

High Acetate Concentration Protects Intestinal Barrier and Exerts Anti-Inflammatory Effects in Organoid-Derived Epithelial Monolayer Cultures from Patients with Ulcerative Colitis

 , , ,
, , ,
Abstract
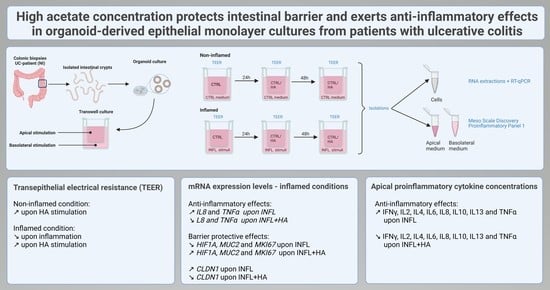
1. Introduction
2. Results
2.1. Impact of High Acetate Supplementation on Barrier Integrity
2.2. High Acetate Administration Downregulates Inflammatory IL 8 and TNFα Expression and Influences Barrier Genes
2.3. Administration of High Acetate Concentrations Decreased Pro-Inflammatory Cytokines in the Apical Transwell Chamber
3. Discussion
4. Materials and Methods
4.1. Human Biopsy Collection and Ethical Statement
4.2. Intestinal Crypt Isolation and Organoid Culture
4.3. Primary Epithelial Monolayer Cultures
4.4. Induction of Inflammation and High Acetate Treatment
4.5. Transepithelial Electrical Resistance Measurements
4.6. Gene Expression Analysis by Quantitative Reverse Transcription
4.7. Cytokine Profiling
4.8. Statistical Analysis
5. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Composition Basal Medium (BM)
- DMEM/F12 1:1 Mixture (Lonza, Basel, Switzerland) supplemented with
- 1× GlutaMAX (Gibco, Thermo Fisher Scientific, Waltham, MA, USA),
- 10 mM HEPES (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and
- 100 μg/mL penicillin/streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA).
Appendix B. Composition Human Expansion Medium (HM)—v/v: Percentage Volume of Total End Volume
| Component | Concentration | Manufacturer | Catalog nr |
| Wnt3A | 50% v/v | In house cell line | / |
| R-spondin | 20% v/v | In house cell line | / |
| Noggin | 10% v/v | In house cell line | / |
| EGF | 50 ng/mL | Life Technologies | PMG8041 |
| A83-01 | 500 nM | Tocris | 2939/10 |
| SB202190 | 10 µM | Sigma-Aldrich | S7067 |
| Nicotinamide | 10 mM | Sigma-Aldrich | N0636 |
| n-Acetylcysteine | 1.25 mM | Sigma-Aldrich | A9165 |
| B27 | 1× | Life Technologies | 17504044 |
Appendix C. Sequence Primers qPCR
| Name | Sequence | Scale | Purification |
| ACTB forward | CCCAGCACAATGAAGATCAAGATC | 25 nm | STD |
| ACTB reverse | CTGATCCACATCTGCTGGAAG | 25 nm | STD |
| B2M forward | TGCTGTCTCCATGTTTGATGTATCT | 25 nm | STD |
| B2M reverse | TCTCTGCTCCCCACCTCTAAGT | 25 nm | STD |
| CLDN1 forward | GCAGATCCAGTGCAAAGTC | 25 nm | STD |
| CLDN1 reverse | CTATCACTCCCAGGAGGATG | 25 nm | STD |
| CLDN2 forward | CACACTGGTTGCCATGCT | 25 nm | STD |
| CLDN2 reverse | ATTCCATCCAGAGGCCCT | 25 nm | STD |
| HIF1A forward | CTAACTAGCCGAGGAAGAACTATGA | 25 nm | STD |
| HIF1A reverse | TGGTTACTGTTGGTATCATATACGTG | 25 nm | STD |
| IL8 forward | ACTGAGAGTGATTGAGAGTGGAC | 25 nm | STD |
| IL8 reverse | AACCCTCTGCACCCAGTTTTC | 25 nm | STD |
| MKI67 forward | AAAGGCAAAGAAGACCTGCTA | 25 nm | STD |
| MKI67 reverse | TTTGCGTGGCCTGTACTAAAT | 25 nm | STD |
| MUC2 forward | CACCAAGACCGTCCTCATG | 25 nm | STD |
| MUC2 reverse | CTTGGCCGAGTACATGACA | 25 nm | STD |
| OCLN forward | TGGCAAAGTGAATGACAAGC | 25 nm | STD |
| OCLN reverse | AGGCGAAGTTAATGGAAGCTC | 25 nm | STD |
| RPS14 forward | TCACCGCCCTACACATCAAAC | 25 nm | STD |
| RPS14 reverse | GCCCGATCTTCATACCCGA | 25 nm | STD |
| TNFA forward | TAGCCCATGTTGTAGCAAACCC | 25 nm | STD |
| TNFA reverse | TATCTCTCAGCTCCACGCCA | 25 nm | STD |
| ZO1 forward | AAAGAAGCAATTCAACAACAGCAA | 25 nm | STD |
| ZO1 reverse | ATCATGCAAATCAAGGTCATCACT | 25 nm | STD |
References
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Hold, G.L.; Smith, M.; Grange, C.; Watt, E.R.; El-Omar, E.M.; Mukhopadhya, I. Role of the Gut Microbiota in Inflammatory Bowel Disease Pathogenesis: What Have We Learnt in the Past 10 Years? World J. Gastroenterol. 2014, 20, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Caenepeel, C.; Sadat Seyed Tabib, N.; Vieira-Silva, S.; Vermeire, S. Review Article: How the Intestinal Microbiota May Reflect Disease Activity and Influence Therapeutic Outcome in Inflammatory Bowel Disease. Aliment. Pharmacol. Ther. 2020, 52, 1453–1468. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative Microbiome Profiling Disentangles Inflammation- and Bile Duct Obstruction-Associated Microbiota Alterations across PSC/IBD Diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Miyoshi, J.; Chang, E.B. The Gut Microbiota and Inflammatory Bowel Diseases. Transl. Res. 2017, 179, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.; Khan, S.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Serban, D.E. Microbiota in Inflammatory Bowel Disease Pathogenesis and Therapy: Is It All about Diet? Nutr. Clin. Pract. 2015, 30, 760–779. [Google Scholar] [CrossRef] [PubMed]
- McIlroy, J.; Ianiro, G.; Mukhopadhya, I.; Hansen, R.; Hold, G.L. Review Article: The Gut Microbiome in Inflammatory Bowel Disease—Avenues for Microbial Management. Aliment. Pharmacol. Ther. 2018, 47, 26–42. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e6. [Google Scholar] [CrossRef]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients with Ulcerative Colitis: A Randomized Clinical Trial. JAMA J. Am. Med. Assoc. 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castaño-Rodríguez, N. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. J. Crohn’s Colitis 2017, 11, 1180–1199. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.A.; Duflou, A.; Löwenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.H.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology 2015, 149, 110–118.e4. [Google Scholar] [CrossRef] [PubMed]
- Danne, C.; Rolhion, N.; Sokol, H. Recipient Factors in Faecal Microbiota Transplantation: One Stool Does Not Fit All. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Pais, P.; Almeida, V.; Yılmaz, M.; Teixeira, M.C. Saccharomyces Boulardii: What Makes It Tick as Successful Probiotic? J. Fungi 2020, 6, 78. [Google Scholar] [CrossRef]
- Bjarnason, I.; Sission, G.; Hayee, B.H. A Randomised, Double-Blind, Placebo-Controlled Trial of a Multi-Strain Probiotic in Patients with Asymptomatic Ulcerative Colitis and Crohn’s Disease. Inflammopharmacology 2019, 27, 465–473. [Google Scholar] [CrossRef]
- Lavelle, A.; Sokol, H. Gut Microbiota-Derived Metabolites as Key Actors in Inflammatory Bowel Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 223–237. [Google Scholar] [CrossRef]
- Deleu, S.; Machiels, K.; Raes, J.; Verbeke, K. Short Chain Fatty Acids and Its Producing Organisms: An Overlooked Therapy for IBD ? EBioMedicine 2021, 66, 8. [Google Scholar] [CrossRef]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Rhodes, J.M. Nutrition and Gut Health: The Impact of Specific Dietary Components–It’s Not Just Five-a-Day. Proc. Nutr. Soc. 2021, 80, 9–18. [Google Scholar] [CrossRef]
- Gill, P.A.; van Zelm, M.C.; Muir, J.G.; Gibson, P.R. Review Article: Short Chain Fatty Acids as Potential Therapeutic Agents in Human Gastrointestinal and Inflammatory Disorders. Aliment. Pharmacol. Ther. 2018, 48, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Offei, B.; Vandecruys, P.; de Graeve, S.; Foulquié-Moreno, M.R.; Thevelein, J.M. Unique Genetic Basis of the Distinct Antibiotic Potency of High Acetic Acid Production in the Probiotic Yeast Saccharomyces Cerevisiae Var. Boulardii. Genome Res. 2019, 29, 1478–1494. [Google Scholar] [CrossRef] [PubMed]
- Li, V.S.W. Modelling Intestinal Inflammation and Infection Using ‘Mini-Gut’ Organoids. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 89–90. [Google Scholar] [CrossRef] [PubMed]
- Poletti, M.; Arnauts, K.; Ferrante, M.; Korcsmaros, T. Organoid-Based Models to Study the Role of Host-Microbiota Interactions in IBD. J. Crohn’s Colitis 2021, 15, 1222. [Google Scholar] [CrossRef] [PubMed]
- Arnauts, K.; Verstockt, B.; Ramalho, A.S.; Vermeire, S.; Verfaillie, C.; Ferrante, M. Ex Vivo Mimicking of Inflammation in Organoids Derived From Patients With Ulcerative Colitis. Gastroenterology 2020, 159, 1564–1567. [Google Scholar] [CrossRef]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and Claudin-2 Expression Is Elevated in Inflammatory Bowel Disease and May Contribute to Early Neoplastic Transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight Junctions in Inflammatory Bowel Diseases and Inflammatory Bowel Disease Associated Colorectal Cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Kao, D.J.; Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; et al. HIF-Dependent Regulation of Claudin-1 Is Central to Intestinal Epithelial Tight Junction Integrity. Mol. Biol. Cell 2015, 26, 2151–2384. [Google Scholar] [CrossRef]
- Kerber, E.L.; Padberg, C.; Koll, N.; Schuetzhold, V.; Fandrey, J.; Winning, S. The Importance of Hypoxia-Inducible Factors (HIF-1 and HIF-2) for the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2020, 21, 8551. [Google Scholar] [CrossRef]
- Zhou, C.; Li, L.; Li, T.; Sun, L.; Yin, J.; Guan, H.; Wang, L.; Zhu, H.; Xu, P.; Fan, X.; et al. SCFAs Induce Autophagy in Intestinal Epithelial Cells and Relieve Colitis by Stabilizing HIF-1α. J. Mol. Med. 2020, 98, 1189–1202. [Google Scholar] [CrossRef]
- Ye, J.; Haskey, N.; Dadlani, H.; Zubaidi, H.; Barnett, J.A.; Ghosh, S.; Gibson, D.L. Deletion of Mucin 2 Induces Colitis with Concomitant Metabolic Abnormalities in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G791–G803. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.N.; McClintock, S.D.; Attili, D.; Pandya, S.; Rehman, H.; Nadeem, D.M.; Jawad-Makki, M.A.H.; Rizvi, A.H.; Berner, M.M.; Dame, M.K.; et al. Ulcerative Colitis-Derived Colonoid Culture: A Multi-Mineral-Approach to Improve Barrier Protein Expression. Front. Cell Dev. Biol. 2020, 8, 1342. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Kaufman, P.D. Ki-67: More than a Proliferation Marker. Chromosoma 2018, 127, 175. [Google Scholar] [CrossRef]
- Cox, M.A.; Jackson, J.; Stanton, M.; Rojas-Triana, A.; Bober, L.; Laverty, M.; Yang, X.; Zhu, F.; Liu, J.; Wang, S.; et al. Short-Chain Fatty Acids Act as Antiinflammatory Mediators by Regulating Prostaglandin E2 and Cytokines. World J. Gastroenterol. 2009, 15, 5549–5557. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Laeremans, T.; Vanhove, W.; Arnauts, K.; Ramalho, A.S.; Farré, R.; Cleynen, I.; Ferrante, M.; Vermeire, S. Butyrate Does Not Protect Against Inflammation-Induced Loss of Epithelial Barrier Function and Cytokine Production in Primary Cell Monolayers From Patients With Ulcerative Colitis. J. Crohn’s Colitis 2019, 13, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Singh, N.P.; Murphy, E.A.; Price, R.L.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S. Chemokine and Cytokine Levels in Inflammatory Bowel Disease Patients. Cytokine 2016, 77, 44–49. [Google Scholar] [CrossRef]
- VanDussen, K.L.; Marinshaw, J.M.; Shaikh, N.; Miyoshi, H.; Moon, C.; Tarr, P.I.; Ciorba, M.A.; Stappenbeck, T.S. Development of an Enhanced Human Gastrointestinal Epithelial Culture System to Facilitate Patient-Based Assays. Gut 2015, 64, 911–920. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | CTRL vs. HA | CTRL vs. INFL | CTRL vs. INFL + HA | HA vs. INFL | HA vs. INFL + HA | INFL vs. INFL + HA | |
|---|---|---|---|---|---|---|---|
| IFNy | p | 0.0007 | 0.119 | 0.0999 | <0.0001 | 0.0833 | 0.0014 |
| Adj. p | 0.0044 | 0.7142 | 0.5993 | <0.0001 | 0.4996 | 0.0081 | |
| IL1b | p | 0.0153 | 0.0056 | 0.729 | <0.0001 | 0.0056 | 0.0153 |
| Adj. p | 0.0919 | 0.0335 | >0.9999 | <0.0001 | 0.0335 | 0.0919 | |
| IL2 | p | 0.001 | 0.119 | 0.0833 | <0.0001 | 0.119 | 0.001 |
| Adj. p | 0.006 | 0.7142 | 0.4996 | <0.0001 | 0.7142 | 0.006 | |
| IL4 | p | 0.0377 | 0.119 | 0.119 | 0.0003 | 0.6033 | 0.0018 |
| Adj. p | 0.226 | 0.7142 | 0.7142 | 0.0017 | >0.9999 | 0.0109 | |
| IL6 | p | 0.0073 | 0.0464 | 0.2987 | <0.0001 | 0.0999 | 0.0024 |
| Adj. p | 0.0436 | 0.2783 | >0.9999 | <0.0001 | 0.5993 | 0.0146 | |
| IL8 | p | 0.0018 | 0.0567 | 0.119 | <0.0001 | 0.119 | 0.0005 |
| Adj. p | 0.0109 | 0.3405 | 0.7142 | <0.0001 | 0.7142 | 0.0032 | |
| IL10 | p | 0.0003 | 0.1659 | 0.0567 | <0.0001 | 0.0833 | 0.001 |
| Adj. p | 0.0017 | 0.9951 | 0.3405 | <0.0001 | 0.4996 | 0.006 | |
| IL12p70 | p | 0.0032 | 0.119 | 0.4884 | <0.0001 | 0.0243 | 0.0243 |
| Adj. p | 0.0194 | 0.7142 | >0.9999 | <0.0001 | 0.1461 | 0.1461 | |
| IL13 | p | 0.0094 | 0.0567 | 0.1659 | <0.0001 | 0.2253 | 0.001 |
| Adj. p | 0.0562 | 0.3405 | 0.9951 | <0.0001 | >0.9999 | 0.006 | |
| TNFα | p | 0.0018 | 0.0567 | 0.3865 | <0.0001 | 0.0243 | 0.0056 |
| Adj. p | 0.0109 | 0.3405 | >0.9999 | <0.0001 | 0.1461 | 0.0335 | |
| Male/female [%] | 5/5 [50%] |
| Age at inclusion in years: Median [IQR] | 47.5 [10.25] |
| Disease duration in years: Median [IQR] | 12.5 [5.25] |
| Total Mayo score: Median [IQR] | 8.5 [3.75] |
| Endoscopic Mayo subscore: [%] | |
| 2 | 5 [50%] |
| 3 | 5 [50%] |
| Medication use [%] | 7 [70%] |
| 5-aminosalicylates | 4 [40%] |
| Corticosteroids | 1 [10%] |
| Immunomodulator | 0 [0%] |
| Biologicals | 3 [30%] |
| Small molecules | 0 [0%] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deleu, S.; Arnauts, K.; Deprez, L.; Machiels, K.; Ferrante, M.; Huys, G.R.B.; Thevelein, J.M.; Raes, J.; Vermeire, S. High Acetate Concentration Protects Intestinal Barrier and Exerts Anti-Inflammatory Effects in Organoid-Derived Epithelial Monolayer Cultures from Patients with Ulcerative Colitis. Int. J. Mol. Sci. 2023, 24, 768. https://doi.org/10.3390/ijms24010768
Deleu S, Arnauts K, Deprez L, Machiels K, Ferrante M, Huys GRB, Thevelein JM, Raes J, Vermeire S. High Acetate Concentration Protects Intestinal Barrier and Exerts Anti-Inflammatory Effects in Organoid-Derived Epithelial Monolayer Cultures from Patients with Ulcerative Colitis. International Journal of Molecular Sciences. 2023; 24(1):768. https://doi.org/10.3390/ijms24010768
Chicago/Turabian StyleDeleu, Sara, Kaline Arnauts, Lowie Deprez, Kathleen Machiels, Marc Ferrante, Geert R. B. Huys, Johan M. Thevelein, Jeroen Raes, and Séverine Vermeire. 2023. "High Acetate Concentration Protects Intestinal Barrier and Exerts Anti-Inflammatory Effects in Organoid-Derived Epithelial Monolayer Cultures from Patients with Ulcerative Colitis" International Journal of Molecular Sciences 24, no. 1: 768. https://doi.org/10.3390/ijms24010768
APA StyleDeleu, S., Arnauts, K., Deprez, L., Machiels, K., Ferrante, M., Huys, G. R. B., Thevelein, J. M., Raes, J., & Vermeire, S. (2023). High Acetate Concentration Protects Intestinal Barrier and Exerts Anti-Inflammatory Effects in Organoid-Derived Epithelial Monolayer Cultures from Patients with Ulcerative Colitis. International Journal of Molecular Sciences, 24(1), 768. https://doi.org/10.3390/ijms24010768