
Extracellular Vesicles from Campylobacter jejuni CDT-Treated Caco-2 Cells Inhibit Proliferation of Tumour Intestinal Caco-2 Cells and Myeloid U937 Cells: Detailing the Global Cell Response for Potential Application in Anti-Tumour Strategies

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
1.1. Significance Statement
1.2. CDT: Structure, Function, and Its Potential Applications
1.3. Extracellular Vesicle Features and Their Role in Host–Bacteria Interactions
2. Results and Discussion
2.1. PART I: Building the Reference Grid for Cell Response to CDT and EV Characterisation
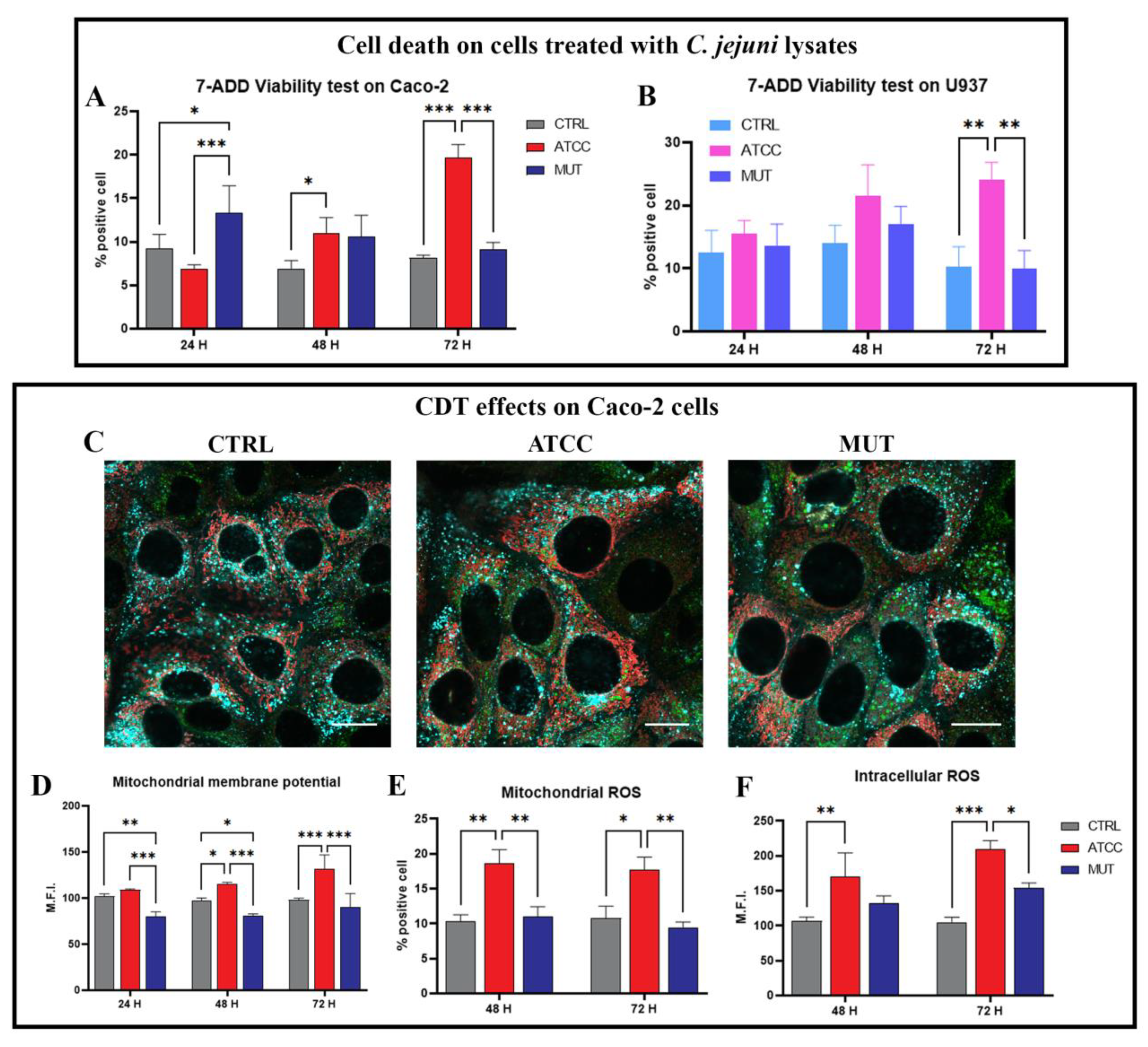
Flow Cytometric Analysis of Viability and Mitochondria Health: Transmembrane Potential and ROS Generation
2.2. Lysosomal and Autophagic-Like Vacuoles Involvement and Aberrant Endocytic Activity Detection
2.3. Lysosomal Exocytosis, EV Release, and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular
2.4. CDT Effects on Caco-2 Cells and U937 Cells: Evaluation of Cell Cycle Blocking
2.5. PART II: Detecting the Global Cell Response Induced by EVs and Their Application as Antiproliferative Signal Carriers
Co-Culture Results: Subcellular Response of the Co-Cultured Cell Line
2.6. Extracellular Vesicles from Caco-2 Infected-Cells on Homologous Caco-2 and Heterologous U937 Cells
2.7. EV Uptake and Evaluation of Specific CDT Effects
3. Materials and Methods
3.1. Bacterial Strains
3.2. C. jejuni Cell Lysate Preparation
3.3. Cell Culture
3.4. Co-Culture
3.5. Pre-Treatment of Cells with C. jejuni Lysates
3.6. Flow Cytometry (FC) and Confocal Microscopy (CM) Staining Assessment of Lysosomal Involvement and Lysosomal Exocytosis
3.7. Autophagic-like Vacuole Detection
3.8. Determination of Mitochondria Membrane Potential (MMP) and Mitochondrial Reactive Oxygen Species (ROS)
3.9. Evaluation of Intracellular ROS Content (Mainly H2O2)
3.10. Cell Cycle Analysis
3.11. Intracellular Detection of Prb, NF-kB, and PCNA
3.12. Extracellular Vesicle FACS Detection
3.13. Scanning Electron Microscopy
3.14. Extracellular Vesicle Isolation
3.15. Nanoparticle Tracking Analysis (NTA)
3.16. miRNA Quantification
3.17. Extracellular Vesicle PKH67 Staining
3.18. Analysis of the In Vitro EV Uptake
3.19. Flow Cytometric Absolute Count
3.20. Cytometric Investigations
3.21. Confocal Microscopy
3.22. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Faïs, T.; Delmas, J.; Serres, A.; Bonnet, R.; Dalmasso, G. Impact of CDT toxin on human diseases. Toxins 2016, 8, 220. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Ge, Z.; Fox, J.G.; Schauer, D.B. Disruption of tight junctions and induction of proinflammatory cytokine responses in colonic epithelial cells by Campylobacter jejuni. Infect. Immun. 2006, 74, 6581–6589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnee, A.E.; Petri, W.A. Campylobacter jejuni and associated immune mechanisms: Short-term effects and long-term implications for infants in low-income countries. Curr. Opin. Infect. Dis. 2017, 30, 322–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwell, C.A.; Dreyfus, L.A. DNase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Lara-Tejero, M.; Galán, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Pons, B.J.; Loiseau, N.; Hashim, S.; Tadrist, S.; Mirey, G.; Vignard, J. Functional Study of Haemophilus ducreyi Cytolethal Distending Toxin Subunit B. Toxins 2020, 12, 530. [Google Scholar] [CrossRef]
- Smith, J.L.; Bayles, D.O. The Contribution of Cytolethal Distending Toxin to Bacterial Pathogenesis. Crit. Rev. Microbiol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Canonico, B.; Campana, R.; Luchetti, F.; Arcangeletti, M.; Betti, M.; Cesarini, E.; Ciacci, C.; Vittoria, E.; Galli, L.; Papa, S.; et al. Campylobacter jejuni cell lysates differently target mitochondria and lysosomes on HeLa cells. Apoptosis 2014, 19, 1225–1242. [Google Scholar] [CrossRef]
- Canonico, B.; Cesarini, E.; Montanari, M.; Di Sario, G.; Campana, R.; Galluzzi, L.; Sola, F.; Gundogdu, O.; Luchetti, F.; Diotallevi, A.; et al. Rapamycin re-directs lysosome network, stimulates er-remodeling, involving membrane CD317 and affecting exocytosis, in Campylobacter Jejuni-lysate-infected U937 cells. Int. J. Mol. Sci. 2020, 21, 2207. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, C.A.; Balbo, P.B.; Pesci, E.C.; Cottle, D.L.; Mirabito, P.M.; Pickett, C.L. Campylobacter jejuni Cytolethal Distending Toxin Causes a G 2 -Phase Cell Cycle Block. Infect. Immun. 1998, 66, 1934–1940. [Google Scholar] [CrossRef]
- Hickey, T.E.; Mcveigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, L.; Guerry, P.; Veigh, A.L.M.C.; Bourgeois, A.L. Campylobacter jejuni Cytolethal Distending Toxin Mediates Release of Interleukin-8 from Intestinal Epithelial Cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gantier, M.P.; Irving, A.T.; Kaparakis-Liaskos, M.; Xu, D.; Evans, V.A.; Cameron, P.U.; Bourne, J.A.; Ferrero, R.L.; John, M.; Behlke, M.A.; et al. Genetic modulation of TLR8 response following bacterial phagocytosis. Hum. Mutat. 2010, 31, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Schauer, D.B.; Fox, J.G. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell. Microbiol. 2008, 10, 1599–1607. [Google Scholar] [CrossRef]
- Canonico, B.; di Sario, G.; Cesarini, E.; Campana, R.; Luchetti, F.; Zamai, L.; Ortolani, C.; Nasoni, M.G.; Baffone, W.; Papa, S. Monocyte response to different Campylobacter jejuni lysates involves endoplasmic reticulum stress and the lysosomal–mitochondrial axis: When cell death is better than cell survival. Toxins 2018, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Jeon, B.; Itoh, K.; Ryu, S. Promoter Analysis of Cytolethal Distending Toxin Genes (cdtA, B, and C) and Effect of a luxS Mutation on CDT Production in Campylobacter jejuni. Microbiol. Immunol. 2005, 49, 599–603. [Google Scholar] [CrossRef]
- Guerra, L.; Guidi, R.; Frisan, T. Do bacterial genotoxins contribute to chronic inflammation, genomic instability and tumor progression? FEBS J. 2011, 278, 4577–4588. [Google Scholar] [CrossRef] [Green Version]
- Billcliff, P.G.; Rollason, R.; Prior, I.; Owen, D.M.; Gaus, K.; Banting, G. CD317/tetherin is an organiser of membrane microdomains. J. Cell Sci. 2013, 126, 1553–1564. [Google Scholar] [CrossRef] [Green Version]
- Pickett, C.L.; Whitehouse, C.A. The cytolethal distending toxin family. Trends Microbiol. 1999, 7, 292–297. [Google Scholar] [CrossRef]
- Shenker, B.J.; Guo, T.L.; Insug, O.; Shapiro, I.M. Induction of apoptosis in human T-cells by methyl mercury: Temporal relationship between mitochondrial dysfunction and loss of reductive reserve. Toxicol. Appl. Pharmacol. 1999, 157, 23–35. [Google Scholar] [CrossRef]
- Bachran, C.; Hasikova, R.; Leysath, C.E.; Sastalla, I.; Zhang, Y.; Fattah, R.J.; Liu, S.; Leppla, S.H. Cytolethal distending toxin B as a cell-killing component of tumor-targeted anthrax toxin fusion proteins. Cell Death Dis. 2014, 5, e1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.A.; Lai, Y.R.; Wu, H.Y.; Lo, Y.J.; Chang, Y.F.; Hung, C.L.; Lin, C.J.; Lo, U.G.; Lin, H.; Hsieh, J.T.; et al. Bacterial genotoxin-coated nanoparticles for radiotherapy sensitization in prostate cancer. Biomedicines 2021, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Vafadar, A.; Taheri-Anganeh, M.; Movahedpour, A.; Jamali, Z.; Irajie, C.; Ghasemi, Y.; Savardashtaki, A. In Silico Design and Evaluation of scFv-CdtB as a Novel Immunotoxin for Breast Cancer Treatment. Int. J. Cancer Manag. 2020, 13, e96094. [Google Scholar] [CrossRef]
- Keshtvarz, M.; Mahboobi, M.; Kieliszek, M.; Miecznikowski, A.; Sedighian, H.; Rezaei, M.; Haghighi, M.A.; Zareh, Z.; Rezaei, E. Engineering of cytolethal distending toxin b by its reducing immunogenicity and maintaining stability as a new drug candidate for tumor therapy; an in silico study. Toxins 2021, 13, 785. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Watson, E.; Sandu, P.; Gundogdu, O.; Mills, D.C.; Inglis, N.F.; Manson, E.; Imrie, L.; Bajaj-Elliott, M.; Wren, B.W.; et al. Campylobacter jejuni Outer Membrane Vesicles Play an Important Role in Bacterial Interactions with Human Intestinal Epithelial Cells. Infect. Immun. 2012, 80, 4089–4098. [Google Scholar] [CrossRef] [Green Version]
- Lindmark, B.; Rompikuntal, P.; Vaitkevicius, K.; Song, T.; Mizunoe, Y.; Uhlin, B.; Guerry, P.; Wai, S. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from Campylobacter jejuni. BMC Microbiol. 2009, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Davies, C.; Taylor, A.J.; Elmi, A.; Winter, J.; Liaw, J.; Grabowska, A.D.; Gundogdu, O.; Wren, B.W.; Kelly, D.J.; Dorrell, N. Sodium Taurocholate Stimulates Campylobacter jejuni Outer Membrane Vesicle Production via Down-Regulation of the Maintenance of Lipid Asymmetry Pathway. Front. Cell. Infect. Microbiol. 2019, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Woith, E.; Fuhrmann, G.; Melzig, M.F. Extracellular Vesicles-Connecting Kingdoms. Int. J. Mol. Sci. 2019, 20, 5695. [Google Scholar] [CrossRef] [Green Version]
- Antonyak, M.A.; Cerione, R.A. The distinct traits of extracellular vesicles generated by transformed cells. Small GTPases 2018, 9, 427–432. [Google Scholar] [CrossRef] [Green Version]
- Ofir-Birin, Y.; Abou Karam, P.; Rudik, A.; Giladi, T.; Porat, Z.; Regev-Rudzki, N. Monitoring extracellular vesicle cargo active uptake by imaging flow cytometry. Front. Immunol. 2018, 9, 1011. [Google Scholar] [CrossRef]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular vesicle heterogeneity: Subpopulations, isolation techniques, and diverse functions in cancer progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Cooper, J.M.; Wiklander, P.B.O.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.V.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, D.; Liu, Z.; Zhou, Y.; Chu, D.; Li, X.; Jiang, X.; Hou, D.; Chen, X.; Chen, Y.; et al. Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse. Sci. Rep. 2015, 5, 17543. [Google Scholar] [CrossRef] [Green Version]
- Murphy, D.E.; de Jong, O.G.; Brouwer, M.; Wood, M.J.; Lavieu, G.; Schiffelers, R.M.; Vader, P. Extracellular vesicle-based therapeutics: Natural versus engineered targeting and trafficking. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kowal, J.; Tkach, M.; Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Statello, L.; Maugeri, M.; Garre, E.; Nawaz, M.; Wahlgren, J.; Papadimitriou, A.; Lundqvist, C.; Lindfors, L.; Collén, A.; Sunnerhagen, P.; et al. Identification of RNA-binding proteins in exosomes capable of interacting with different types of RNA: RBP-facilitated transport of RNAs into exosomes. PLoS ONE 2018, 13, e0195969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, A.; Sampey, G.; Chung, M.C.; Bailey, C.; van Hoek, M.L.; Kashanchi, F.; Hakami, R.M. The carrying pigeons of the cell: Exosomes and their role in infectious diseases caused by human pathogens. Pathog. Dis. 2014, 71, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanpour, M.; Rezaie, J.; Nouri, M.; Panahi, Y. The role of extracellular vesicles in COVID-19 virus infection. Infect. Genet. Evol. 2020, 85, 104422. [Google Scholar] [CrossRef] [PubMed]
- González, M.F.; Díaz, P.; Sandoval-Bórquez, A.; Herrera, D.; Quest, A.F.G.G. Helicobacter pylori outer membrane vesicles and extracellular vesicles from helicobacter pylori-infected cells in gastric disease development. Int. J. Mol. Sci. 2021, 22, 4823. [Google Scholar] [CrossRef]
- Pant, S.; Hilton, H.; Burczynski, M.E. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharmacol. 2012, 83, 1484–1494. [Google Scholar] [CrossRef]
- Hüser, J.; Rechenmacher, C.E.; Blatter, L.A. Imaging the permeability pore transition in single mitochondria. Biophys. J. 1998, 74, 2129–2137. [Google Scholar] [CrossRef] [Green Version]
- Krippeit-Drews, P.; Düfer, M.; Drews, G. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem. Biophys. Res. Commun. 2000, 267, 179–183. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [Green Version]
- Slodzinski, M.K.; Aon, M.A.; O’Rourke, B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J. Mol. Cell. Cardiol. 2008, 45, 650–660. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.J.; Nguyen, J.A.; Cheung, S.M.; Arnold, C.R.; Balce, D.R.; Wang, Y.T.; Soderholm, A.; McKenna, N.; Aggarwal, D.; Campden, R.I.; et al. Macrophages disseminate pathogen associated molecular patterns through the direct extracellular release of the soluble content of their phagolysosomes. Nat. Commun. 2022, 13, 3072. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratta, S.; Tancini, B.; Sagini, K.; Delo, F.; Chiaradia, E.; Urbanelli, L.; Emiliani, C. Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 2020, 21, 2576. [Google Scholar] [CrossRef] [Green Version]
- Harting, M.T.; Srivastava, A.K.; Zhaorigetu, S.; Bair, H.; Prabhakara, K.S.; Toledano Furman, N.E.; Vykoukal, J.V.; Ruppert, K.A.; Cox, C.S.; Olson, S.D. Inflammation-Stimulated Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Inflammation. Stem Cells 2018, 36, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Wäster, P.; Eriksson, I.; Vainikka, L.; Öllinger, K. Extracellular vesicles released by melanocytes after UVA irradiation promote intercellular signaling via miR21. Pigment Cell Melanoma Res. 2020, 33, 542–555. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Orjalo, A.V.; Rodier, F.; Lithgow, G.J.; Campisi, J. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 2009, 1, 402–411. [Google Scholar] [CrossRef]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Campisi, J.; Benz, C.C. Expression of microRNA-146 suppresses NF-κB activity with reduction of metastatic potential in breast cancer cells. Oncogene 2008, 27, 5643–5647. [Google Scholar] [CrossRef] [Green Version]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Caggiu, E.; Paulus, K.; Mameli, G.; Arru, G.; Sechi, G.P.; Sechi, L.A. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci 2018, 13, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Wang, Y.; Liu, X.; Wang, S.; Wu, S.; Yuan, Z.; Zhu, X. miRNA-146a-5p mitigates stress-induced premature senescence of D-galactose-induced primary thymic stromal cells. Cytokine 2021, 137, 155314. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y. LncRNA CAIF suppresses LPS-induced inflammation and apoptosis of cardiomyocytes through regulating miR-16 demethylation. Immunity Inflamm. Dis. 2021, 9, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Liang, H.; Sun, W.; Li, J.; Liu, Y.; Fan, Q.; Zhang, H.; Yue, X.; Li, J.; Chen, X.; et al. Deregulation of the miR-16-KRAS axis promotes colorectal cancer. Sci. Rep. 2016, 6, 37459. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef]
- Quereda, V.; Porlan, E.; Canãmero, M.; Dubus, P.; Malumbres, M. An essential role for Ink4 and Cip/Kip cell-cycle inhibitors in preventing replicative stress. Cell Death Differ. 2016, 23, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Johmura, Y.; Nakanishi, M. Multiple facets of p53 in senescence induction and maintenance. Cancer Sci. 2016, 107, 1550–1555. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi Cytolethal Distending Toxin Induces Cell Cycle Arrest and Apoptosis via the DNA Damage Checkpoint Pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar] [CrossRef]
- Ohara, M.; Oswald, E.; Sugai, M. Cytolethal Distending Toxin: A Bacterial Bullet Targeted to Nucleus. J. Biochem. 2004, 136, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Taieb, F.; Petit, C.; Nougayrède, J.-P.; Oswald, E. The Enterobacterial Genotoxins: Cytolethal Distending Toxin and Colibactin. EcoSal Plus 2016, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, G.; Ji, C. Inhibition of human cervical cancer cell growth by Salviolone is mediated via autophagy induction, cell migration and cell invasion suppression, G2/M cell cycle arrest and downregulation of Nf-kB/m-TOR/PI3K/AKT pathway. JBUON 2018, 23, 1739–1744. [Google Scholar] [PubMed]
- Zhang, Y.; Zhao, Y.; Ran, Y.; Guo, J.; Cui, H.; Liu, S. Alantolactone exhibits selective antitumor effects in HELA human cervical cancer cells by inhibiting cell migration and invasion, G2/M cell cycle arrest, mitochondrial mediated apoptosis and targeting Nf-kB signalling pathway. JBUON 2019, 24, 2310–2315. [Google Scholar] [PubMed]
- Dubreuil, V.; Marzesco, A.M.; Corbeil, D.; Huttner, W.B.; Wilsch-Bräuninger, M. Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J. Cell Biol. 2007, 176, 483–495. [Google Scholar] [CrossRef]
- Ferrandina, G.; Petrillo, M.; Bonanno, G.; Scambia, G. Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets 2009, 13, 823–837. [Google Scholar] [CrossRef]
- Ren, F.; Sheng, W.Q.; Du, X. CD133: A cancer stem cells marker, is used in colorectal cancers. World J. Gastroenterol. 2013, 19, 2603–2611. [Google Scholar] [CrossRef]
- Corbeil, D.; Roandper, K.; Fargeas, C.A.; Joester, A.; Huttner, W.B. Prominin: A story of cholesterol, plasma membrane protrusions and human pathology. Traffic 2001, 2, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Jászai, J.; Fargeas, C.A.; Graupner, S.; Tanaka, E.M.; Brand, M.; Huttner, W.B.; Corbeil, D. Distinct and conserved prominin-1/CD133-positive retinal cell populations identified across species. PLoS ONE 2011, 6, e17590. [Google Scholar] [CrossRef] [Green Version]
- Barzegar Behrooz, A.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2019, 27, 257–269. [Google Scholar] [CrossRef]
- Vu, L.T.; Gong, J.; Pham, T.T.; Kim, Y.; Le, M.T.N. microRNA exchange via extracellular vesicles in cancer. Cell Prolif. 2020, 53, e12877. [Google Scholar] [CrossRef] [PubMed]
- Marzesco, A.M.; Wilsch-Bräuninger, M.; Dubreuil, V.; Janich, P.; Langenfeld, K.; Thiele, C.; Huttner, W.B.; Corbeil, D. Release of extracellular membrane vesicles from microvilli of epithelial cells is enhanced by depleting membrane cholesterol. FEBS Lett. 2009, 583, 897–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taïeb, N.; Maresca, M.; Guo, X.J.; Garmy, N.; Fantini, J.; Yahi, N. The first extracellular domain of the tumour stem cell marker CD133 contains an antigenic ganglioside-binding motif. Cancer Lett. 2009, 278, 164–173. [Google Scholar] [CrossRef]
- Corbeil, D.; Santos, M.F.; Karbanová, J.; Kurth, T.; Rappa, G.; Lorico, A. Uptake and Fate of Extracellular Membrane Vesicles: Nucleoplasmic Reticulum-Associated Late Endosomes as a New Gate to Intercellular Communication. Cells 2020, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Kim, S.; Ko, J. Roles of CD133 in microvesicle formation and oncoprotein trafficking in colon cancer. FASEB J. 2019, 33, 4248–4260. [Google Scholar] [CrossRef]
- El-Aouar Filho, R.A.; Nicolas, A.; De Paula Castro, T.L.; Deplanche, M.; De Carvalho Azevedo, V.A.; Goossens, P.L.; Taieb, F.; Lina, G.; Le Loir, Y.; Berkova, N. Heterogeneous family of cyclomodulins: Smart weapons that allow bacteria to hijack the eukaryotic cell cycle and promote infections. Front. Cell. Infect. Microbiol. 2017, 7, 208. [Google Scholar] [CrossRef] [Green Version]
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dörsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43. [Google Scholar] [CrossRef]
- Mantel, P.Y.; Hjelmqvist, D.; Walch, M.; Kharoubi-Hess, S.; Nilsson, S.; Ravel, D.; Ribeiro, M.; Grüring, C.; Ma, S.; Padmanabhan, P.; et al. Infected erythrocyte-derived extracellular vesicles alter vascular function via regulatory Ago2-miRNA complexes in malaria. Nat. Commun. 2016, 7, 12727. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, G.; Dentelli, P.; Togliatto, G.; Rosso, A.; Gili, M.; Gallo, S.; Deregibus, M.C.; Camussi, G.; Brizzi, M.F. Activated Stat5 trafficking Via Endothelial Cell-derived Extracellular Vesicles Controls IL-3 Pro-angiogenic Paracrine Action. Sci. Rep. 2016, 6, 25689. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.S.; Gadegaard, I.S.E.; Arnspang, E.C.; Blans, K.; Nejsum, L.N.; Rasmussen, J.T. Specific and non-invasive fluorescent labelling of extracellular vesicles for evaluation of intracellular processing by intestinal epithelial cells. Biomedicines 2020, 8, 211. [Google Scholar] [CrossRef]
- Joshi, B.S.; de Beer, M.A.; Giepmans, B.N.G.; Zuhorn, I.S. Endocytosis of Extracellular Vesicles and Release of Their Cargo from Endosomes. ACS Nano 2020, 14, 4444–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Qiao, Y.; Li, X.; Chen, J.; Ding, J.; Bai, L.; Shen, F.; Shi, B.; Liu, J.; Peng, L.; et al. Exosomes Exploit the Virus Entry Machinery and Pathway to Transmit Alpha Interferon-Induced Antiviral Activity. J. Virol. 2018, 92, e01578-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zong, S.; Wang, Z.; Lu, J.; Zhu, D.; Zhang, Y.; Zhang, R.; Cui, Y. Visualization and intracellular dynamic tracking of exosomes and exosomal miRNAs using single molecule localization microscopy. Nanoscale 2018, 10, 5154–5162. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Okada, K.; Hayashi, O. Immuno-regulatory and anti-inflammatory actions of phycocyanin on Caco-2/U937 cells co-culture as a model of the intestinal barrier. Funct. Foods Heal. Dis. 2019, 9, 466. [Google Scholar] [CrossRef] [Green Version]
- Chazotte, B. Labeling lysosomes in live cells with LysoTracker. Cold Spring Harb. Protoc. 2011, 2011, pdb.prot5571. [Google Scholar] [CrossRef]
- Canonico, B.; Cesarini, E.; Salucci, S.; Luchetti, F.; Falcieri, E.; Di Sario, G.; Palma, F.; Papa, S. Defective autophagy, mitochondrial clearance and lipophagy in niemann-pick type B lymphocytes. PLoS ONE 2016, 11, e0165780. [Google Scholar] [CrossRef] [Green Version]
- Traganos, F.; Darzynkiewicz, Z. Lysosomal Proton Pump Activity: Supravital Cell Staining with Acridine Orange Differentiates Leukocyte Subpopulations. Methods Cell Biol. 1994, 41, 185–194. [Google Scholar] [CrossRef]
- Andrews, N.W. Detection of Lysosomal Exocytosis by Surface Exposure of Lamp1 Luminal Epitopes. Methods Mol. Biol. 2017, 1594, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, C.L.; Colombo, M.I. Assays to Assess Autophagy Induction and Fusion of Autophagic Vacuoles with a Degradative Compartment, Using Monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol. 2009, 452, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.; Kusmartsev, S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 2010, 594, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Brocco, D.; Lanuti, P.; Simeone, P.; Bologna, G.; Pieragostino, D.; Cufaro, M.C.; Graziano, V.; Peri, M.; Di Marino, P.; De Tursi, M.; et al. Circulating Cancer Stem Cell-Derived Extracellular Vesicles as a Novel Biomarker for Clinical Outcome Evaluation. J. Oncol. 2019, 2019, 5879616. [Google Scholar] [CrossRef]
- Simeone, P.; Celia, C.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Cilurzo, F.; Grande, R.; Diomede, F.; Vespa, S.; Canonico, B.; et al. Diameters and Fluorescence Calibration for Extracellular Vesicle Analyses by Flow Cytometry. Int. J. Mol. Sci. 2020, 21, 7885. [Google Scholar] [CrossRef]
- Burattini, S.; Battistelli, M.; Codenotti, S.; Falcieri, E.; Fanzani, A.; Salucci, S. Melatonin action in tumor skeletal muscle cells: An ultrastructural study. Acta Histochem. 2016, 118, 278–285. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Burwinkel, B. Extracellular miRNAs: The mystery of their origin and function. Trends Biochem. Sci. 2012, 37, 460–465. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Guescini, M.; Canonico, B.; Lucertini, F.; Maggio, S.; Annibalini, G.; Barbieri, E.; Luchetti, F.; Papa, S.; Stocchi, V. Muscle Releases Alpha-Sarcoglycan Positive Extracellular Vesicles Carrying miRNAs in the Bloodstream. PLoS ONE 2015, 10, e0125094. [Google Scholar] [CrossRef] [Green Version]
- Gai, C.; Gomez, Y.; Tetta, C.; Felice Brizzi, M.; Camussi, G. Protective Role of Stem Cell Derived Extracellular Vesicles in an In Vitro Model of Hyperglycemia-Induced Endothelial Injury. J. Cell Sci. Ther. 2017, 8, 264. [Google Scholar] [CrossRef]
- Busatto, S.; Yang, Y.; Walker, S.A.; Davidovich, I.; Lin, W.H.; Lewis-Tuffin, L.; Anastasiadis, P.Z.; Sarkaria, J.; Talmon, Y.; Wurtz, G.; et al. Brain metastases-derived extracellular vesicles induce binding and aggregation of low-density lipoprotein. J. Nanobiotechnology 2020, 18, 162. [Google Scholar] [CrossRef] [PubMed]
- Brando, B.; Barnett, D.; Janossy, G.; Mandy, F.; Autran, B.; Rothe, G.; Scarpati, B.; D’Avanzo, G.; D’Hautcourt, J.-L.; Lenkei, R.; et al. Cytofluorometric methods for assessing absolute numbers of cell subsets in blood. Cytometry 2000, 42, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S. Innovative Approaches of Engineering Tumor-Targeting Bacteria with Different Therapeutic Payloads to Fight Cancer: A Smart Strategy of Disease Management. Int. J. Nanomed. 2021, 16, 8159–8184. [Google Scholar] [CrossRef] [PubMed]
- Mammes, A.; Pasquier, J.; Mammes, O.; Conti, M.; Douard, R.; Loric, S. Extracellular vesicles: General features and usefulness in diagnosis and therapeutic management of colorectal cancer. World J. Gastrointest. Oncol. 2021, 13, 1561–1598. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C. jejuni Strains | Description | References |
|---|---|---|
| 11168H cdtA mutant | Isogenic mutant of Cj0079 obtained from the Campylobacter mutant bank (LSHTM) | LSHTM mutant bank |
| ATCC 33291 | Campylobacter jejuni subsp. jejuni (ATCC® 33291™) | ATCC®, Rockville, ND, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montanari, M.; Guescini, M.; Gundogdu, O.; Luchetti, F.; Lanuti, P.; Ciacci, C.; Burattini, S.; Campana, R.; Ortolani, C.; Papa, S.; et al. Extracellular Vesicles from Campylobacter jejuni CDT-Treated Caco-2 Cells Inhibit Proliferation of Tumour Intestinal Caco-2 Cells and Myeloid U937 Cells: Detailing the Global Cell Response for Potential Application in Anti-Tumour Strategies. Int. J. Mol. Sci. 2023, 24, 487. https://doi.org/10.3390/ijms24010487
Montanari M, Guescini M, Gundogdu O, Luchetti F, Lanuti P, Ciacci C, Burattini S, Campana R, Ortolani C, Papa S, et al. Extracellular Vesicles from Campylobacter jejuni CDT-Treated Caco-2 Cells Inhibit Proliferation of Tumour Intestinal Caco-2 Cells and Myeloid U937 Cells: Detailing the Global Cell Response for Potential Application in Anti-Tumour Strategies. International Journal of Molecular Sciences. 2023; 24(1):487. https://doi.org/10.3390/ijms24010487
Chicago/Turabian StyleMontanari, Mariele, Michele Guescini, Ozan Gundogdu, Francesca Luchetti, Paola Lanuti, Caterina Ciacci, Sabrina Burattini, Raffaella Campana, Claudio Ortolani, Stefano Papa, and et al. 2023. "Extracellular Vesicles from Campylobacter jejuni CDT-Treated Caco-2 Cells Inhibit Proliferation of Tumour Intestinal Caco-2 Cells and Myeloid U937 Cells: Detailing the Global Cell Response for Potential Application in Anti-Tumour Strategies" International Journal of Molecular Sciences 24, no. 1: 487. https://doi.org/10.3390/ijms24010487