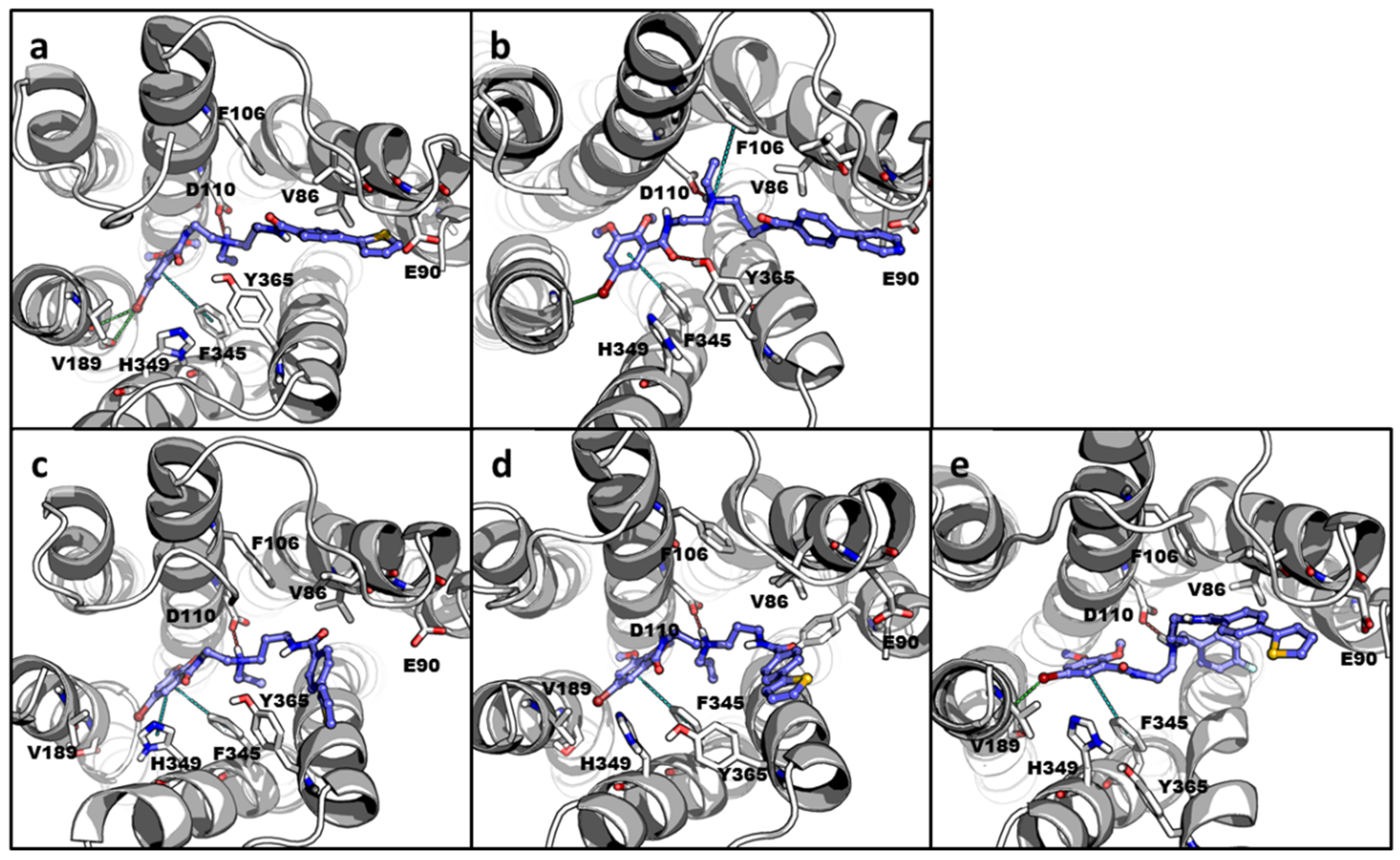
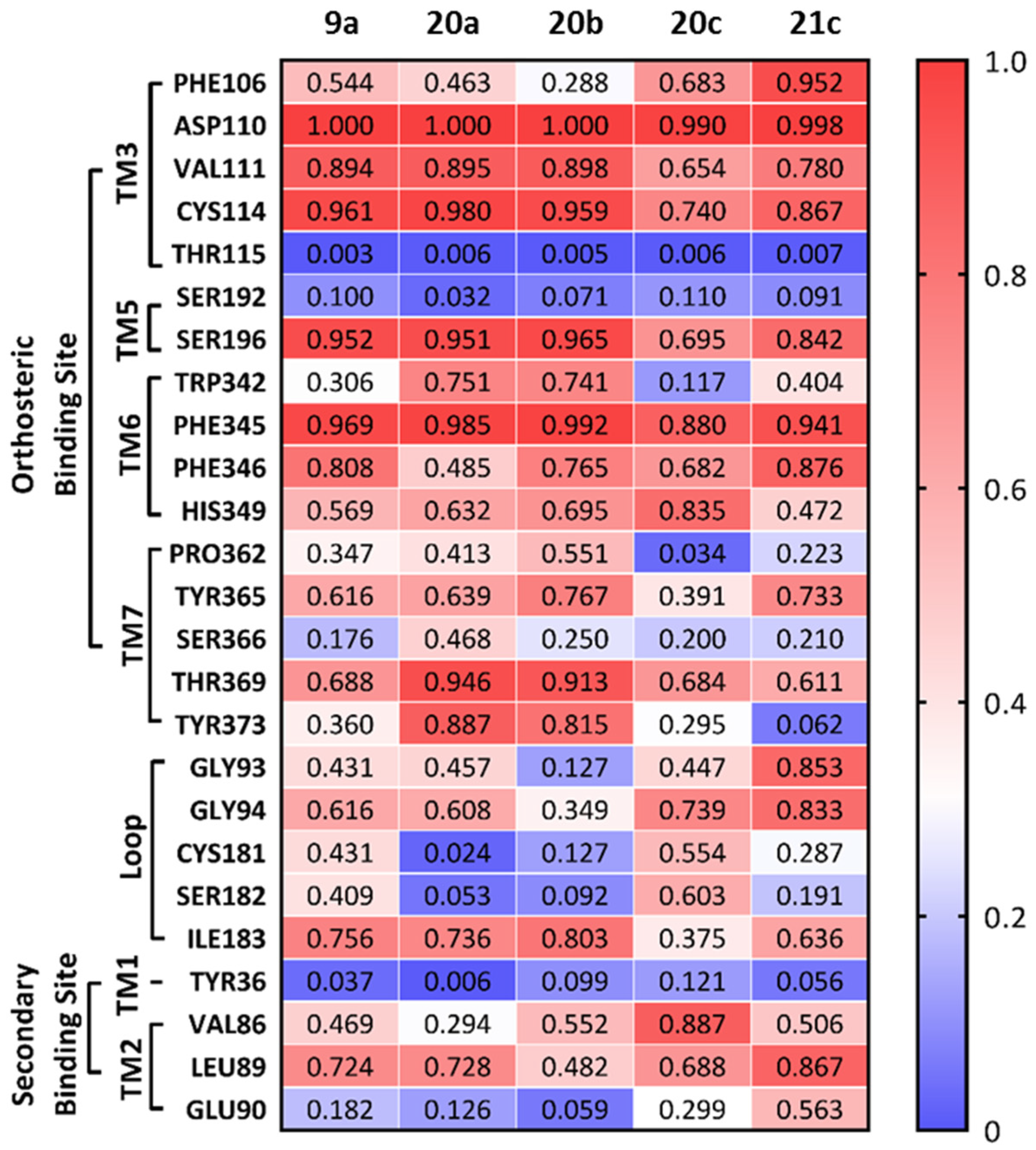
2.3. Molecular Docking and Molecular Dynamics Simulations (MDS)
2.4. Comprehensive Screening for Other GPCRs
3. Discussion
4. Materials and Methods
4.1. General
4.2. Chemistry
4.3. Statistical Analysis
4.3.1. Radioligand Binding Assays
4.3.2. β-Arrestin Recruitment Assay
4.3.3. Molecular Docking and Molecular Dynamics Simulations (MDS)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Luedtke, R.R.; Rangel-Barajas, C.; Malik, M.; Reichert, D.E.; Mach, R.H. Bitropic D3 Dopamine Receptor Selective Compounds as Potential Antipsychotics. Curr. Pharm. Des. 2015, 21, 3700–3724. [Google Scholar] [CrossRef]
- Strange, P.G. Antipsychotic drug action: Antagonism, inverse agonism or partial agonism. Trends Pharmacol. Sci. 2008, 29, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Fowler, J.S.; Wang, G.J.; Swanson, J.M. Dopamine in drug abuse and addiction: Results from imaging studies and treatment implications. Mol. Psychiatry 2004, 9, 557–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.G.; Newman, A.H.; Gardner, E.L.; Ashby, C.R., Jr.; Heidbreder, C.A.; Pak, A.C.; Peng, X.Q.; Xi, Z.X. Acute administration of SB-277011A, NGB 2904, or BP 897 inhibits cocaine cue-induced reinstatement of drug-seeking behavior in rats: Role of dopamine D3 receptors. Synapse 2005, 57, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, P.; Le Foll, B. The dopamine D3 receptor, a quarter century later. Eur. J. Neurosci. 2017, 45, 2–19. [Google Scholar] [CrossRef]
- Sokoloff, P.; Giros, B.; Martres, M.P.; Bouthenet, M.L.; Schwartz, J.C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 1990, 347, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.H.; Blaylock, B.L.; Nader, M.A.; Bergman, J.; Sibley, D.R.; Skolnick, P. Medication discovery for addiction: Translating the dopamine D3 receptor hypothesis. Biochem. Pharmacol. 2012, 84, 882–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.H.; Grundt, P.; Nader, M.A. Dopamine D3 receptor partial agonists and antagonists as potential drug abuse therapeutic agents. J. Med. Chem. 2005, 48, 3663–3679. [Google Scholar] [CrossRef]
- You, Z.B.; Gao, J.T.; Bi, G.H.; He, Y.; Boateng, C.; Cao, J.; Gardner, E.L.; Newman, A.H.; Xi, Z.X. The novel dopamine D3 receptor antagonists/partial agonists CAB2-015 and BAK4-54 inhibit oxycodone-taking and oxycodone-seeking behavior in rats. Neuropharmacology 2017, 126, 190–199. [Google Scholar] [CrossRef]
- Newman, A.H.; Xi, Z.X.; Heidbreder, C. Current Perspectives on Selective Dopamine D(3) Receptor Antagonists/Partial Agonists as Pharmacotherapeutics for Opioid and Psychostimulant Use Disorders; Springer: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- You, Z.B.; Bi, G.H.; Galaj, E.; Kumar, V.; Cao, J.; Gadiano, A.; Rais, R.; Slusher, B.S.; Gardner, E.L.; Xi, Z.X.; et al. Dopamine D(3)R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 2019, 44, 1415–1424. [Google Scholar] [CrossRef]
- Boeckler, F.; Gmeiner, P. Dopamine D3 receptor ligands: Recent advances in the control of subtype selectivity and intrinsic activity. Biochim. Biophys. Acta 2007, 1768, 871–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Mach, R.H.; Luedtke, R.R.; Reichert, D.E. Subtype selectivity of dopamine receptor ligands: Insights from structure and ligand-based methods. J. Chem. Inf. Model. 2010, 50, 1970–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.H.; Beuming, T.; Banala, A.K.; Donthamsetti, P.; Pongetti, K.; LaBounty, A.; Levy, B.; Cao, J.; Michino, M.; Luedtke, R.R.; et al. Molecular determinants of selectivity and efficacy at the dopamine D3 receptor. J. Med. Chem. 2012, 55, 6689–6699. [Google Scholar] [CrossRef] [PubMed]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Chen, X.; Brodbeck, R.; Primus, R.; Braun, J.; Wasley, J.W.; Thurkauf, A. NGB 2904 and NGB 2849: Two highly selective dopamine D3 receptor antagonists. Bioorg. Med. Chem. Lett. 1998, 8, 2715–2718. [Google Scholar] [CrossRef]
- Reavill, C.; Taylor, S.G.; Wood, M.D.; Ashmeade, T.; Austin, N.E.; Avenell, K.Y.; Boyfield, I.; Branch, C.L.; Cilia, J.; Coldwell, M.C.; et al. Pharmacological actions of a novel, high-affinity, and selective human dopamine D(3) receptor antagonist, SB-277011-A. J. Pharmacol. Exp. Ther. 2000, 294, 1154–1165. [Google Scholar]
- Mach, R.H.; Huang, Y.; Freeman, R.A.; Wu, L.; Vangveravong, S.; Luedtke, R.R. Conformationally-flexible benzamide analogues as dopamine D3 and sigma 2 receptor ligands. Bioorg. Med. Chem. Lett. 2004, 14, 195–202. [Google Scholar] [CrossRef]
- Pilla, M.; Perachon, S.; Sautel, F.; Garrido, F.; Mann, A.; Wermuth, C.G.; Schwartz, J.C.; Everitt, B.J.; Sokoloff, P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature 1999, 400, 371–375. [Google Scholar] [CrossRef]
- Stemp, G.; Ashmeade, T.; Branch, C.L.; Hadley, M.S.; Hunter, A.J.; Johnson, C.N.; Nash, D.J.; Thewlis, K.M.; Vong, A.K.; Austin, N.E.; et al. Design and synthesis of trans-N-[4-[2-(6-cyano-1,2,3, 4-tetrahydroisoquinolin-2-yl)ethyl]cyclohexyl]-4-quinolinecarboxamide (SB-277011): A potent and selective dopamine D(3) receptor antagonist with high oral bioavailability and CNS penetration in the rat. J. Med. Chem. 2000, 43, 1878–1885. [Google Scholar] [CrossRef]
- Mach, R.H.; Tu, Z.; Xu, J.; Li, S.; Jones, L.A.; Taylor, M.; Luedtke, R.R.; Derdeyn, C.P.; Perlmutter, J.S.; Mintun, M.A. Endogenous dopamine (DA) competes with the binding of a radiolabeled D(3) receptor partial agonist in vivo: A positron emission tomography study. Synapse 2011, 65, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Maramai, S.; Gemma, S.; Brogi, S.; Campiani, G.; Butini, S.; Stark, H.; Brindisi, M. Dopamine D3 Receptor Antagonists as Potential Therapeutics for the Treatment of Neurological Diseases. Front. Neurosci. 2016, 10, 451. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.B.; Kumar, V.; Bonifazi, A.; Guerrero, A.M.; Cemaj, S.L.; Gadiano, A.; Lam, J.; Xi, Z.X.; Rais, R.; Slusher, B.S.; et al. Investigation of Novel Primary and Secondary Pharmacophores and 3-Substitution in the Linking Chain of a Series of Highly Selective and Bitopic Dopamine D3 Receptor Antagonists and Partial Agonists. J. Med. Chem. 2019, 62, 9061–9077. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Taylor, M.; Griffin, S.A.; McInnis, T.; Sumien, N.; Mach, R.H.; Luedtke, R.R. Evaluation of Substituted N-Phenylpiperazine Analogs as D3 vs. D2 Dopamine Receptor Subtype Selective Ligands. Molecules 2021, 26, 3182. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, A.; Newman, A.H.; Keck, T.M.; Gervasoni, S.; Vistoli, G.; Del Bello, F.; Giorgioni, G.; Pavletic, P.; Quaglia, W.; Piergentili, A. Scaffold Hybridization Strategy Leads to the Discovery of Dopamine D(3) Receptor-Selective or Multitarget Bitopic Ligands Potentially Useful for Central Nervous System Disorders. ACS Chem. Neurosci. 2021, 12, 3638–3649. [Google Scholar] [CrossRef]
- Reilly, S.W.; Riad, A.A.; Hsieh, C.J.; Sahlholm, K.; Jacome, D.A.; Griffin, S.; Taylor, M.; Weng, C.C.; Xu, K.; Kirschner, N.; et al. Leveraging a Low-Affinity Diazaspiro Orthosteric Fragment to Reduce Dopamine D3 Receptor (D3R) Ligand Promiscuity across Highly Conserved Aminergic G-Protein-Coupled Receptors (GPCRs). J. Med. Chem. 2019, 62, 5132–5147. [Google Scholar] [CrossRef]
- Wassouf, Z.; Schulze-Hentrich, J.M. Alpha-synuclein at the nexus of genes and environment: The impact of environmental enrichment and stress on brain health and disease. J. Neurochem. 2019, 150, 591–604. [Google Scholar] [CrossRef]
- Zingales, V.; Torrisi, S.A.; Leggio, G.M.; Bucolo, C.; Drago, F.; Salomone, S. Pharmacological and Genetic Evidence of Dopamine Receptor 3-Mediated Vasoconstriction in Isolated Mouse Aorta. Biomolecules 2021, 11, 418. [Google Scholar] [CrossRef]
- Hsieh, C.J.; Riad, A.; Lee, J.Y.; Sahlholm, K.; Xu, K.; Luedtke, R.R.; Mach, R.H. Interaction of Ligands for PET with the Dopamine D3 Receptor: In Silico and In Vitro Methods. Biomolecules 2021, 11, 529. [Google Scholar] [CrossRef]
- Micheli, F.; Arista, L.; Bonanomi, G.; Blaney, F.E.; Braggio, S.; Capelli, A.M.; Checchia, A.; Damiani, F.; Di-Fabio, R.; Fontana, S.; et al. 1,2,4-Triazolyl azabicyclo [3.1.0]hexanes: A new series of potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2010, 53, 374–391. [Google Scholar] [CrossRef]
- Mugnaini, M.; Iavarone, L.; Cavallini, P.; Griffante, C.; Oliosi, B.; Savoia, C.; Beaver, J.; Rabiner, E.A.; Micheli, F.; Heidbreder, C.; et al. Occupancy of brain dopamine D3 receptors and drug craving: A translational approach. Neuropsychopharmacology 2013, 38, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.; Nestor, L.J.; McGonigle, J.; Paterson, L.; Boyapati, V.; Ersche, K.D.; Flechais, R.; Kuchibatla, S.; Metastasio, A.; Orban, C.; et al. Acute D3 Antagonist GSK598809 Selectively Enhances Neural Response During Monetary Reward Anticipation in Drug and Alcohol Dependence. Neuropsychopharmacology 2017, 42, 1925–1926. [Google Scholar] [CrossRef] [PubMed]
- Micheli, F.; Bacchi, A.; Braggio, S.; Castelletti, L.; Cavallini, P.; Cavanni, P.; Cremonesi, S.; Cin, M.D.; Feriani, A.; Gehanne, S.; et al. 1,2,4-Triazolyl 5-Azaspiro[2.4]heptanes: Lead Identification and Early Lead Optimization of a New Series of Potent and Selective Dopamine D3 Receptor Antagonists. J. Med. Chem. 2016, 59, 8549–8576. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.W.; Griffin, S.; Taylor, M.; Sahlholm, K.; Weng, C.C.; Xu, K.; Jacome, D.A.; Luedtke, R.R.; Mach, R.H. Highly Selective Dopamine D3 Receptor Antagonists with Arylated Diazaspiro Alkane Cores. J. Med. Chem. 2017, 60, 9905–9910. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Levant, B.; Jiang, C.; Keck, T.M.; Newman, A.H.; Wang, S. Tranylcypromine substituted cis-hydroxycyclobutylnaphthamides as potent and selective dopamine D(3) receptor antagonists. J. Med. Chem. 2014, 57, 4962–4968. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Zhou, Q.; Yan, W.; Sun, J.; Kozikowski, A.P.; Zhao, S.; Huang, X.P.; Cheng, J. Design and Synthesis of Bitopic 2-Phenylcyclopropylmethylamine (PCPMA) Derivatives as Selective Dopamine D3 Receptor Ligands. J. Med. Chem. 2020, 63, 4579–4602. [Google Scholar] [CrossRef]
- Bennacef, I.; Salinas, C.A.; Bonasera, T.A.; Gunn, R.N.; Audrain, H.; Jakobsen, S.; Nabulsi, N.; Weinzimmer, D.; Carson, R.E.; Huang, Y.; et al. Dopamine D3 receptor antagonists: The quest for a potentially selective PET ligand. Part 3: Radiosynthesis and in vivo studies. Bioorg. Med. Chem. Lett. 2009, 19, 5056–5059. [Google Scholar] [CrossRef]
- Keck, T.M.; John, W.S.; Czoty, P.W.; Nader, M.A.; Newman, A.H. Identifying Medication Targets for Psychostimulant Addiction: Unraveling the Dopamine D3 Receptor Hypothesis. J. Med. Chem. 2015, 58, 5361–5380. [Google Scholar] [CrossRef] [Green Version]
- Appel, N.M.; Li, S.H.; Holmes, T.H.; Acri, J.B. Dopamine D3 Receptor Antagonist (GSK598809) Potentiates the Hypertensive Effects of Cocaine in Conscious, Freely-Moving Dogs. J. Pharmacol. Exp. Ther. 2015, 354, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Battiti, F.O.; Cemaj, S.L.; Guerrero, A.M.; Shaik, A.B.; Lam, J.; Rais, R.; Slusher, B.S.; Deschamps, J.R.; Imler, G.H.; Newman, A.H.; et al. The Significance of Chirality in Drug Design and Synthesis of Bitopic Ligands as D3 Receptor (D3R) Selective Agonists. J. Med. Chem. 2019, 62, 6287–6314. [Google Scholar] [CrossRef]
- Shaik, A.B.; Boateng, C.A.; Battiti, F.O.; Bonifazi, A.; Cao, J.; Chen, L.; Chitsazi, R.; Ravi, S.; Lee, K.H.; Shi, L.; et al. Structure Activity Relationships for a Series of Eticlopride-Based Dopamine D2/D3 Receptor Bitopic Ligands. J. Med. Chem. 2021, 64, 15313–15333. [Google Scholar] [CrossRef]
- Mukherjee, J.; Yang, Z.Y.; Das, M.K.; Brown, T. Fluorinated benzamide neuroleptics--III. Development of (S)-N-[(1-allyl-2-pyrrolidinyl)methyl]-5-(3-[18F]fluoropropyl)-2, 3-dimethoxybenzamide as an improved dopamine D-2 receptor tracer. Nucl. Med. Biol. 1995, 22, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, J.; Constantinescu, C.C.; Hoang, A.T.; Jerjian, T.; Majji, D.; Pan, M.L. Dopamine D3 receptor binding of (18)F-fallypride: Evaluation using in vitro and in vivo PET imaging studies. Synapse 2015, 69, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halldin, C.; Farde, L.; Hogberg, T.; Mohell, N.; Hall, H.; Suhara, T.; Karlsson, P.; Nakashima, Y.; Swahn, C.G. Carbon-11-FLB 457: A radioligand for extrastriatal D2 dopamine receptors. J. Nucl. Med. 1995, 36, 1275–1281. [Google Scholar] [PubMed]
- Yang, D.; Kefi, S.; Audinot, V.; Millan, M.J.; Langlois, M. Benzamides derived from 1,2-diaminocyclopropane as novel ligands for human D2 and D3 dopamine receptors. Bioorg. Med. Chem. 2000, 8, 321–327. [Google Scholar] [CrossRef]
- Chen, P.J.; Taylor, M.; Griffin, S.A.; Amani, A.; Hayatshahi, H.; Korzekwa, K.; Ye, M.; Mach, R.H.; Liu, J.; Luedtke, R.R.; et al. Design, synthesis, and evaluation of N-(4-(4-phenyl piperazin-1-yl)butyl)-4-(thiophen-3-yl)benzamides as selective dopamine D3 receptor ligands. Bioorg. Med. Chem. Lett. 2019, 29, 2690–2694. [Google Scholar] [CrossRef]
- Tu, Z.; Li, S.; Cui, J.; Xu, J.; Taylor, M.; Ho, D.; Luedtke, R.R.; Mach, R.H. Synthesis and pharmacological evaluation of fluorine-containing D(3) dopamine receptor ligands. J. Med. Chem. 2011, 54, 1555–1564. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Barajas, C.; Malik, M.; Taylor, M.; Neve, K.A.; Mach, R.H.; Luedtke, R.R. Characterization of [(3) H]LS-3-134, a novel arylamide phenylpiperazine D3 dopamine receptor selective radioligand. J. Neurochem. 2014, 131, 418–431. [Google Scholar] [CrossRef] [Green Version]
- Luedtke, R.R.; Freeman, R.A.; Boundy, V.A.; Martin, M.W.; Huang, Y.; Mach, R.H. Characterization of (125)I-IABN, a novel azabicyclononane benzamide selective for D2-like dopamine receptors. Synapse 2000, 38, 438–449. [Google Scholar] [CrossRef]
- Wang, T.; Li, Z.; Cvijic, M.E.; Krause, C.; Zhang, L.; Sum, C.S. Measurement of beta-Arrestin Recruitment for GPCR Targets. In Assay Guidance Manual; Markossian, S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C., Baell, J., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Bethesda: Rockville, MD, USA, 2004. [Google Scholar]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Zhao, X.; Jones, A.; Olson, K.R.; Peng, K.; Wehrman, T.; Park, A.; Mallari, R.; Nebalasca, D.; Young, S.W.; Xiao, S.H. A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J. Biomol. Screen 2008, 13, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Hayatshahi, H.S.; Xu, K.; Griffin, S.A.; Taylor, M.; Mach, R.H.; Liu, J.; Luedtke, R.R. Analogues of Arylamide Phenylpiperazine Ligands To Investigate the Factors Influencing D3 Dopamine Receptor Bitropic Binding and Receptor Subtype Selectivity. ACS Chem. Neurosci. 2018, 9, 2972–2983. [Google Scholar] [CrossRef] [PubMed]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.; Tu, Z.; McElveen, E.; Xu, J.; Taylor, M.; Luedtke, R.R.; Mach, R.H. Synthesis and in vitro binding of N-phenyl piperazine analogs as potential dopamine D3 receptor ligands. Bioorg. Med. Chem. 2005, 13, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Vangveravong, S.; Zhang, Z.; Taylor, M.; Bearden, M.; Xu, J.; Cui, J.; Wang, W.; Luedtke, R.R.; Mach, R.H. Synthesis and characterization of selective dopamine D(2) receptor ligands using aripiprazole as the lead compound. Bioorg. Med. Chem. 2011, 19, 3502–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Vangveravong, S.; Li, S.; Fan, J.; Jones, L.A.; Cui, J.; Wang, R.; Tu, Z.; Chu, W.; Perlmutter, J.S.; et al. Positron emission tomography imaging of dopamine D2 receptors using a highly selective radiolabeled D2 receptor partial agonist. Neuroimage 2013, 71, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Wang, Q.; Mishra, Y.; Xu, J.; Reichert, D.E.; Malik, M.; Taylor, M.; Luedtke, R.R.; Mach, R.H. Synthesis, pharmacological evaluation and molecular modeling studies of triazole containing dopamine D3 receptor ligands. Bioorg. Med. Chem. Lett. 2015, 25, 519–523. [Google Scholar] [CrossRef] [Green Version]
- Vilkman, H.; Kajander, J.; Aalto, S.; Vahlberg, T.; Nagren, K.; Allonen, T.; Syvalahti, E.; Hietala, J. The effects of lorazepam on extrastriatal dopamine D(2/3)-receptors-A double-blind randomized placebo-controlled PET study. Psychiatry Res. 2009, 174, 130–137. [Google Scholar] [CrossRef]
- Lober, S.; Hubner, H.; Tschammer, N.; Gmeiner, P. Recent advances in the search for D3- and D4-selective drugs: Probes, models and candidates. Trends Pharmacol. Sci. 2011, 32, 148–157. [Google Scholar] [CrossRef]
- Gao, M.; Wang, M.; Mock, B.H.; Glick-Wilson, B.E.; Yoder, K.K.; Hutchins, G.D.; Zheng, Q.H. An improved synthesis of dopamine D2/D3 receptor radioligands [11C]fallypride and [18F]fallypride. Appl. Radiat. Isot. 2010, 68, 1079–1086. [Google Scholar] [CrossRef]
- Pettit, G.R.; Piatak, D.M. Hydrogen Bromide-Acetic Acid Demethylation of 2,3-Dimethoxy-6-bromobenzoic Acid. An Example of Concomitant Bromine Migration1,2. J. Org. Chem. 1960, 25, 721–725. [Google Scholar] [CrossRef]
- Komiya, C.; Aihara, K.; Morishita, K.; Ding, H.; Inokuma, T.; Shigenaga, A.; Otaka, A. Development of an Intein-Inspired Amide Cleavage Chemical Device. J. Org. Chem. 2016, 81, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Varseev, G.N.; Maier, M.E. A novel palladium-catalyzed arylation-dehydroaromatization reaction: Synthesis of 7-aryltetralones. Org. Lett. 2005, 7, 3881–3884. [Google Scholar] [CrossRef]
- O'Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, J.; Jo, S.; Brooks, C.L., 3rd; Lee, H.S.; Im, W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem. 2017, 38, 1879–1886. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O'Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Venable, R.M.; Sodt, A.J.; Rogaski, B.; Rui, H.; Hatcher, E.; MacKerell, A.D., Jr.; Pastor, R.W.; Klauda, J.B. CHARMM all-atom additive force field for sphingomyelin: Elucidation of hydrogen bonding and of positive curvature. Biophys. J. 2014, 107, 134–145. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Case, D.B.-S.I.; Brozell, S.; Cerutti, D.; Cheatham III, T.; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; Gohlke, H.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
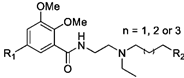
| Cmpd | R1 | R2 | n | Ki ± SEM (nM) b | D2/D3 | IC50 (nM) c | Imax (% Control) d | |
|---|---|---|---|---|---|---|---|---|
| D3 | D2 | |||||||
| 6a |  | 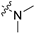 | 2 | 1763 ± 942 | 4564 ± 2445 | 2.6 | >1000 | 92.7 ± 5.4 |
| 6b |  | 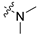 | 3 | 1236 ± 438 | 2810 ± 571 | 2.3 | 430 ± 467 | 98.4 ± 2.9 |
| 6c |  | 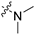 | 2 | 1627 ± 165 | 5416 ± 1043 | 3.3 | >1000 | 98.0 ± 5.6 |
| 6d |  | 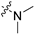 | 3 | 141 ± 26 | 449 ± 90 | 3.2 | 152 ± 185 | 94.0 ± 6.4 |
| 9a | 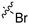 | 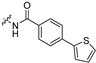 | 1 | 1.0 ± 0.01 | 169 ± 4 | 169 | 14.0 ± 7.4 | 67.3 ± 18.9 |
| 9b | 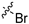 |  | 2 | 8.0 ± 0.8 | 103 ± 14 | 12.9 | 104 ± 136 | 70.8 ± 7.3 |
| 11a | 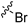 | 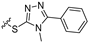 | 2 | 125 ± 2 | 342 ± 23 | 2.7 | NT e | NT |
| 11b | 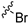 | 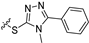 | 3 | 22.2 ± 1.8 | 89.7 ± 3.6 | 4.6 | NT | NT |

| Cmpd | R3 | Ki ± SEM (nM) b | D2/D3 | IC50 (nM) c | Imax (% Control) d | |
|---|---|---|---|---|---|---|
| D3 | D2 | |||||
| 9a | 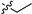 | 1.0 ± 0.01 | 169 ± 4 | 169 | 14.0 ± 7.4 | 67.3 ± 18.9 |
| 20a |  | 2.7 ± 0.4 | 259 ± 23 | 110 | 26.5 ± 12.9 | 88.1 ± 20.4 |
| 20b |  | 5.8 ± 1.0 | 243 ± 15 | 46.7 | 51.6 ± 40.8 | 100.6 ± 7.9 |
| 20c | 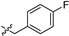 | 299 ± 101 | 574 ± 170 | 1.9 | NT e | NT |

| Cmpd | Ar | Ki ± SEM (nM) b | D2/D3 | IC50 (nM) c | Imax (% Control) d | cLogP e | |
|---|---|---|---|---|---|---|---|
| D3 | D2 | ||||||
| 21a |  | 1.2 ± 0.2 | 169 ± 12 | 141 | 16.4 ± 7.7 | 66.1 ± 17.6 | 4.12 |
| 21b |  | 13.2 ± 0.5 | 525 ± 72 | 39.8 | 19.6 ± 23.7 | 75.2 ± 4.8 | 3.21 |
| 21c | 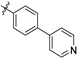 | 1.1 ± 0.1 | 107 ± 5 | 97.3 | 1.3 ± 1.0 | 78.2 ± 18.0 | 3.47 |
| 21d | 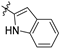 | 0.8 ± 0.2 | 148 ± 9 | 180.5 | 9.3 ± 12.0 | 57.7 ± 26.9 | 2.72 |
| 21e | 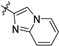 | 2.8 ± 0.5 | 142 ± 16 | 50.7 | 4.3 ± 2.5 | 85.4 ± 6.0 | 2.41 |
| 21f | 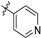 | 6.1 ± 0.6 | 327 ± 32 | 53.7 | 2.7 ± 0.6 | 85.7 ± 13.1 | 1.79 |
| 21g | 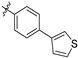 | 2.5 ± 0.3 | 312 ± 18 | 125 | 36.8 ± 35.7 | 50.1 ± 20.9 | 4.78 |
| 21h | 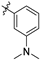 | 11.2 ± 1.4 | 248 ± 19 | 22.1 | 4.1 ± 2.1 | 73.8 ± 14.9 | 3.41 |
| 21i | 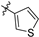 | 7.0 ± 0.5 | 304 ± 7 | 43.4 | 2.7 ± 0.2 | 99.6 ± 20.7 | 3.05 |
| 21j | 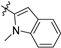 | 3.1 ± 0.5 | 192 ± 22 | 61.9 | 4.8 ± 0.8 | 81.5 ± 10.1 | 2.96 |
| Cmpd | Docking | MDS | |
|---|---|---|---|
| Distance to ASP110 (Å) | Binding Energy (kcal/mol) | Ligand RMSD (Å) | |
| fallypride a | 2.7 | −7.71 | 2.08 ± 0.33 |
| 9a | 2.6 | −9.74 | 3.18 ± 0.54 |
| 20a | 2.9 | −10.11 | 2.18 ± 0.74 |
| 20b | 2.8 | −10.00 | 2.29 ± 0.60 |
| 20c | 2.9 | −10.22 | 3.00 ± 0.82 |
| 21c | 2.7 | −10.10 | 2.45 ± 0.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.Y.; Lee, J.Y.; Hsieh, C.-J.; Taylor, M.; Luedtke, R.R.; Mach, R.H. Design and Synthesis of Conformationally Flexible Scaffold as Bitopic Ligands for Potent D3-Selective Antagonists. Int. J. Mol. Sci. 2023, 24, 432. https://doi.org/10.3390/ijms24010432
Kim HY, Lee JY, Hsieh C-J, Taylor M, Luedtke RR, Mach RH. Design and Synthesis of Conformationally Flexible Scaffold as Bitopic Ligands for Potent D3-Selective Antagonists. International Journal of Molecular Sciences. 2023; 24(1):432. https://doi.org/10.3390/ijms24010432
Chicago/Turabian StyleKim, Ho Young, Ji Youn Lee, Chia-Ju Hsieh, Michelle Taylor, Robert R. Luedtke, and Robert H. Mach. 2023. "Design and Synthesis of Conformationally Flexible Scaffold as Bitopic Ligands for Potent D3-Selective Antagonists" International Journal of Molecular Sciences 24, no. 1: 432. https://doi.org/10.3390/ijms24010432