
Peptide Drug Conjugates and Their Role in Cancer Therapy

 , , , , , and
, , , , , and
Abstract
:1. Introduction
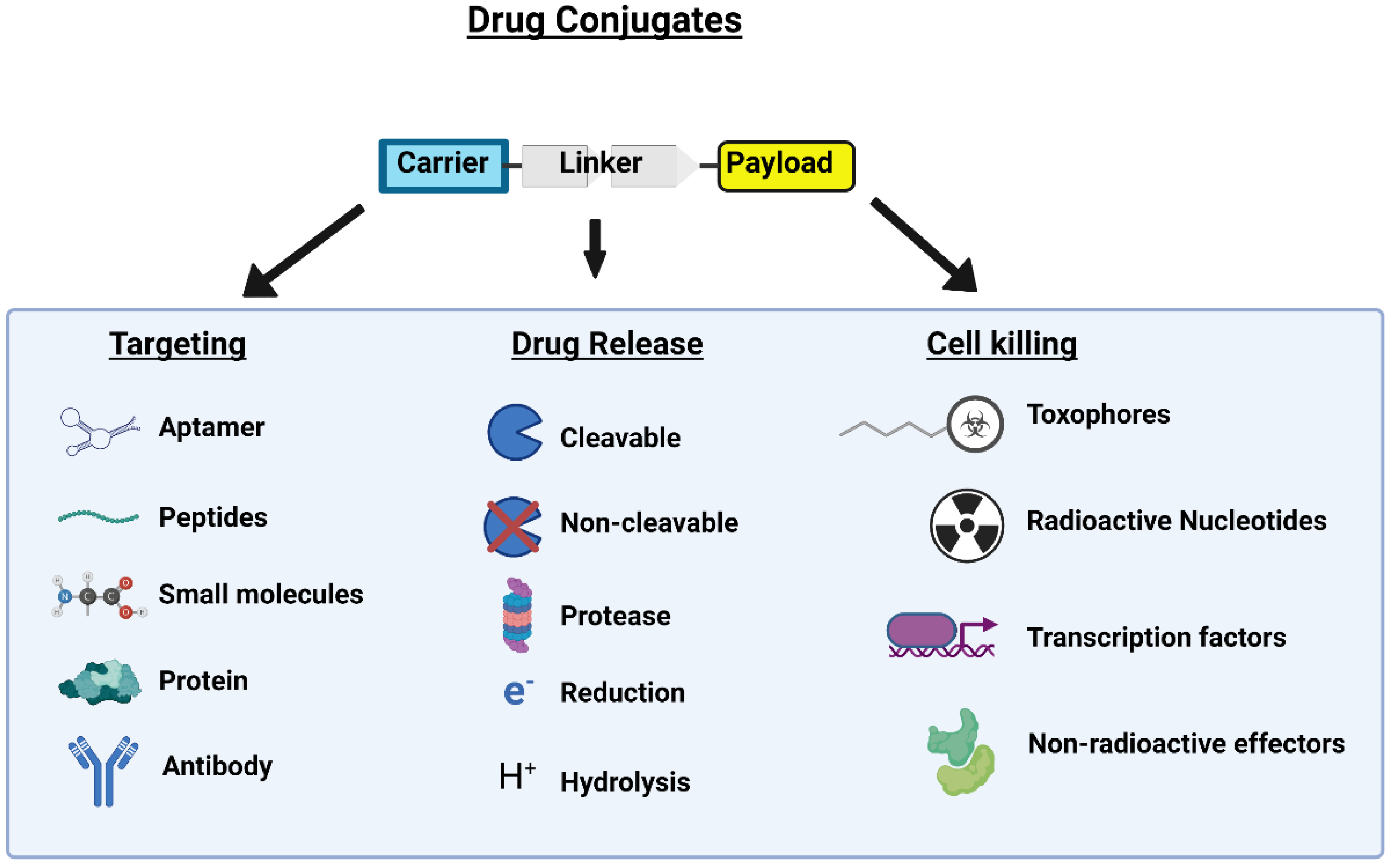
2. Peptide Drug Conjugates
2.1. Peptides for Specific Organ Targeting
2.2. Cell-Penetrating Peptides
2.3. Cell-Targeting Peptides
3. Linker Region
3.1. Non-Cleavable Linkers
3.2. pH-Sensitive Linkers
3.3. Enzyme-Sensitive Linkers
3.4. Redox-Sensitive Linkers
4. Payload
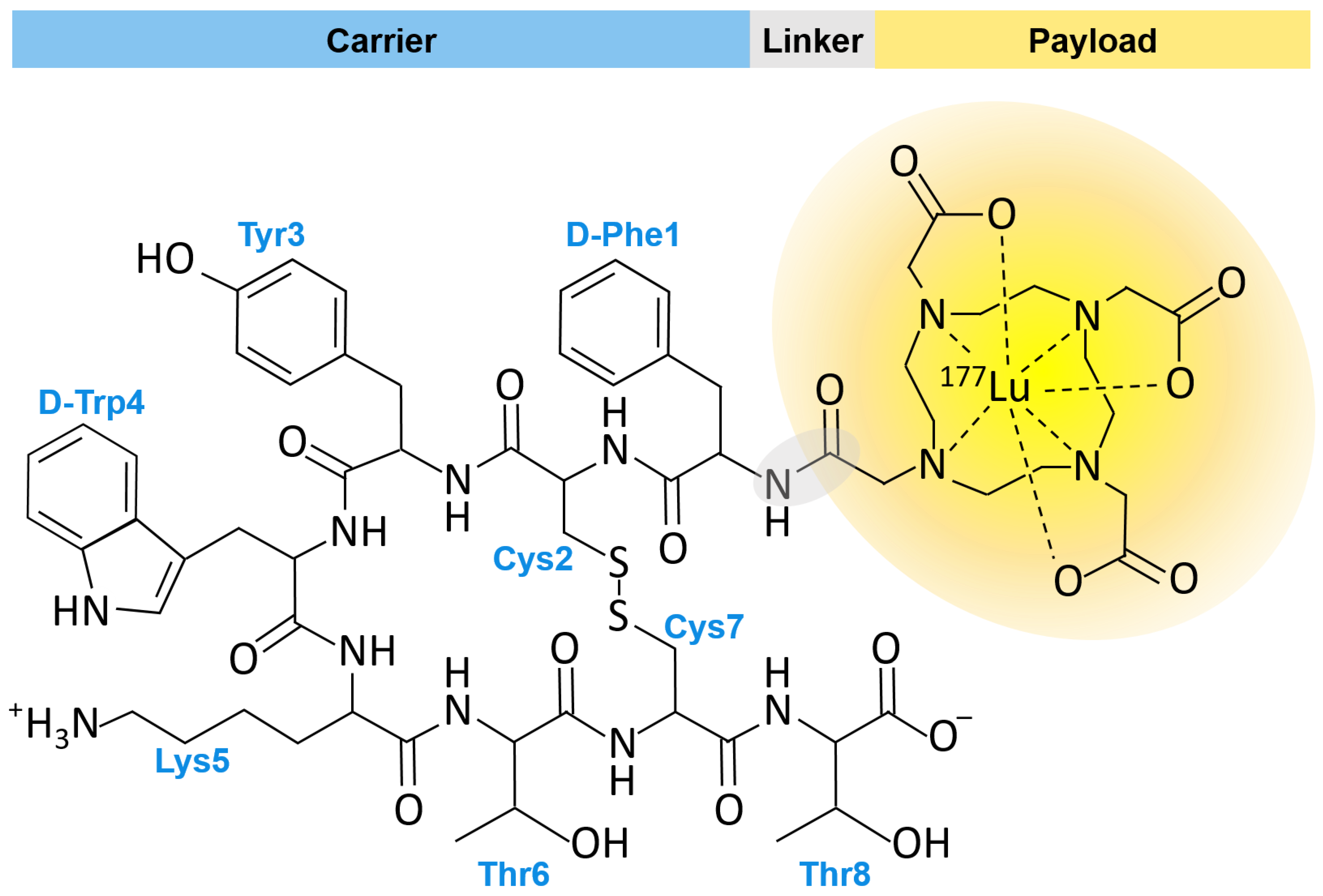
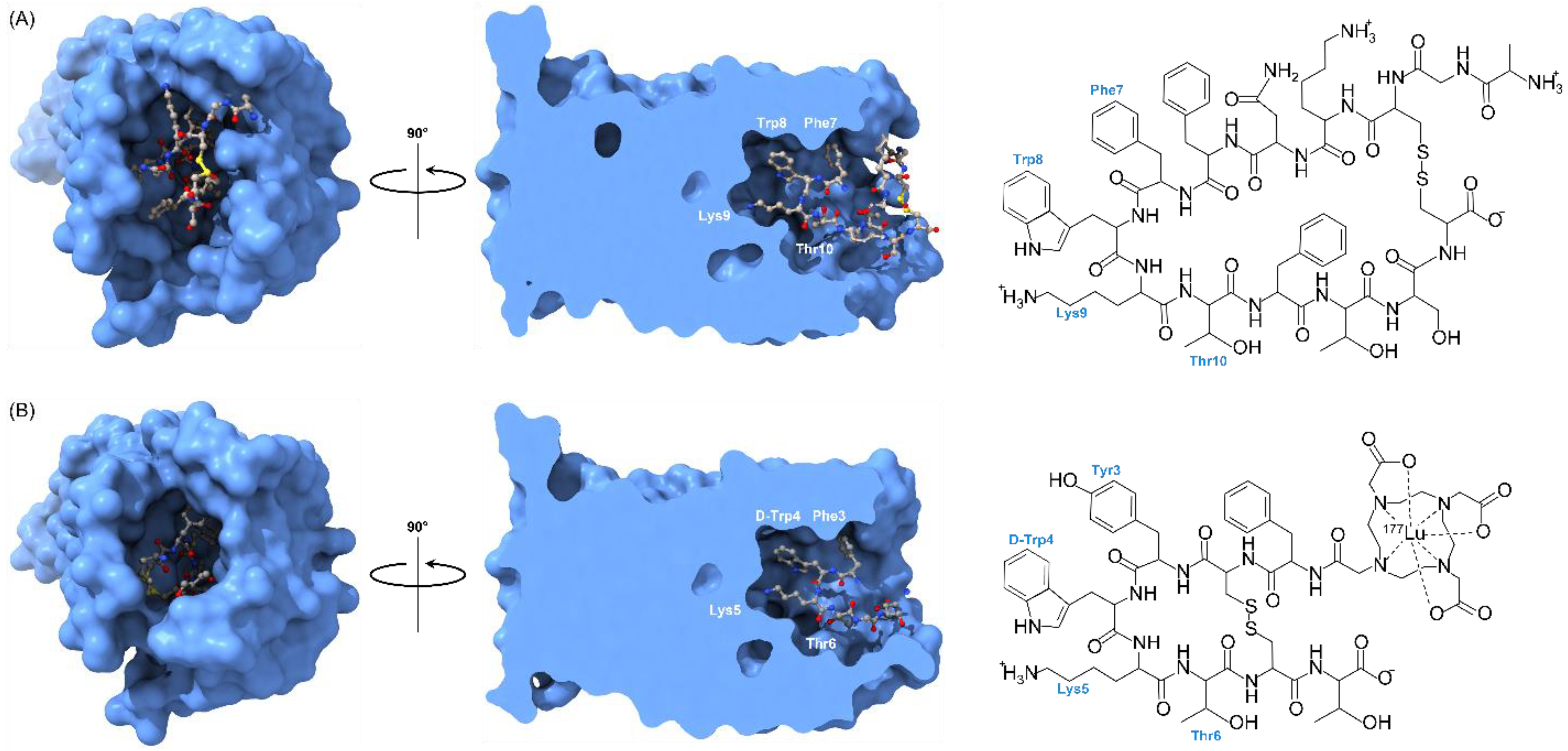
5. Lu-177 Current Clinical Application and Trials
5.1. FDA Approved PDC: Lu-177 DOTA-TATE
5.2. Examples of PDC Clinical Trials Utilziing Lu-177
5.3. PDC Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACPP | Activatable cell penetrating peptide |
| ADA | Anti drug Antibody |
| ADC | Antibody drug conjugate |
| ANG | Angiopep-2 |
| AuNPs | Gold nanoparticles |
| BBB | Blood–brain barrier |
| CCRCC | Clear cell renal cell carcinoma |
| CPP | Cell-penetrating peptide |
| CPT | Camptothecin |
| CRPC | Castration resistant prostate cancer |
| CTP | Cell-targeting peptide |
| DCR | Disease Control Rate |
| DOTA | Tetraazacyclododecane-tetraacetic acid |
| DMA | 2,3-dimethylmaleic anhydride |
| ES-SCLC | Extensive Stage Small Cell Lung Cancer |
| FAP | Fibroblast Activation Protein |
| FDA | Food and Drug Administration |
| GEP-NETs | Gastroenteropancreatic neuroendocrine tumors |
| HR | Hormone Receptor |
| IRIT | Intracavitary radioimmunotherapy |
| IV | Intravenous |
| mAb | Monoclonal antibody |
| MCRPC | Metastatic Castration-resistant Prostate Cancer |
| MHNPC | Metastatic hormone-naive prostate cancer |
| MOA | Mechanism of action |
| MT1-MMP | Membrane type 1 metalloproteinase |
| NET | Neuroendocrine tumor |
| NSCLC | Non-Small Cell Lung Cancer |
| ODD | Oxygen-dependent degradation |
| OHSPC | Oligometastatic hormone-sensitive prostate cancer |
| PABC | Valine-citrulline-p-aminobenzyloxycarbonyl |
| PDC | Peptide drug conjugate |
| PEG | Polyethylene glycol |
| P.K. | Pharmacokinetics |
| PSMA | Prostate-specific membrane antigen |
| PSar | Polysarcosine |
| PTX | Paclitaxel |
| RLT | Radioligand therapy |
| RMT | Radiometabolic Therapy |
| SBRT | Stereotactic body radiotherapy |
| SCLC | Small cell lung cancer |
| siRNA | short interfering RNA |
| SORT1 | sortilin1 |
| STTR | Somatostatin receptor |
| TATE | Tyr3-octreotate |
| T-DM1 | Trastuzumab |
| TNBC | Triple Negative Breast Cancer |
| VEGF | Vascular Endothelial Growth Factor |
| WDpNET | Well differentiated pancreatic neuroendocrine tumors |
References
- Mattiuzzi, C.; Lippi, G. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, W.R.; McGee-Avila, J.K.; Vo, J.B.; Luo, Q.; Chen, Y.; Inoue-Choi, M.; Berrington de Gonzalez, A.; Freedman, N.D.; Shiels, M.S. Trends in Cancer Mortality Among Black Individuals in the US From 1999 to 2019. JAMA Oncol. 2022, 8, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, P.S.; Callahan, K.E.; Gomez, S.L.; Marcos-Gragera, R.; Cobb, T.R.; Roca-Barcelo, A.; Ramirez, A.G. High cancer mortality for US-born Latinos: Evidence from California and Texas. BMC Cancer 2017, 17, 478. [Google Scholar] [CrossRef] [PubMed]
- Dieleman, J.L.; Baral, R.; Birger, M.; Bui, A.L.; Bulchis, A.; Chapin, A.; Hamavid, H.; Horst, C.; Johnson, E.K.; Joseph, J.; et al. US Spending on Personal Health Care and Public Health, 1996–2013. JAMA 2016, 316, 2627–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippman, S.M.; Hawk, E.T. Cancer prevention: From 1727 to milestones of the past 100 years. Cancer Res. 2009, 69, 5269–5284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide-drug conjugates (PDCs): A novel trend of research and development on targeted therapy, hype or hope? Acta Pharm. Sin. B 2022, in press. [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Pettinato, M.C. Introduction to Antibody-Drug Conjugates. Antibodies 2021, 10, 42. [Google Scholar] [CrossRef]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cheetham, A.G.; Angacian, G.; Su, H.; Xie, L.; Cui, H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 112–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokeshwar, B.L.; Kallifatidis, G.; Hoy, J.J. Atypical chemokine receptors in tumor cell growth and metastasis. Adv. Cancer Res. 2020, 145, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruoslahti, E. Tumor penetrating peptides for improved drug delivery. Adv. Drug Deliv. Rev. 2017, 110–111, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Al-Toubah, T.; El-Haddad, G.; Strosberg, J. (177)Lu-DOTATATE for the treatment of gastroenteropancreatic neuroendocrine tumors. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1023–1031. [Google Scholar] [CrossRef]
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting cancer with antibody-drug conjugates: Promises and challenges. MAbs 2021, 13, 1951427. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, S.; Liu, J.; Li, Q.; Zheng, L.; Xie, L.; Li, H.; Huang, D. Novel small peptides derived from VEGF(125-136): Potential drugs for radioactive diagnosis and therapy in A549 tumor-bearing nude mice. Sci. Rep. 2017, 7, 4278. [Google Scholar] [CrossRef] [Green Version]
- Watt, H.L.; Kharmate, G.; Kumar, U. Biology of somatostatin in breast cancer. Mol. Cell. Endocrinol. 2008, 286, 251–261. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Zeng, C.; Li, B.; Zhang, C.; Li, W.; Hou, X.; Dong, Y. Targeted delivery of atorvastatin via asialoglycoprotein receptor (ASGPR). Bioorg. Med. Chem. 2019, 27, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hu, J.; Wu, W.; Qin, Y.; Fu, J.; Zhou, J.; Liu, C.; Yin, J. N-acetyl-galactosamine modified metal-organic frameworks to inhibit the growth and pulmonary metastasis of liver cancer stem cells through targeted chemotherapy and starvation therapy. Acta Biomater. 2022, 151, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Jafari, B.; Pourseif, M.M.; Barar, J.; Rafi, M.A.; Omidi, Y. Peptide-mediated drug delivery across the blood-brain barrier for targeting brain tumors. Expert Opin. Drug Deliv. 2019, 16, 583–605. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Wang, C.; He, Z.; Cheng, B.; Zheng, L.; Huang, K. Peptide-Drug Conjugate: A Novel Drug Design Approach. Curr. Med. Chem. 2017, 24, 3373–3396. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Regberg, J.; Srimanee, A.; Langel, U. Applications of cell-penetrating peptides for tumor targeting and future cancer therapies. Pharmaceuticals 2012, 5, 991–1007. [Google Scholar] [CrossRef] [Green Version]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Hu, G.; Guo, M.; Xu, J.; Wu, F.; Fan, J.; Huang, Q.; Yang, G.; Lv, Z.; Wang, X.; Jin, Y. Nanoparticles Targeting Macrophages as Potential Clinical Therapeutic Agents Against Cancer and Inflammation. Front. Immunol. 2019, 10, 1998. [Google Scholar] [CrossRef]
- Cheng, H.; Zhu, J.Y.; Xu, X.D.; Qiu, W.X.; Lei, Q.; Han, K.; Cheng, Y.J.; Zhang, X.Z. Activable Cell-Penetrating Peptide Conjugated Prodrug for Tumor Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 16061–16069. [Google Scholar] [CrossRef]
- Vives, E.; Schmidt, J.; Pelegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheetham, A.G.; Keith, D.; Zhang, P.; Lin, R.; Su, H.; Cui, H. Targeting Tumors with Small Molecule Peptides. Curr. Cancer Drug Targets 2016, 16, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, U.B.; Schreiber, V.; Schultz, H.; Mischler, F.; Schughart, K. Tumor cell-targeting by phage-displayed peptides. Cancer Gene Ther. 2002, 9, 606–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolhassani, A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim. Biophys. Acta 2011, 1816, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Alas, M.; Saghaeidehkordi, A.; Kaur, K. Peptide-Drug Conjugates with Different Linkers for Cancer Therapy. J. Med. Chem. 2021, 64, 216–232. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Muller, T.D.; Yi, C.X.; Habegger, K.; Schriever, S.C.; Garcia-Caceres, C.; Kabra, D.G.; et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lin, Y.; Gillies, R.J. Tumor pH and its measurement. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2010, 51, 1167–1170. [Google Scholar] [CrossRef] [Green Version]
- Travis, J.; Salvesen, G.S. HUMAN PLASMA PROTEINASE INHIBITORS. Annu. Rev. Biochem. 1983, 52, 655–709. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Rompicharla, S.V.K.; Kumari, P.; Ghosh, B.; Biswas, S. Octa-arginine modified poly(amidoamine) dendrimers for improved delivery and cytotoxic effect of paclitaxel in cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lu, L.; Yan, S.; Yi, H.; Yao, H.; Wu, D.; He, G.; Tao, X.; Deng, X. Autophagy and doxorubicin resistance in cancer. Anti-Cancer. Drugs 2018, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Cheetham, A.G.; Lock, L.L.; Cui, H. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site. Bioconjug. Chem. 2013, 24, 604–613. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Cen, B.; Liao, L.; Wang, Z.; Qin, Y.; Wu, Z.; Liao, W.; Zhang, Z.; Ji, A. A tumor-targeting cRGD-EGFR siRNA conjugate and its anti-tumor effect on glioblastoma in vitro and in vivo. Drug Deliv. 2017, 24, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Farmer, F.T.; Haggith, J.W.; Hill, M. The treatment of myelomatosis with lutecium 177. Br. J. Radiol. 1960, 33, 374–378. [Google Scholar] [CrossRef]
- Banerjee, S.; Pillai, M.R.; Knapp, F.F. Lutetium-177 therapeutic radiopharmaceuticals: Linking chemistry, radiochemistry, and practical applications. Chem. Rev. 2015, 115, 2934–2974. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Q.; Cai, L.; Xie, Y.; Chen, Y. The efficacy of (177)Lu-DOTATATE peptide receptor radionuclide therapy (PRRT) in patients with metastatic neuroendocrine tumours: A systematic review and meta-analysis. J. Cancer Res. Clin. Oncol. 2020, 146, 1533–1543. [Google Scholar] [CrossRef]
- Robertson, M.J.; Meyerowitz, J.G.; Panova, O.; Borrelli, K.; Skiniotis, G. Plasticity in ligand recognition at somatostatin receptors. Nat. Struct. Mol. Biol. 2022, 29, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.M.; Iegre, J.; DH, O.D.; Olwegard Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Kalimuthu, K.; Lubin, B.C.; Bazylevich, A.; Gellerman, G.; Shpilberg, O.; Luboshits, G.; Firer, M.A. Gold nanoparticles stabilize peptide-drug-conjugates for sustained targeted drug delivery to cancer cells. J. Nanobiotechnol. 2018, 16, 34. [Google Scholar] [CrossRef]
- Wu, H.; Huang, J. Optimization of Protein and Peptide Drugs Based on the Mechanisms of Kidney Clearance. Protein Pept. Lett. 2018, 25, 514–521. [Google Scholar] [CrossRef]
- Sorolla, A.; Wang, E.; Golden, E.; Duffy, C.; Henriques, S.T.; Redfern, A.D.; Blancafort, P. Precision medicine by designer interference peptides: Applications in oncology and molecular therapeutics. Oncogene 2020, 39, 1167–1184. [Google Scholar] [CrossRef] [Green Version]
- Werle, M.; Bernkop-Schnurch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Wenande, E.; Garvey, L.H. Immediate-type hypersensitivity to polyethylene glycols: A review. Clin. Exp. Allergy 2016, 46, 907–922. [Google Scholar] [CrossRef]
- Hong, L.; Wang, Z.; Wei, X.; Shi, J.; Li, C. Antibodies against polyethylene glycol in human blood: A literature review. J. Pharmacol. Toxicol. Methods 2020, 102, 106678. [Google Scholar] [CrossRef]
- Avramis, V.I.; Sencer, S.; Periclou, A.P.; Sather, H.; Bostrom, B.C.; Cohen, L.J.; Ettinger, A.G.; Ettinger, L.J.; Franklin, J.; Gaynon, P.S.; et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: A Children’s Cancer Group study. Blood 2002, 99, 1986–1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinincalTrials.gov. This Study Collects Information on the Safety of Inhaled Pegylated Adrenomedullin (PEG-ADM), How the Drug Is Tolerated and How It Affects Patients Suffering From a Type of Lung Failure That Cause Fluid to Build up in the Lungs Making Breathing Difficult (ARDS). Available online: https://clinicaltrials.gov/ct2/show/NCT04417036 (accessed on 17 November 2022).
- Hoang Thi, T.T.; Pilkington, E.H.; Nguyen, D.H.; Lee, J.S.; Park, K.D.; Truong, N.P. The Importance of Poly(ethylene glycol) Alternatives for Overcoming PEG Immunogenicity in Drug Delivery and Bioconjugation. Polymers 2020, 12, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Hou, Y.; Wang, H.; Lu, H. Polysarcosine as an Alternative to PEG for Therapeutic Protein Conjugation. Bioconjug. Chem. 2018, 29, 2232–2238. [Google Scholar] [CrossRef] [PubMed]
- Son, K.; Ueda, M.; Taguchi, K.; Maruyama, T.; Takeoka, S.; Ito, Y. Evasion of the accelerated blood clearance phenomenon by polysarcosine coating of liposomes. J. Control. Release 2020, 322, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Podust, V.N.; Balan, S.; Sim, B.C.; Coyle, M.P.; Ernst, U.; Peters, R.T.; Schellenberger, V. Extension of in vivo half-life of biologically active molecules by XTEN protein polymers. J. Control. Release 2016, 240, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Song, M.; Sim, B.C.; Gu, C.; Podust, V.N.; Wang, C.W.; McLaughlin, B.; Shah, T.P.; Lax, R.; Gast, R.; et al. Multivalent antiviral XTEN-peptide conjugates with long in vivo half-life and enhanced solubility. Bioconjug. Chem. 2014, 25, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Schlapschy, M.; Binder, U.; Borger, C.; Theobald, I.; Wachinger, K.; Kisling, S.; Haller, D.; Skerra, A. PASylation: A biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 2013, 26, 489–501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, S.; Wu, W.; Xia, X.; Zhang, J. PASylation improves pharmacokinetic of liposomes and attenuates anti-PEG IgM production: An alternative to PEGylation. Nanomedicine 2023, 47, 102622. [Google Scholar] [CrossRef]
- Olivier, T.; Prasad, V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022, 18, 101374. [Google Scholar] [CrossRef]
- Lamson, N.G.; Berger, A.; Fein, K.C.; Whitehead, K.A. Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nat. Biomed. Eng. 2020, 4, 84–96. [Google Scholar] [CrossRef]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Intervention | ClinicalTrials.gov Identifier | Phase | Indication | Target |
|---|---|---|---|---|
| 177Lu-PNT2002 versus abiraterone or enzalutamide | NCT04647526 | 3 | Metastatic Castration-resistant Prostate Cancer (mCRPC) | PSMA |
| 177Lu-PSMA-I&T versus Hormone Therapy | NCT05204927 | 3 | mCRPC | PSMA |
| 177Lu-Ludotadipep | NCT05579184 | 2 | mCRPC | PSMA |
| 177Lu-PSMA-617 | NCT05114746 | 2 | mCRPC | PSMA |
| 177Lu-PSMA (+/−) Ipilimumab and Nivolumab | NCT05150236 | 2 | mCRPC | PSMA |
| 177Lu-PSMA and enzalutamide (nonsteroidal antiandrogen) | NCT04419402 | 2 | mCRPC | PSMA |
| 177Lu-PSMA (DGUL) and Ga-68-NGUL | NCT05547061 | 1/2 | mCRPC | PSMA |
| 177Lu-PSMA-I&T | NCT05383079 | 1/2 | mCRPC | PSMA |
| Cabazitaxel in combination with 177Lu-PSMA-617 | NCT05340374 | 1/2 | mCRPC | PSMA |
| Abemaciclib and 177Lu-PSMA-617 | NCT05113537 | 1/2 | mCRPC | PSMA |
| 177Lu-rhPSMA-10.1 | NCT05413850 | 1/2 | mCRPC | PSMA |
| 177Lu-EB-PSMA-617 | NCT03780075 | 1 | mCRPC | PSMA |
| 177Lu-PSMA-EB-01 (+/−) radioligand therapy (RLT) | NCT05613738 | 1 | mCRPC | PSMA |
| 177Lu-PSMA + olaparib (PARP inhibitor) | NCT03874884 | 1 | mCRPC | PSMA |
| 177Lu-EB-PSMA (55 mCi) | NCT04996602 | 1 | mCRPC | PSMA |
| 177Lu-Ludotadipep | NCT05458544 | 1 | mCRPC | PSMA |
| 177Lu-DOTA-TLX591 | NCT04786847 | 1 | mCRPC | PSMA |
| Radiometabolic Therapy (RMT) with 177Lu PSMA 617 | NCT03454750 | 2 | Castration Resistant Prostate Cancer (CRPC) | PSMA |
| 177Lu-PSMA-617 | NCT04443062 | Oligo-metastatic Hormone Sensitive Prostate Cancer (mHSP) | PSMA | |
| Standard of Care (SOC) (+/−) 177Lu-PSMA-617 | NCT04720157 | mHSPC | PSMA | |
| Docetaxel +/− 177Lu-PSMA | NCT04343885 | 2 | metastatic hormone-naive prostate cancer (mHNPC) | PSMA |
| 177Lu-TLX591 | NCT05146973 | 2 | PSMA-expressing prostate cancer | PSMA |
| 225Ac-J591 and 177Lu-PSMA-I&T | NCT04886986 | 1/2 | Prostate cancer | PSMA |
| 177Lu-PSMA | NCT05230251 | 2 | Prostate cancer | PSMA |
| 177Lu PSMA 617 | NCT04663997 | 2 | Prostate cancer | PSMA |
| 177-Lu-PSMA given before stereotactic body radiotherapy (SBRT) | NCT04597411 | 2 | Prostate cancer | PSMA |
| 177Lu-PSMA-617 | NCT05613842 | 2 | Hormone-sensitive disease (cohort A) castrate-resistant Disease (Cohort B) | PSMA |
| 177Lu-PSMA radioligand therapy | NCT05162573 | 1 | node-positive prostate cancer | PSMA |
| 177Lu-PP-F11N | NCT02088645 | 1 | Advanced medullary thyroid carcinoma GEP-NET | cholecystokinin-2 receptors |
| 177Lu-AB-3PRGD2 | NCT05013086 | 1 | Non-Small Cell Lung Cancer (NSCLC) | Integrin αvβ3 |
| 177Lu-DOTA-TATE in combination with carboplatin, etoposide, and tislelizumab | NCT05142696 | 1 | Extensive Stage Small Cell Lung Cancer (ES-SCLC) | STTR |
| GD2-SADA:177Lu-DOTA complex | NCT05130255 | 1 | GD2 expressing solid tumors (Small Cell Lung Cancer, Sarcoma and Malignant Melanoma) | GD2 |
| Standard of Care (+/−) 177Lu-DOTA-TATE | NCT05109728 | 1 | Glioblastoma | STTR |
| Intracavitary radioimmunotherapy (iRIT) with a newly developed radioimmunoconjugate 177Lu labeled 6A10-Fab-fragments | NCT05533242 | 1 | Glioblastoma | carbonic anhydrase XII |
| Combination of 177Lu-girentuximab and nivolumab | NCT05239533 | 2 | Advanced clear cell renal cell carcinoma/ccRCC | Carbonic Anhydrase IX |
| 68Ga-PSMA PET-CT with 177Lu-EB-PSMA-617 | NCT05170555 | NA | Renal Cell Carcinoma | PSMA |
| 177Lu-PNT6555 | NCT05432193 | 1 | Fibroblast Activation Protein (FAP) overexpressing tumors (Colorectal Cancer; Esophageal Cancer; Melanoma; Soft Tissue Sarcoma | FAP |
| [68Ga]Ga DOTA-5G and 177Lu DOTA-ABM-5G theranostic | NCT04665947 | 1 | Locally advanced or metastatic pancreatic adenocarcinoma (PDAC) | - |
| 177Lu-octreotate versus sunitinib | NCT02230176 | 2 | Progressive pancreatic, inoperable, somatostatin receptor positive, well differentiated pancreatic neuroendocrine tumors (WDpNET). | STTR |
| 177Lu-DOTATATE versus capecitabine and temozolomide | NCT05247905 | 2 | Metastatic Pancreatic Neuroendocrine Tumor and Unresectable Pancreatic Neuroendocrine Carcinoma | STTR |
| 177Lu-DOTATATE hepatic intraarterial infusion | NCT04544098 | 1 | Neuroendocrine Tumors Liver-Dominant Metastatic Pancreatic Neuroendocrine Tumors | STTR |
| 177Lu-DOTATOC | NCT04276597 | 2 | Somatostatin receptor-expressing Pulmonary, Pheochromocytoma, Paraganglioma, and Thymus neuroendocrine tumors | STTR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heh, E.; Allen, J.; Ramirez, F.; Lovasz, D.; Fernandez, L.; Hogg, T.; Riva, H.; Holland, N.; Chacon, J. Peptide Drug Conjugates and Their Role in Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 829. https://doi.org/10.3390/ijms24010829
Heh E, Allen J, Ramirez F, Lovasz D, Fernandez L, Hogg T, Riva H, Holland N, Chacon J. Peptide Drug Conjugates and Their Role in Cancer Therapy. International Journal of Molecular Sciences. 2023; 24(1):829. https://doi.org/10.3390/ijms24010829
Chicago/Turabian StyleHeh, Ethan, Jesse Allen, Fabiola Ramirez, Daniel Lovasz, Lorena Fernandez, Tanis Hogg, Hannah Riva, Nathan Holland, and Jessica Chacon. 2023. "Peptide Drug Conjugates and Their Role in Cancer Therapy" International Journal of Molecular Sciences 24, no. 1: 829. https://doi.org/10.3390/ijms24010829
APA StyleHeh, E., Allen, J., Ramirez, F., Lovasz, D., Fernandez, L., Hogg, T., Riva, H., Holland, N., & Chacon, J. (2023). Peptide Drug Conjugates and Their Role in Cancer Therapy. International Journal of Molecular Sciences, 24(1), 829. https://doi.org/10.3390/ijms24010829