
Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates
 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Results
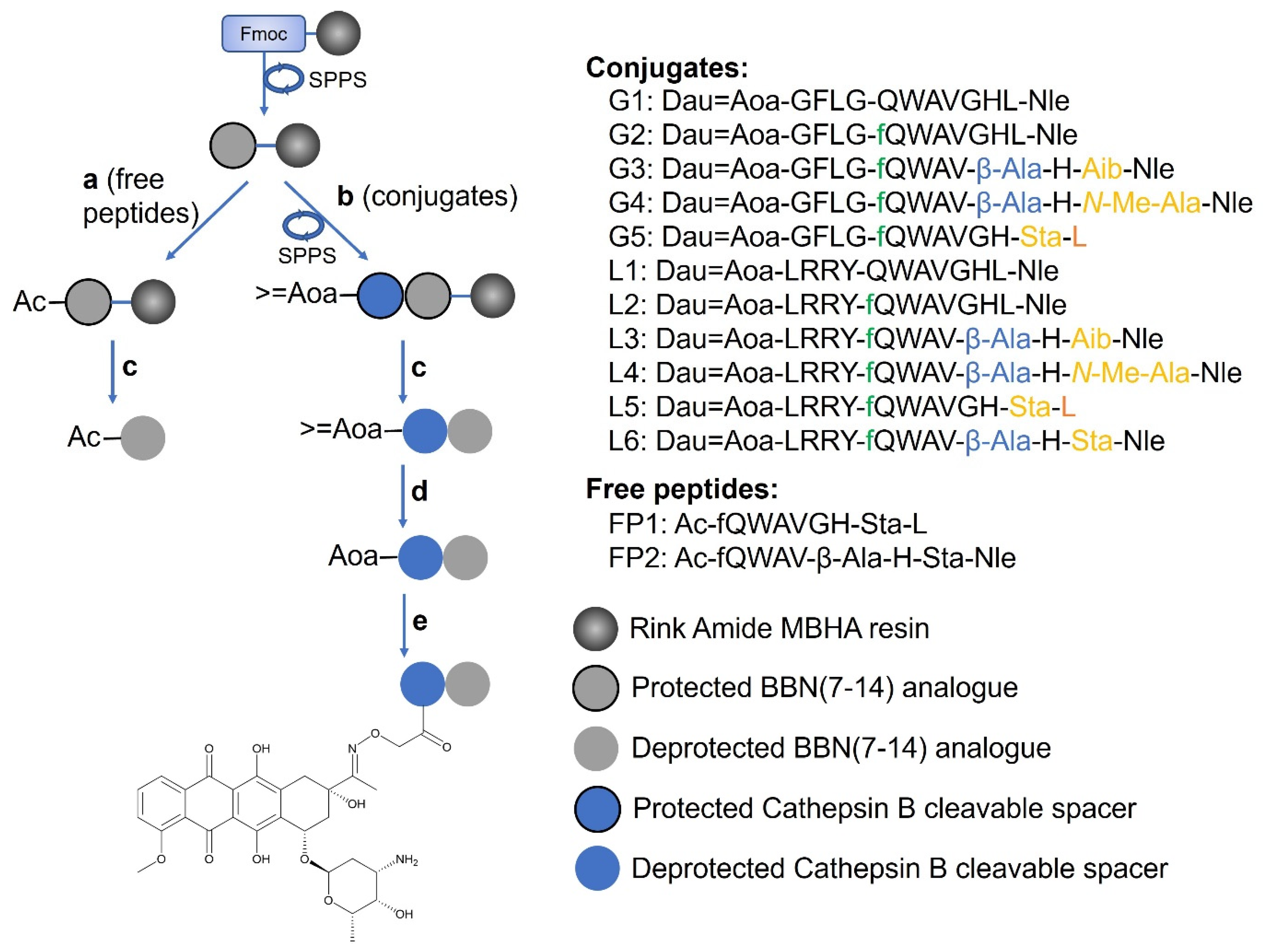
2.1. Synthesis of the Peptide–Drug Conjugates
2.2. GRP-R Expression in Selected Cell Lines
2.3. In Vitro Cytostatic Effect of Dau-BBN(7-14) Conjugates
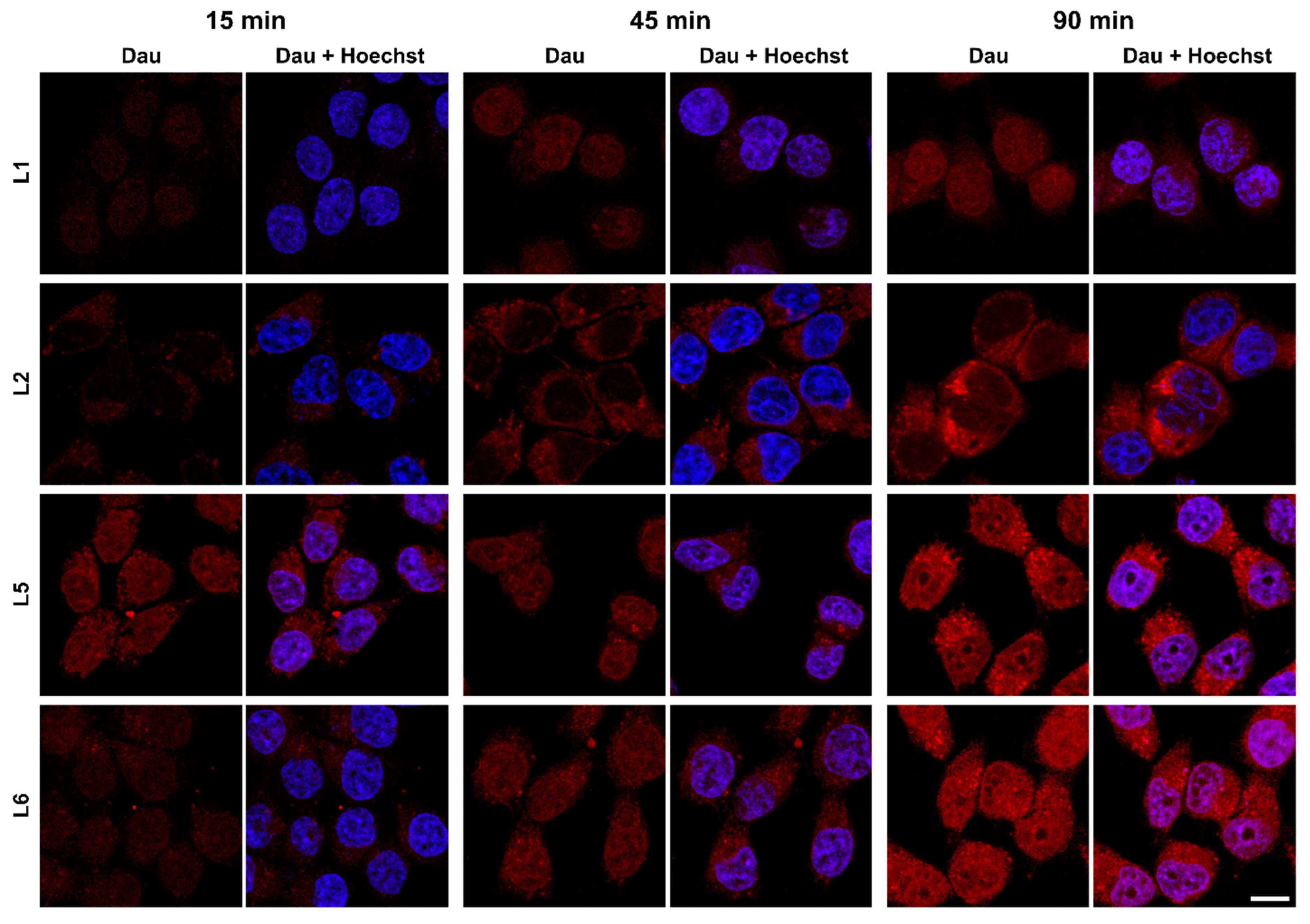
2.4. Cellular Uptake and Localisation of Dau–BBN (7-14) Conjugates
2.5. Metabolism and Release of Dau=Aoa-Leu-OH in Lysosomal Environment
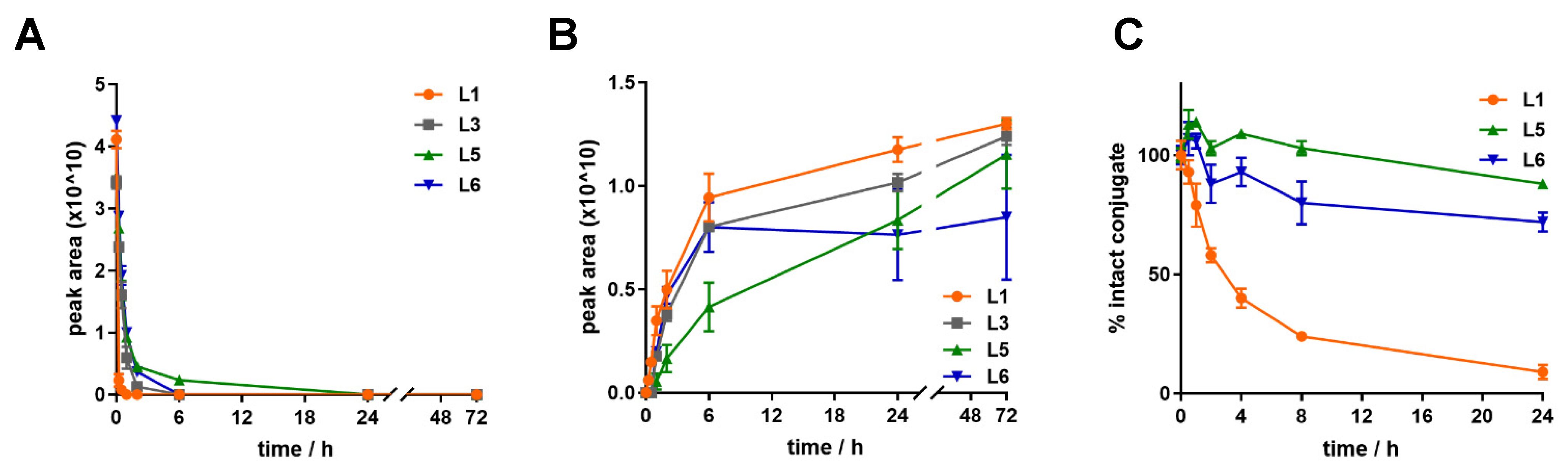
2.6. Stability of Dau–BBN (7-14) Conjugates
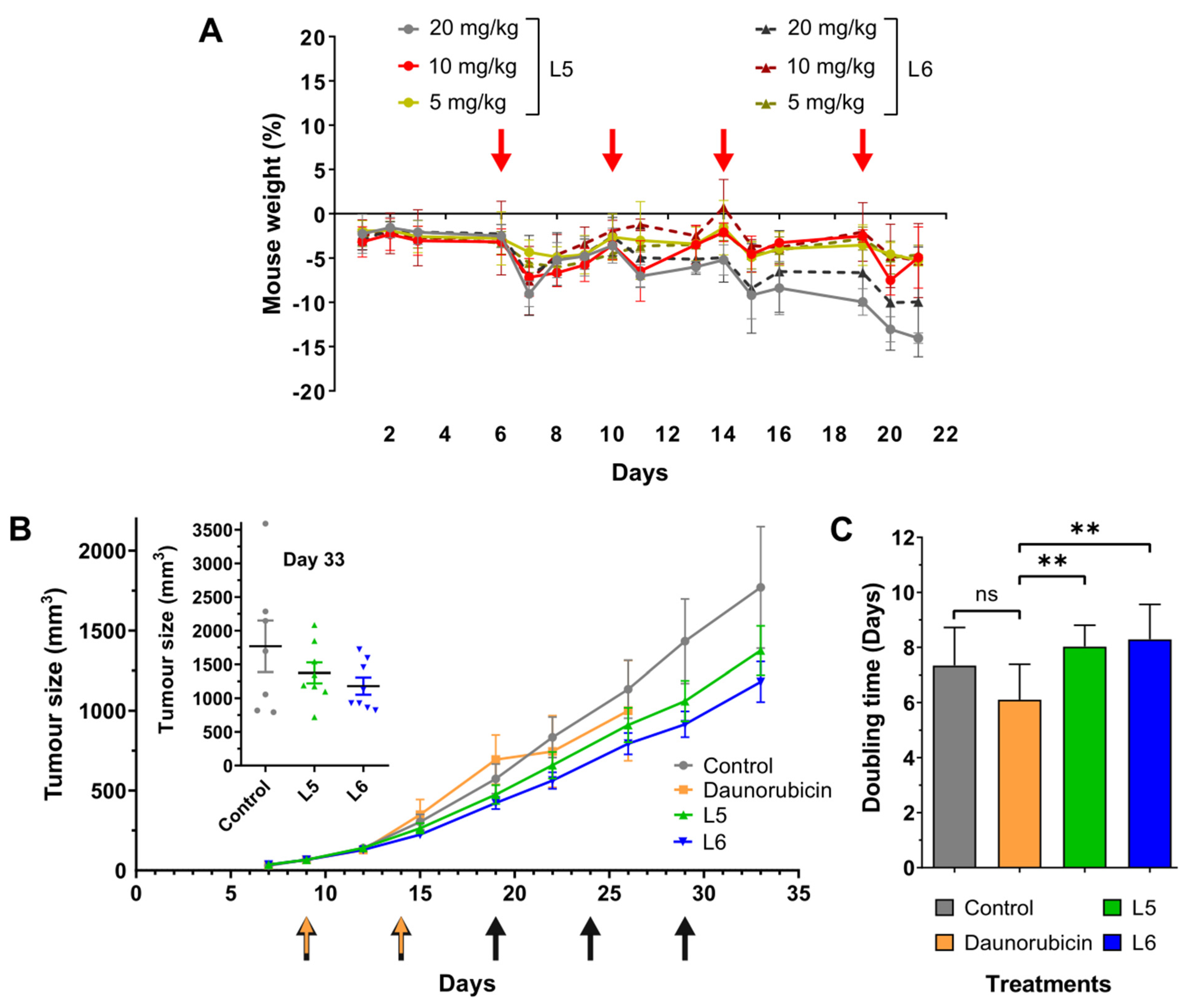
2.7. In Vivo Chronic Toxicity Studies of L5 and L6
2.8. In Vivo Tumour Growth Inhibition by L5 and L6
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents
4.2. Synthesis of Peptide Sequences
4.3. Isopropylidene Deprotection and Conjugation to Daunorubicin
4.4. RP-HPLC
4.5. Liquid Chromatography–Mass Spectrometry (LC–MS)
4.6. Cell Lines and Culturing
4.7. MTT Assays
4.8. Stability of Bombesin-Based Bioconjugates in Mouse Plasma
4.9. Stability of Bombesin-Based Bioconjugates in Cell Culture Media
4.10. Isolation of Lysosomes from Rat Liver
4.11. Metabolism in Rat Liver Lysosomal Homogenate
4.12. RT-qPCR Measurements
4.13. Western Blot
4.14. Flow Cytometry
4.15. Confocal Microscopy
4.16. Materials and Animals
4.17. In Vivo Toxicity and Antitumour Efficacy of the Bombesin-Based Bioconjugates
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody Drug Conjugate: The “Biological Missile” for Targeted Cancer Therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Ölwegård Halvarsson, M.; Spring, D.R. Peptides as a Platform for Targeted Therapeutics for Cancer: Peptide–Drug Conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.; Nilvebrant, J.; Nygren, P.-Å.; Lehmann, F. Progress and Future Directions with Peptide-Drug Conjugates for Targeted Cancer Therapy. Molecules 2021, 26, 6042. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S.; Siahaan, T.J. Peptide-Mediated Targeted Drug Delivery. Med. Res. Rev. 2012, 32, 637–658. [Google Scholar] [CrossRef]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Pepaxti®—Melphalan Flufenamide. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/pepaxti (accessed on 10 December 2022).
- Dhillon, S. Melphalan Flufenamide (Melflufen): First Approval. Drugs 2021, 81, 963–969. [Google Scholar] [CrossRef]
- Mittra, E.S. Neuroendocrine Tumor Therapy: 177 Lu-DOTATATE. Am. J. Roentgenol. 2018, 211, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Solanki, H.K.; Davidson, M.; Apostolopoulos, V.; Bojarska, J. Peptide-Drug Conjugates: A New Hope for Cancer Management. Molecules 2022, 27, 7232. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Yu, L.; Miao, Y.; Liu, X.; Yu, Z.; Wei, M. Peptide–Drug Conjugates (PDCs): A Novel Trend of Research and Development on Targeted Therapy, Hype or Hope? Sin. B, 2022, in press. [CrossRef]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Farkas, V.; Buday, L.; Szabó, Z.; Halmos, G.; Mező, G. Synthesis and in Vitro Biochemical Evaluation of Oxime Bond-Linked Daunorubicin–GnRH-III Conjugates Developed for Targeted Drug Delivery. Beilstein J. Org. Chem. 2018, 14, 756–771. [Google Scholar] [CrossRef]
- Schuster, S.; Biri-Kovács, B.; Szeder, B.; Buday, L.; Gardi, J.; Szabó, Z.; Halmos, G.; Mező, G. Enhanced In Vitro Antitumor Activity of GnRH-III-Daunorubicin Bioconjugates Influenced by Sequence Modification. Pharmaceutics 2018, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Ranđelović; Schuster; Kapuvári; Fossati; Steinkühler; Mező; Tóvári Improved In Vivo Anti-Tumor and Anti-Metastatic Effect of GnRH-III-Daunorubicin Analogs on Colorectal and Breast Carcinoma Bearing Mice. Int. J. Mol. Sci. 2019, 20, 4763. [CrossRef]
- Schuster, S.; Juhász, É.; Halmos, G.; Neundorf, I.; Gennari, C.; Mező, G. Development and Biochemical Characterization of Self-Immolative Linker Containing GnRH-III-Drug Conjugates. Int. J. Mol. Sci. 2022, 23, 5071. [Google Scholar] [CrossRef]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.-C.; Gugger, M. Bombesin Receptor Subtypes in Human Cancers: Detection with the Universal Radioligand 125I-[D-TYR6, b-ALA11, PHE13, NLE14] Bombesin(6–14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International Union of Pharmacology. LXVIII. Mammalian Bombesin Receptors: Nomenclature, Distribution, Pharmacology, Signaling, and Functions in Normal and Disease States. Pharm. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Preston, S.R.; Miller, G.V.; Primrose, J.N. Bombesin-like Peptides and Cancer. Crit. Rev. Oncol. Hematol. 1996, 23, 225–238. [Google Scholar] [CrossRef]
- Ramos-Álvarez, I.; Moreno, P.; Mantey, S.A.; Nakamura, T.; Nuche-Berenguer, B.; Moody, T.W.; Coy, D.H.; Jensen, R.T. Insights into Bombesin Receptors and Ligands: Highlighting Recent Advances. Peptides 2015, 72, 128–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajo, A.M.; Schally, A.V.; Krupa, M.; Hebert, F.; Groot, K.; Szepeshazi, K. Bombesin Antagonists Inhibit Growth of MDA-MB-435 Estrogen-Independent Breast Cancers and Decrease the Expression of the ErbB-2/HER-2 Oncoprotein and c- Jun and c- Fos Oncogenes. Proc. Natl. Acad. Sci. USA 2002, 99, 3836–3841. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.A. Investigation of Bombesin Peptide as a Targeting Ligand for the Gastrin Releasing Peptide (GRP) Receptor. Bioorg. Med. Chem. 2016, 8, 5834–5841. [Google Scholar] [CrossRef] [PubMed]
- Begum, A.A.; Wan, Y.; Toth, I.; Moyle, P.M. Bombesin/Oligoarginine Fusion Peptides for Gastrin Releasing Peptide Receptor (GRPR) Targeted Gene Delivery. Bioorg. Med. Chem. 2018, 26, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin Receptor Antagonists May Be Preferable to Agonists for Tumor Targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Identification and Stabilization of a Highly Selective Gastrin-releasing Peptide Receptor Agonist. J. Pep. Sci. 2019, 25, e3224. [Google Scholar] [CrossRef]
- Llinares, M.; Devin, C.; Chaloin, O.; Azay, J.; Noel-Artis, A.M.; Bernad, N.; Fehrentz, J.A.; Martinez, J. Syntheses and Biological Activities of Potent Bombesin Receptor Antagonists: Bombesin Receptor Antagonists. J. Pept. Res. 1999, 53, 275–283. [Google Scholar] [CrossRef]
- Valverde, I.E.; Bauman, A.; Kluba, C.A.; Vomstein, S.; Walter, M.A.; Mindt, T.L. 1,2,3-Triazoles as Amide Bond Mimics: Triazole Scan Yields Protease-Resistant Peptidomimetics for Tumor Targeting. Angew. Chem. Int. Ed. 2013, 52, 8957–8960. [Google Scholar] [CrossRef]
- Nagy, A.; Armatis, P.; Cai, R.-Z.; Szepeshazi, K.; Halmos, G.; Schally, A.V. Design, Synthesis, and in Vitro Evaluation of Cytotoxic Analogs of Bombesin-like Peptides Containing Doxorubicin or Its Intensely Potent Derivative, 2-Pyrrolinodoxorubicin. Proc. Natl. Acad. Sci. USA 1997, 94, 652–656. [Google Scholar] [CrossRef]
- Accardo, A.; Galli, F.; Mansi, R.; Del Pozzo, L.; Aurilio, M.; Morisco, A.; Ringhieri, P.; Signore, A.; Morelli, G.; Aloj, L. Pre-Clinical Evaluation of Eight DOTA Coupled Gastrin-Releasing Peptide Receptor (GRP-R) Ligands for in Vivo Targeting of Receptor-Expressing Tumors. EJNMMI Res. 2016, 6, 17. [Google Scholar] [CrossRef]
- De, K.; Banerjee, I.; Sinha, S.; Ganguly, S. Synthesis and Exploration of Novel Radiolabeled Bombesin Peptides for Targeting Receptor Positive Tumor. Peptides 2017, 89, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Lymperis, E.; Kaloudi, A.; Kanellopoulos, P.; de Jong, M.; Krenning, E.; Nock, B.; Maina, T. Comparing Gly11/DAla11-Replacement vs. the in-Situ Neprilysin-Inhibition Approach on the Tumor-Targeting Efficacy of the 111In-SB3/111In-SB4 Radiotracer Pair. Molecules 2019, 24, 1015. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lau, J.; Rousseau, E.; Zhang, Z.; Uribe, C.F.; Kuo, H.-T.; Zeisler, J.; Zhang, C.; Kwon, D.; Lin, K.-S.; Bénard, F. Positron Emission Tomography Imaging of the Gastrin-Releasing Peptide Receptor with a Novel Bombesin Analogue. ACS Omega 2019, 4, 1470–1478. [Google Scholar] [CrossRef]
- Schroeder, R.P.J.; Müller, C.; Reneman, S.; Melis, M.L.; Breeman, W.A.P.; de Blois, E.; Bangma, C.H.; Krenning, E.P.; van Weerden, W.M.; de Jong, M. A Standardised Study to Compare Prostate Cancer Targeting Efficacy of Five Radiolabelled Bombesin Analogues. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1386–1396. [Google Scholar] [CrossRef]
- Tu, Y.; Tao, J.; Wang, F.; Liu, P.; Han, Z.; Li, Z.; Ma, Y.; Gu, Y. A Novel Peptide Targeting Gastrin Releasing Peptide Receptor for Pancreatic Neoplasm Detection. Biomater. Sci. 2020, 8, 2682–2693. [Google Scholar] [CrossRef]
- Höhne, A.; Mu, L.; Honer, M.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Borkowski, S.; Berndorff, D.; Klar, U.; et al. Synthesis, 18F-Labeling, and in Vitro and in Vivo Studies of Bombesin Peptides Modified with Silicon-Based Building Blocks. Bioconjugate Chem. 2008, 19, 1871–1879. [Google Scholar] [CrossRef]
- Mansi, R.; Nock, B.A.; Dalm, S.U.; Busstra, M.B.; van Weerden, W.M.; Maina, T. Radiolabeled Bombesin Analogs. Cancers 2021, 13, 5766. [Google Scholar] [CrossRef]
- Abiraj, K.; Mansi, R.; Tamma, M.-L.; Fani, M.; Forrer, F.; Nicolas, G.; Cescato, R.; Reubi, J.C.; Maecke, H.R. Bombesin Antagonist–Based Radioligands for Translational Nuclear Imaging of Gastrin-Releasing Peptide Receptor–Positive Tumors. J. Nucl. Med. 2011, 52, 1970–1978. [Google Scholar] [CrossRef]
- Accardo, A.; Mansi, R.; Salzano, G.; Morisco, A.; Aurilio, M.; Parisi, A.; Maione, F.; Cicala, C.; Ziaco, B.; Tesauro, D.; et al. Bombesin Peptide Antagonist for Target-Selective Delivery of Liposomal Doxorubicin on Cancer Cells. J. Drug Target. 2013, 21, 240–249. [Google Scholar] [CrossRef]
- Akbar, M.J.; Lukasewicz Ferreira, P.C.; Giorgetti, M.; Stokes, L.; Morris, C.J. Bombesin Receptor-Targeted Liposomes for Enhanced Delivery to Lung Cancer Cells. Beilstein J. Nanotechnol. 2019, 10, 2553–2562. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Mantey, S.A.; Pradhan, T.K.; Schumann, M.; Nakagawa, T.; Martinez, A.; Fuselier, J.; Coy, D.H.; Jensen, R.T. Development of High Affinity Camptothecin-Bombesin Conjugates That Have Targeted Cytotoxicity for Bombesin Receptor-Containing Tumor Cells. J. Biol. Chem. 2004, 279, 23580–23589. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.-C.; Luo, J.; Mackey, V.L.; Fuselier, J.A.; Coy, D.H. Effects of Camptothecin on Tumor Cell Proliferation and Angiogenesis When Coupled to a Bombesin Analog Used as a Targeted Delivery Vector. Anti-Cancer Drugs 2007, 18, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Morelli, G.; Accardo, A.; Tesauro, D.; Cicala, C.; Salzano, G.; De Rosa, G.; Morisco, A.; Aloj, L.; Aurilio, M.; Maione, F.; et al. Peptide-Modified Liposomes for Selective Targeting of Bombesin Receptors Overexpressed by Cancer Cells: A Potential Theranostic Agent. Int. J. Nanomed. 2012, 7, 2007–2017. [Google Scholar] [CrossRef]
- Accardo, A.; Mannucci, S.; Nicolato, E.; Vurro, F.; Diaferia, C.; Bontempi, P.; Marzola, P.; Morelli, G. Easy Formulation of Liposomal Doxorubicin Modified with a Bombesin Peptide Analogue for Selective Targeting of GRP Receptors Overexpressed by Cancer Cells. Drug Deliv. Transl. Res. 2019, 9, 215–226. [Google Scholar] [CrossRef]
- Safavy, A.; Raisch, K.P.; Matusiak, D.; Bhatnagar, S.; Helson, L. Single-Drug Multiligand Conjugates: Synthesis and Preliminary Cytotoxicity Evaluation of a Paclitaxel−Dipeptide “Scorpion” Molecule. Bioconjugate Chem. 2006, 17, 565–570. [Google Scholar] [CrossRef]
- Dheer, D.; Nicolas, J.; Shankar, R. Cathepsin-Sensitive Nanoscale Drug Delivery Systems for Cancer Therapy and Other Diseases. Adv. Drug Deliv. Rev. 2019, 151–152, 130–151. [Google Scholar] [CrossRef]
- Zhong, Y.-J.; Shao, L.-H.; Li, Y. Cathepsin B-Cleavable Doxorubicin Prodrugs for Targeted Cancer Therapy. Int. J. Oncol. 2013, 42, 373–383. [Google Scholar] [CrossRef]
- Coley, H.; Amos, W.; Twentyman, P.; Workman, P. Examination by Laser Scanning Confocal Fluorescence Imaging Microscopy of the Subcellular Localisation of Anthracyclines in Parent and Multidrug Resistant Cell Lines. Br. J. Cancer 1993, 67, 1316–1323. [Google Scholar] [CrossRef]
- Schally, A.V.; Nagy, A. New Approaches to Treatment of Various Cancers Based on Cytotoxic Analogs of LHRH, Somatostatin and Bombesin. Life Sci. 2003, 72, 2305–2320. [Google Scholar] [CrossRef]
- Bősze, S.; Zsila, F.; Biri-Kovács, B.; Szeder, B.; Majer, Z.; Hudecz, F.; Uray, K. Tailoring Uptake Efficacy of HSV-1 GD Tailoring Uptake Efficacy of Hsv-1 GD Derived Carrier Peptides. Biomolecules 2020, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Kroll, C.; Mansi, R.; Braun, F.; Dobitz, S.; Maecke, H.R.; Wennemers, H. Hybrid Bombesin Analogues: Combining an Agonist and an Antagonist in Defined Distances for Optimized Tumor Targeting. J. Am. Chem. Soc. 2013, 135, 16793–16796. [Google Scholar] [CrossRef] [PubMed]
- Shirmardi, S.P.; Gandomkar, M.; Maragheh, M.G.; Shamsaei, M. Preclinical Evaluation of a New Bombesin Analog for Imaging of Gastrin-Releasing Peptide Receptors. Cancer Biother. Radiopharm. 2011, 26, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cai, W.; Cao, F.; Schreibmann, E.; Wu, Y.; Wu, J.C.; Xing, L.; Chen, X. 18F-Labeled Bombesin Analogs for Targeting GRP Receptor-Expressing Prostate Cancer. J. Nucl. Med. 2006, 47, 492–501. [Google Scholar]
- Darker, J.G.; Brough, S.J.; Heath, J.; Smart, D. Discovery of Potent and Selective Peptide Agonists at the GRP-Preferring Bombesin Receptor (BB2). J. Pept. Sci. 2001, 7, 598–605. [Google Scholar] [CrossRef]
- Mansi, R.; Wang, X.; Forrer, F.; Waser, B.; Cescato, R.; Graham, K.; Borkowski, S.; Reubi, J.C.; Maecke, H.R. Development of a Potent DOTA-Conjugated Bombesin Antagonist for Targeting GRPr-Positive Tumours. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 97–107. [Google Scholar] [CrossRef]
- Abouzayed, A.; Tano, H.; Nagy, Á.; Rinne, S.S.; Wadeea, F.; Kumar, S.; Westerlund, K.; Tolmachev, V.; Eriksson Karlström, A.; Orlova, A. Preclinical Evaluation of the GRPR-Targeting Antagonist RM26 Conjugated to the Albumin-Binding Domain for GRPR-Targeting Therapy of Cancer. Pharmaceutics 2020, 12, 977. [Google Scholar] [CrossRef]
- Jamous, M.; Tamma, M.L.; Gourni, E.; Waser, B.; Reubi, J.C.; Maecke, H.R.; Mansi, R. PEG Spacers of Different Length Influence the Biological Profile of Bombesin-Based Radiolabeled Antagonists. Nucl. Med. Biol. 2014, 41, 464–470. [Google Scholar] [CrossRef]
- Schlage, P.; Mező, G.; Orbán, E.; Bősze, S.; Manea, M. Anthracycline-GnRH Derivative Bioconjugates with Different Linkages: Synthesis, in Vitro Drug Release and Cytostatic Effect. J. Control. Release 2011, 156, 170–178. [Google Scholar] [CrossRef]
- Orbán, E.; Manea, M.; Marquadt, A.; Bánóczi, Z.; Csík, G.; Fellinger, E.; Bősze, S.; Hudecz, F. A New Daunomycin–Peptide Conjugate: Synthesis, Characterization and the Effect on the Protein Expression Profile of HL-60 Cells in Vitro. Bioconjugate Chem. 2011, 22, 2154–2165. [Google Scholar] [CrossRef]
- Orbán, E.; Mező, G.; Schlage, P.; Csík, G.; Kulić, Ž.; Ansorge, P.; Fellinger, E.; Möller, H.M.; Manea, M. In Vitro Degradation and Antitumor Activity of Oxime Bond-Linked Daunorubicin–GnRH-III Bioconjugates and DNA-Binding Properties of Daunorubicin–Amino Acid Metabolites. Amino Acids 2011, 41, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Flecknell, P. Replacement, Reduction and Refinement. ALTEX Altern. Anim. Exp. 2002, 19, 73–78. [Google Scholar]
- Cailleau, R.; Olivé, M.; Cruciger, Q. Long-Term Human Breast Carcinoma Cell Lines of Metastatic Origin: Preliminary Characterization. In vitro 1978, 14, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Kaighn, M.; Narayan, K.; Ohnuki, Y.; Lechner, J.; Jones, L. Establishment and Characterization of a Human Prostatic Carcinoma Cell Line (PC-3). Investig. Urol. 1979, 17, 16–23. [Google Scholar]
- Price, J.E.; Polyzos, A.; Zhang, R.D.; Daniels, L.M. Tumorigenicity and Metastasis of Human Breast Carcinoma Cell Lines in Nude Mice. Cancer Res. 1990, 50, 717–721. [Google Scholar]
- Slater, T.F.; Sawyer, B.; Sträuli, U. Studies on Succinate-Tetrazolium Reductase Systems: III. Points of Coupling of Four Different Tetrazolium Salts III. Points of Coupling of Four Different Tetrazolium Salts. Biochim. Biophys. Acta 1963, 77, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peterson, D.; Kimura, H.; Schubert, D. Mechanism of Cellular 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide (MTT) Reduction. J. Neurochem. 1997, 69, 581–593. [Google Scholar] [CrossRef]
- Altman, F.P. Tetrazolium Salts and Formazans. Prog. Histochem. Cytochem. 1976, 9, 1–56. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid Colorimetric Assay for Cell Growth and Survival: Modifications to the Tetrazolium Dye Procedure Giving Improved Sensitivity and Reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Reed, J.; Reed, T.A. A Set of Constructed Type Spectra for the Practical Estimation of Peptide Secondary Structure from Circular Dichroism. Anal. Biochem. 1997, 254, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Pethő, L.; Mező, G.; Schlosser, G. Overcharging Effect in Electrospray Ionization Mass Spectra of Daunomycin-Tuftsin Bioconjugates. Molecules 2019, 24, 2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | (Dau=Aoa)-BBN(7-14) Compound/BBN(7-14) Compound | RP-HPLC Rt (min) a | ESI-MS MWmeas/MWcal (g/mol) b |
|---|---|---|---|
| G1 | [Nle14] | 24.35 | 1878.8988/1879.1050 |
| G2 | [D-Phe6, Nle14] | 24.75 | 2025.9836/2026.2820 |
| G3 | [D-Phe6, β-Ala11, Aib13, Nle14] | 23.45 | 2011.9672/2012.2550 |
| G4 | [D-Phe6, β-Ala11, N-Me-Ala13, Nle14] | 23.42 | 2011.9494/2012.2550 |
| G5 | [D-Phe6, Sta13, Leu14] | 24.25 | 2069.9928/2070.3350 |
| L1 | [Nle14] | 20.93 | 2093.0528/2093.3780 |
| L2 | [D-Phe6, Nle14] | 22.22 | 2240.1192/2240.5550 |
| L3 | [D-Phe6, β-Ala11, Aib13, Nle14] | 21.23 | 2226.1036/2226.5280 |
| L4 | [D-Phe6, β-Ala11, N-Me-Ala13, Nle14] | 20.58 | 2226.1018/2226.5280 |
| L5 | [D-Phe6, Sta13, Leu14] | 22.35 | 2284.1462/2284.6080 |
| L6 | [D-Phe6, β-Ala11, Sta13, Nle14] | 21.30 | 2298.1598/2298.6350 |
| FP1 | [D-Phe6, Sta13, Leu14] | 21.76 | 1155.6368/1155.3690 |
| FP2 | [D-Phe6, β-Ala11, Sta13, Nle14] | 21.17 | 1168.6534/1169.3960 |
| Conjugate/Free Peptide | MDA-MB-231 IC50 (µM) | MDA-MB-453 IC50 (µM) | PC-3 IC50 (µM) |
|---|---|---|---|
| G1 | 22.80 ± 3.12 | 11.45 ± 1.53 | 11.83 ± 2.50 |
| G2 | 7.29 ± 3.41 | 12.72 ± 1.22 | 5.98 ± 2.10 |
| G3 | 9.28 ± 0.13 | 8.78 ± 0.97 | 4.55 ± 0.76 |
| G4 | 20.98 ± 0.55 | 7.37 ± 1.28 | 5.73 ± 0.24 |
| G5 | 18.29 ± 1.46 | 11.62 ± 3.33 | 9.69 ± 0.17 |
| L1 | 4.15 ± 0.18 | 7.87 ± 0.09 | 4.38 ± 0.33 |
| L2 | 11.74 ± 0.09 | 19.14 ± 0.49 | 8.57 ± 1.61 |
| L3 | 18.96 ± 3.23 | >25 | >25 |
| L4 | 5.31 ± 0.01 | 21.21 ± 5.36 | 4.08 ± 0.09 |
| L5 | 3.35 ± 0.32 | 5.86 ± 0.75 | 2.22 ± 0.19 |
| L6 | 9.88 ± 2.82 | 9.64 ± 0.25 | 18.04 ± 3.01 |
| FP1 | >100 | >100 | >100 |
| FP2 | >100 | >100 | >100 |
| Dau | 0.90 ± 0.06 | 0.81 ± 0.07 | 0.75 ± 0.01 |
| Conjugate | MDA-MB-231 UC50 (µM) | MDA-MB-453 UC50 (µM) | PC-3 UC50 (µM) |
|---|---|---|---|
| G1 | >25 | >25 | >25 |
| G2 | >25 | 17.04 | 10.50 |
| G3 | >25 | 22.59 | >25 |
| G4 | 23.33 | 20.22 | >25 |
| G5 | >25 | >25 | >25 |
| L1 | 18.67 | 11.63 | 22.92 |
| L2 | 18.27 | 21.58 | 20.87 |
| L3 | >25 | 24.59 | >25 |
| L4 | >25 | >25 | >25 |
| L5 | 15.87 | 8.96 | 12.35 |
| L6 | 15.47 | 4.15 | 16.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomena, J.; Vári, B.; Oláh-Szabó, R.; Biri-Kovács, B.; Bősze, S.; Borbély, A.; Soós, Á.; Ranđelović, I.; Tóvári, J.; Mező, G. Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates. Int. J. Mol. Sci. 2023, 24, 3400. https://doi.org/10.3390/ijms24043400
Gomena J, Vári B, Oláh-Szabó R, Biri-Kovács B, Bősze S, Borbély A, Soós Á, Ranđelović I, Tóvári J, Mező G. Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates. International Journal of Molecular Sciences. 2023; 24(4):3400. https://doi.org/10.3390/ijms24043400
Chicago/Turabian StyleGomena, Jacopo, Balázs Vári, Rita Oláh-Szabó, Beáta Biri-Kovács, Szilvia Bősze, Adina Borbély, Ádám Soós, Ivan Ranđelović, József Tóvári, and Gábor Mező. 2023. "Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates" International Journal of Molecular Sciences 24, no. 4: 3400. https://doi.org/10.3390/ijms24043400
APA StyleGomena, J., Vári, B., Oláh-Szabó, R., Biri-Kovács, B., Bősze, S., Borbély, A., Soós, Á., Ranđelović, I., Tóvári, J., & Mező, G. (2023). Targeting the Gastrin-Releasing Peptide Receptor (GRP-R) in Cancer Therapy: Development of Bombesin-Based Peptide–Drug Conjugates. International Journal of Molecular Sciences, 24(4), 3400. https://doi.org/10.3390/ijms24043400